
PREFMoDeL: A Systematic Review and Proposed Taxonomy of Biomolecular Features for Deep Learning

Abstract
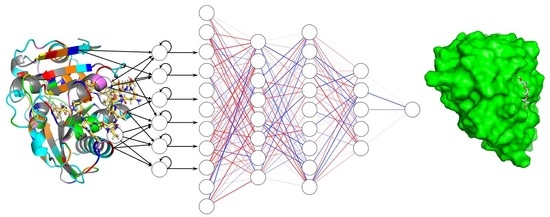
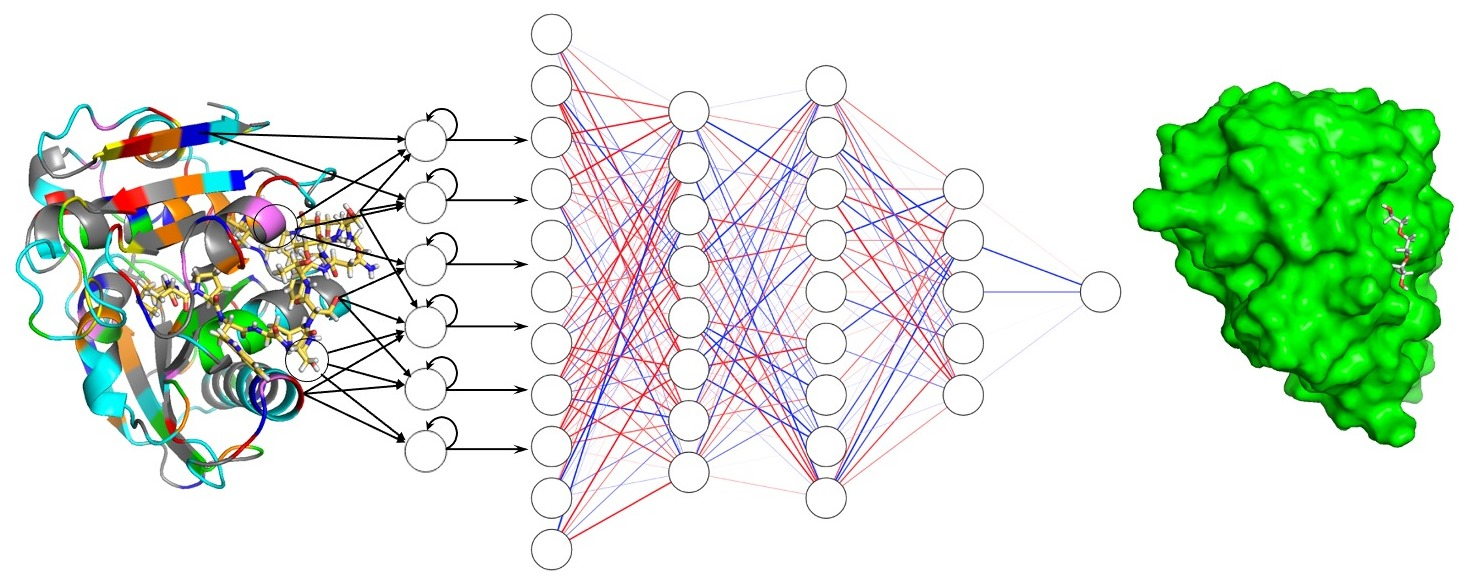
:
1. Introduction
2. Background
2.1. Vocabulary
2.2. Parts of a Machine Learning Model
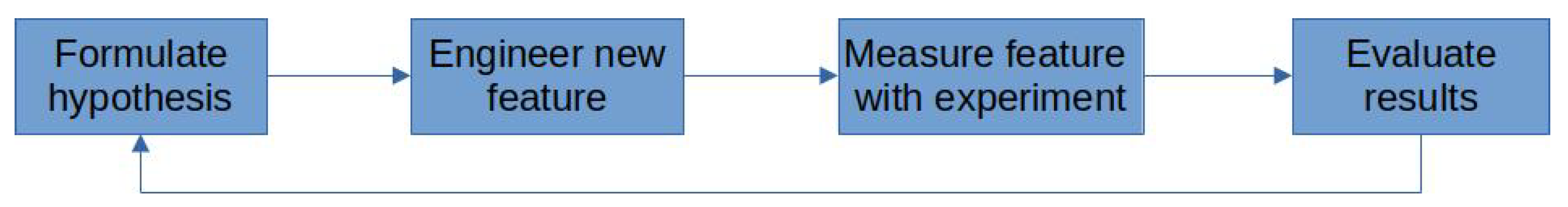
2.3. Features and Feature Engineering as Analogues to the Scientific Method
2.3.1. The Importance of Feature Relevance
2.3.2. Desirable Feature Characteristics
- Representativeness: f accurately represents the underlying phenomenon.
- Fixed dimension: f is of a fixed size, such as an appropriately shaped NumPy array.
- Continuity: f takes continuous values.
- Differentiability: f is differentiable.
- Normalizability: f is normalized or normalizable to the interval (0,1) or the interval (−1,1).
- Linearizability: If f is exponential or nonlinear, f can be linearized.
- Reversibility: f is generated by a lossless transform that can be inverted, such that .
- Example uniqueness: f is unique for a given input i. Similar to the concept of one-to-one in linear algebra.
- SE(3) invariance or SE(3) equivariance: A rotation or translation in space either does not change (invariant) or evenly changes (equivariant) the values in f, respectively.
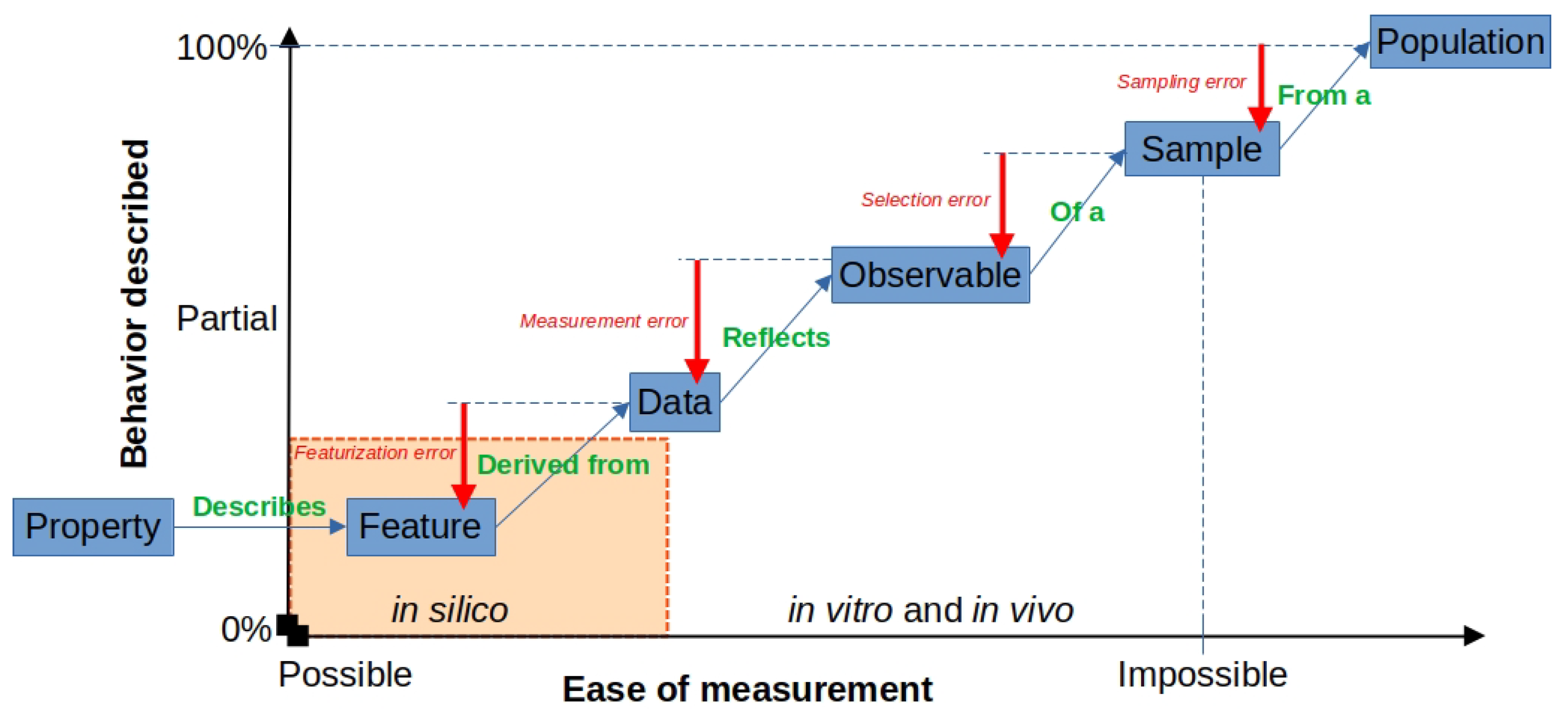
2.3.3. Errors in Feature Engineering from Physical and Computational Sources
- When a sample is measured, the particular sample chosen may not be representative of the population from which it is picked, incurring sampling error.
- The phenomena and its associated observable(s) are necessarily incomplete, because not all observables can be measured, and those that can may not be measurable in isolation from others. Therefore, the selection of which observables to measure incurs selection error.
- The data may suffer from a low signal-to-noise ratio or reflect measurement error in a sample.
- During featurization, the scientist might unknowingly neglect useful information, which further convolutes the expected behavior described in the original data, incurring featurization error. This is because the featurization process is dependent on the scientist’s discretion and bias (intended or not) to select valuable information from that dataset.
2.4. Circumventing Feature-Associated Errors by Systematic Testing
- Existing features are redesigned without insight from previous investigation(s).
- Features are applied in a context that experts in the field know to be inappropriate.
- Errors are made in feature selection that impede network training or human insight.
- Features are implemented in a CPU- or memory-inefficient manner that prevents replication.
3. Methods
3.1. Literature Curation
3.2. Feature Extraction
3.3. Analysis Tools
4. Results
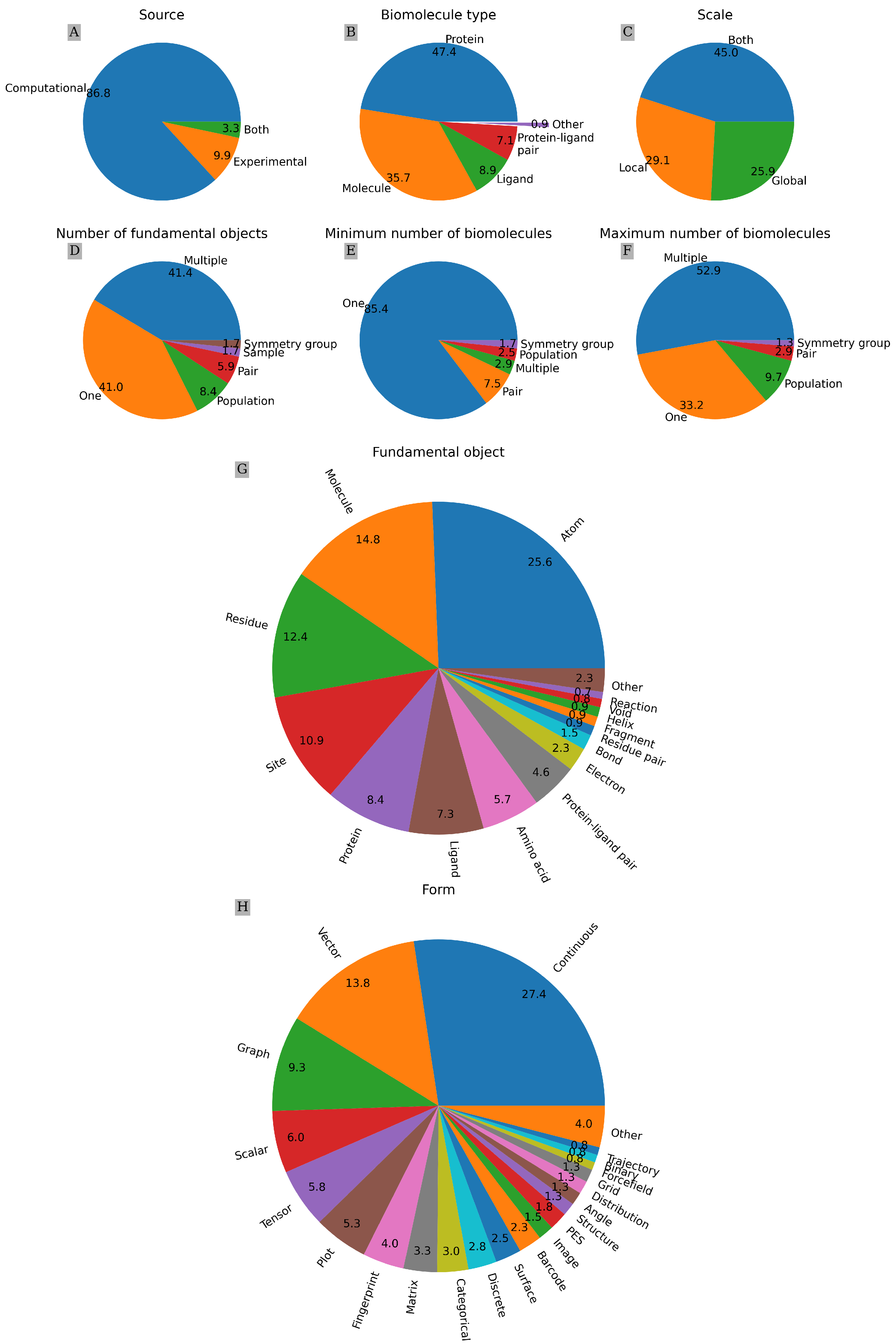
4.1. Taxonomic Charts
4.2. Categorical Properties
- Source: Either physical experiment or computational model.
- Biomolecule type: Protein, ligand, protein–ligand pair, generic molecule, or other.
- Number of biomolecules: One, pair, multiple, or population. Minimum and maximum number possible to be represented by each feature are counted as separate properties.
- Fundamental object: Smallest object considered explicitly, e.g., an atom or an amino acid.
- Number of fundamental objects: One, pair, multiple, symmetry group, sample, or population
- Form: Data structure, such as a scalar, matrix, or graph.
- Scale: Local, global, or both.
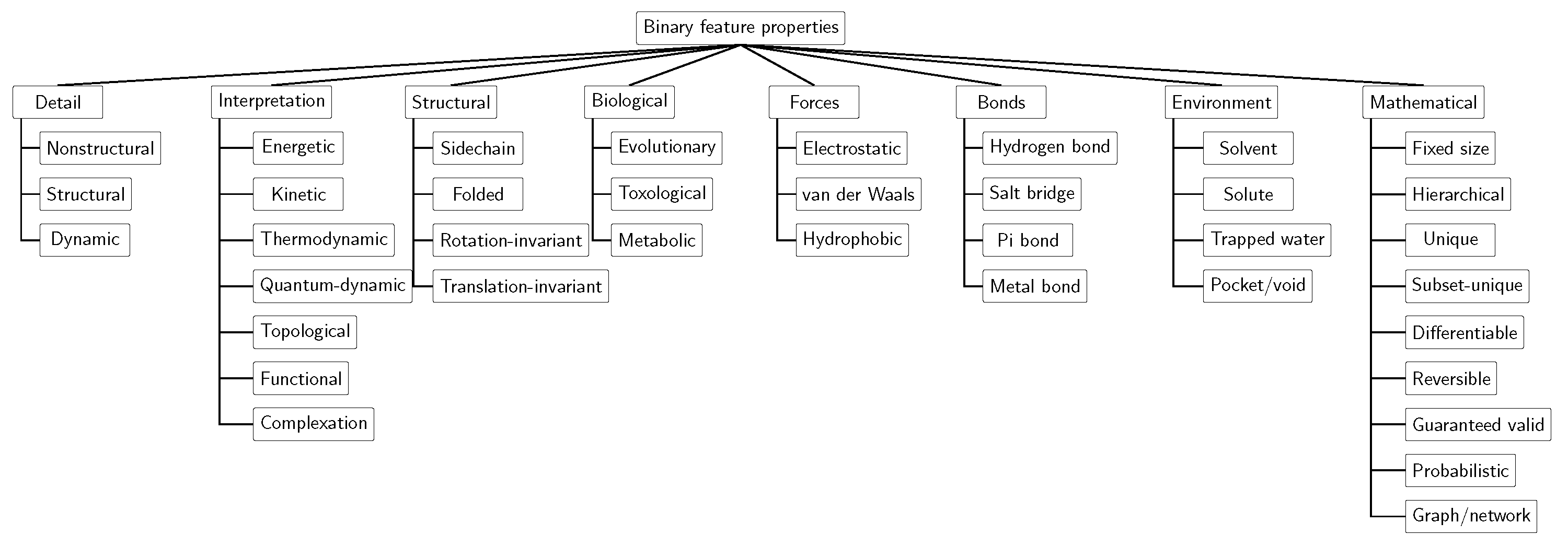
4.3. Binary Properties
- Detail: Nonstructural, structural, and dynamical.
- Interpretation: Energetic, kinetic, thermodynamic, quantum dynamic, topological, functional, and complexation.
- Structural: Sidechain atoms, macromolecular folding, rotation invariance, and translation equivariance.
- Biological interpretation: Evolutionary, toxological, and metabolic.
- Forces: van der Waals, electrostatic, and hydrophobic.
- Chemical bond: Hydrogen bond, salt bridge, pi bond, and metal bond.
- Environment: Solvent, solute, trapped waters, and pockets/voids.
- Mathematical features: Fixed size, hierarchical, unique, subset-unique, differentiable, reversible, guaranteed valid, probabilistic, and graph/network.
4.4. Structural-Feature-Specific Properties
- Structural comparison: Generated by the comparison of two structures, e.g., RMSD.
4.5. Dynamical-Feature-Specific Properties
- Dynamic perspective: State-based or transition-based.
- Structure set type: Ensemble or trajectory.
- Probability of dynamics: Most-probable dynamics or rare-event dynamics.
- Considered states: All states or only relevant states.
- Period timescale: Thermal noise, functional motion, or evolutionary.
- Trajectory bias: From a biased simulation, or an unbiased one.
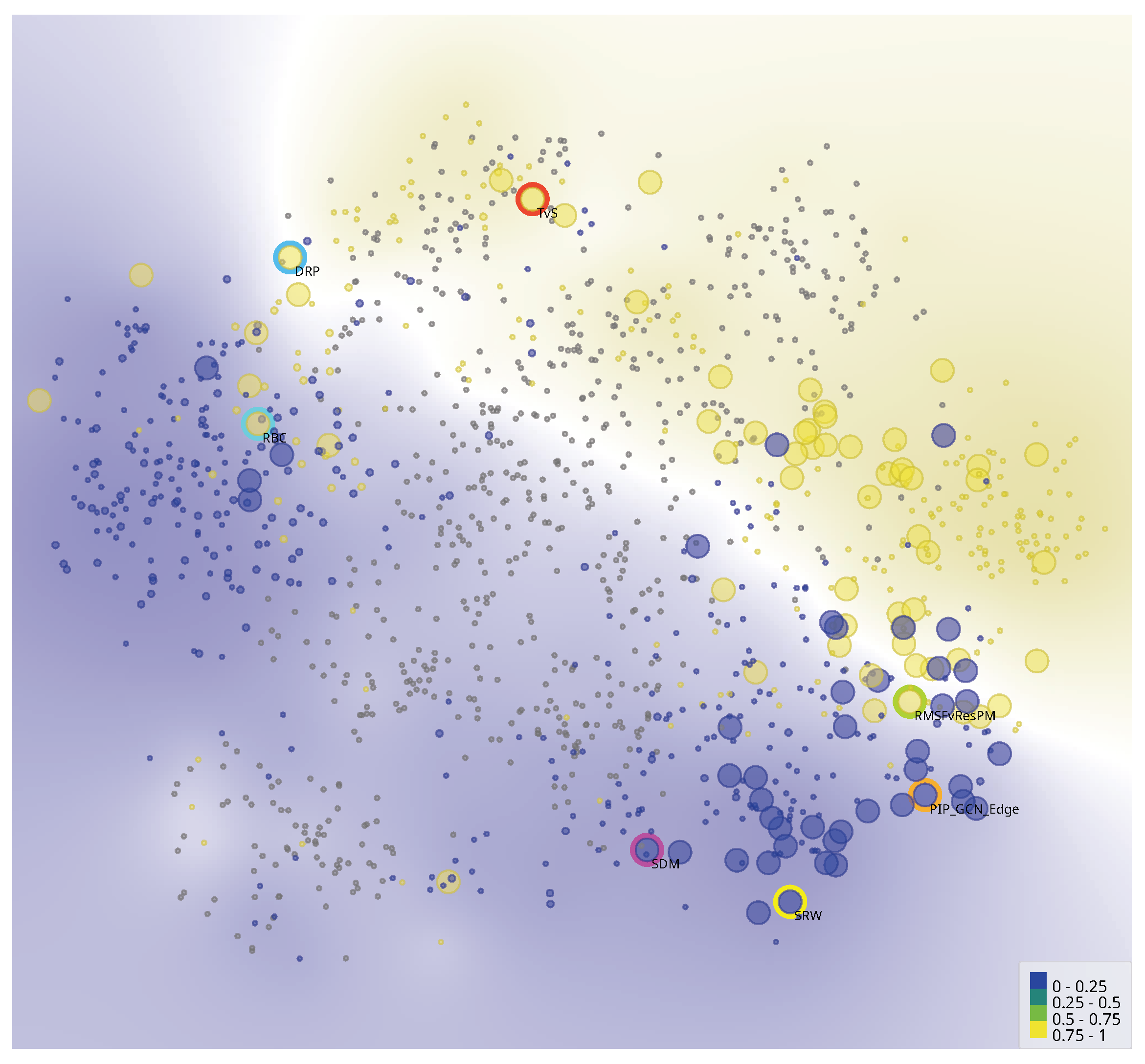
5. Analysis
6. Discussion
6.1. A Note on Feature Characterization Error
6.2. Directions for Future Work
6.2.1. Feature Relevance
6.2.2. Underrepresented Taxa
- Residue-Specific Atomic Subsets. Previous work by Cang and Wei included three atomic structural partitions [54]. In one representation, they included the entire structure for atom partitioning, in a second representation they only included primary and secondary shell residues in the binding site, and in a third representation they selected subsets representing the interactions between two atomic elements. However, this approach excludes allosteric networks and amino acid level properties. To alleviate this unintended bias, scientists should partition structures by residue properties and by computationally or experimentally elucidated allosteric residue interaction networks.
- Amino acid specific features. Currently, few amino acid specific representations exist, thus this is a major growth area for protein-specific structural dynamic features. As mentioned, amino acid motion (and their restriction, such as in active site preorganization) is essential for protein function, although without the tools to experimentally measure amino acid motion, insights cannot be drawn from amino acid dynamics. New features should address amino acid specific dynamics, and extend these dynamic models beyond the common but rudimentary Principal Component Analysis (PCA) to describe more complex motions with simple equations.
- Structural trajectories: “dynamicalization” of structural features. It would be computationally trivial to expand all structural features to dynamic structural features through molecular dynamics simulation. Recent work has improved the efficiency and analysis of these simulations by integration with DL modeling [55,56] and given guidance to practitioners for which models they should use with their systems of study [57].
- Evolutionary trajectories. In order to make evolutionary methods more accessible and useful and to take advantage of existing evolutionary knowledge, many desired features can be computed along an evolutionary trajectory. This is accessible for most computational methods and may be powerfully applied to evolutionary design tasks.
6.2.3. Deduplication and Expansion of Feature Properties
6.2.4. Potential New Features for Molecular Deep Learning
- Kinetics-Weighted and Multi-Transition State Sequences. The consensus structure method described by Smith et al. [58] is an excellent advance in enzyme design, but can be slightly modified to incorporate more dynamic detail. Each transition state’s contribution can be weighted by the inverse of the experimentally determined forward kinetic constant for that particular reaction step. This adjustment would enable the slowest reaction barriers to exert the greatest influence on the consensus structure. Secondly, multiple transition states might be considered rather than creating a consensus structure. This can be accomplished by optimizing multiple states and their transitions by using fast protein motions, such as sidechain rearrangements, cofactor vibrations, and other atom-scale dynamics. These two features, coupled with methods developed to utilize them to the fullest extent, would enhance structure- and dynamics-based enzyme engineering efforts.
- Bond strain vector. A bond strain vector would describe bond strain for every bond in a protein or ligand from theoretically ideal values. Such a feature can be visualized as a one-dimensional vector where each index in the vector holds the energy of the indexed bond in kilojoules per mole. Through application of Hooke’s law and idealized bond geometries, bond energy deviations are easily computed. Then, they can be visualized on a protein structure, or for a ligand, bound in an active site. Such a feature is extensible to analogues for angular strain (between three atoms) or torsional strain (between four atoms) which together describe the system in more dynamic and mechanical detail. This feature would illustrate where strain is present in a protein or a ligand structure and may prove important in deep learning tasks concerning function such as recognition, binding, and catalysis [59].
- Amino acid sidechain normal modes. Conformational plasticity, from high-frequency rate-promoting vibrations to sidechain rotamer sampling to large domain motion, is often required to understand protein function [60,61,62,63]. This is especially relevant on the scale of individual amino acid sidechains within a binding pocket, as they are often subject to rapid structural rearrangements to enable precise biochemical function. Bonk et al. simulated enzyme active site dynamics with restricted conformational mobility to determine which structural features activate catalysis [64]. The ability to compute other such elementary features may lead to widely interpretable insight when applied to novel systems. For example, single amino acid motions are critical for the substrate IP7 to enter the active site of PPIP5K2. Here, a single residue glutamine-192 utilizes spontaneous motion and the irreversibility of reverse substrate diffusion to ratchet IP7 from a capture site to the active site [65]. These case studies demonstrate the value of the automatic extraction of this detail to sense amino acid motions for analysis or design purposes. While alpha carbon PCA is a popular model to enable quick visually interpretable dynamic structural features for a gross protein structure traveling through an ensemble or trajectory [66], no analogous feature exists for substructures such as sidechains within the protein. In order to compute such a feature, one would fix a reference frame at each alpha carbon and measure wagging or spinning modes for each amino acid along with their frequencies using PCA or nonlinear dimensionality reduction techniques. Next, these modes could be projected onto the larger protein structure and analyzed for their correspondence with the more slowly relaxing backbone modes produced by alpha carbon PCA. These collections of principal components can then be used to construct features assuming either independence of motion or any particular dependence, based on correlations in molecular dynamics trajectories or NMR-derived ensembles. By repeating this process for all non-glycine amino acids in a structure, these “amino acid sidechain normal modes” could reveal how sums of large and small dynamic principal components enable protein function on multiple scales.
- Docking strength for interaction specificity and uniqueness. Specificity in biomolecular recognition and binding is a composite problem involving the maximization of on-target interactions and minimization of off-target ones. Because so many off-target molecules exist in the cell, this is challenging to solve analytically. However, methods that measure the strength of binding between two proteins raise the potential to initiate examination of this space. Such a method could be docking-based, but may be efficiently implemented by using DL to reduce docking strength determination to a sequence of matrix multiplications. Using such a method, ostensibly a “kinetic association constant regressor network”, off-target interactions could be minimized for each non-target endogenous protein, while on-target interactions would be maximized. Such a solution would be a first step towards minimizing unintentional drug side effects without requiring manual testing of all possible cross interactions in the cell.
6.2.5. Automated Feature Engineering and the Need for Functional Data
6.2.6. Unsupervised Learning Visualization for Dataset Exploration
6.2.7. Feature Implementations and Future Integrations
6.2.8. Publicly Available Biophysical Datasets
7. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AI | Artificial Intelligence |
| API | Application Programming Interface |
| BMR | Binding Mode Representation |
| BMRB | Biological Magnetic Resonance Database |
| CASF | Comparative Assessment of Scoring Functions |
| CCE | Categorical Cross Entropy |
| CNN | Convolutional Neural Network |
| CPU | Central Processing Unit |
| CSV | Comma Separated Value |
| DL | Deep Learning |
| DRIN | Dynamic Residue Interaction Network |
| EMDB | Electron Microscopy Database |
| EMPIAR | Electron microscopy public image archive |
| EPR | Electron Paramagnetic Resonance |
| FRET | Förster Resonance Energy Transfer |
| GNN | Graph Neural Network |
| MD | Molecular Dynamics |
| ML | Machine Learning |
| ND | n-Dimensional |
| PCA | Principal Component Analysis |
| PDB | Protein Data Bank |
| Portable Document Format | |
| PREFMoDeL | Public Repository of Engineered Features for Molecular Deep Learning |
| SASBDB | Small Angle Scattering Biological Data Bank |
| SGD | Stochastic Gradient Descent |
| SWIF | Surface-Weighted Interaction Fingerprint |
| t-SNE | t-distributed Stochastic Neighbor Embedding |
| URL | Uniform Resource Locator |
References
- Drews, J. Drug Discovery: A Historical Perspective. Science 2000, 287, 1960–1964. [Google Scholar] [CrossRef]
- Vincent, F.; Nueda, A.; Lee, J.; Schenone, M.; Prunotto, M.; Mercola, M. Phenotypic drug discovery: Recent successes, lessons learned and new directions. Nat. Rev. Drug Discov. 2022, 21, 899–914. [Google Scholar] [CrossRef]
- Dara, S.; Dhamercherla, S.; Jadav, S.S.; Babu, C.M.; Ahsan, M.J. Machine Learning in Drug Discovery: A Review. Artif. Intell. Rev. 2022, 55, 1947–1999. [Google Scholar] [CrossRef] [PubMed]
- Schneider, P.; Walters, W.P.; Plowright, A.T.; Sieroka, N.; Listgarten, J.; Goodnow, R.A.; Fisher, J.; Jansen, J.M.; Duca, J.S.; Rush, T.S.; et al. Rethinking drug design in the artificial intelligence era. Nat. Rev. Drug Discov. 2020, 19, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Long, A.a.W.; Nayler, J.H.C.; Smith, H.; Taylor, T.; Ward, N. Derivatives of 6-aminopenicillanic acid. Part XI. α-Amino-p-hydroxybenzylpenicillin. J. Chem. Soc. Org. 1971, 1920–1922. [Google Scholar] [CrossRef]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef]
- Jumper, J.; Hassabis, D. Protein structure predictions to atomic accuracy with AlphaFold. Nat. Methods 2022, 19, 11–12. [Google Scholar] [CrossRef]
- Dauparas, J.; Anishchenko, I.; Bennett, N.; Bai, H.; Ragotte, R.J.; Milles, L.F.; Wicky, B.I.M.; Courbet, A.; Haas, R.J.d.; Bethel, N.; et al. Robust deep learning based protein sequence design using ProteinMPNN. Science 2022, 378, 49–56. [Google Scholar] [CrossRef]
- Arunachalam, P.S.; Walls, A.C.; Golden, N.; Atyeo, C.; Fischinger, S.; Li, C.; Aye, P.; Navarro, M.J.; Lai, L.; Edara, V.V.; et al. Adjuvanting a subunit COVID-19 vaccine to induce protective immunity. Nature 2021, 594, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Peón, A.; Naulaerts, S.; Ballester, P.J. Predicting the Reliability of Drug-target Interaction Predictions with Maximum Coverage of Target Space. Sci. Rep. 2017, 7, 3820. [Google Scholar] [CrossRef]
- Cerisier, N.; Petitjean, M.; Regad, L.; Bayard, Q.; Réau, M.; Badel, A.; Camproux, A.C. High Impact: The Role of Promiscuous Binding Sites in Polypharmacology. Molecules 2019, 24, 2529. [Google Scholar] [CrossRef] [PubMed]
- Blaschke, T.; Feldmann, C.; Bajorath, J. Prediction of Promiscuity Cliffs Using Machine Learning. Mol. Informatics 2021, 40, 2000196. [Google Scholar] [CrossRef] [PubMed]
- Feldmann, C.; Bajorath, J. Machine learning reveals that structural features distinguishing promiscuous and non-promiscuous compounds depend on target combinations. Sci. Rep. 2021, 11, 7863. [Google Scholar] [CrossRef]
- Gilberg, E.; Gütschow, M.; Bajorath, J. Promiscuous Ligands from Experimentally Determined Structures, Binding Conformations, and Protein Family-Dependent Interaction Hotspots. ACS Omega 2019, 4, 1729–1737. [Google Scholar] [CrossRef] [PubMed]
- Wigh, D.S.; Goodman, J.M.; Lapkin, A.A. A review of molecular representation in the age of machine learning. Comput. Mol. Sci. 2022, 12, e1603. [Google Scholar] [CrossRef]
- Friederich, P.; Krenn, M.; Tamblyn, I.; Aspuru-Guzik, A. Scientific intuition inspired by machine learning-generated hypotheses. Mach. Learn. Sci. Technol. 2021, 2, 025027. [Google Scholar] [CrossRef]
- Gu, J.; Wang, Z.; Kuen, J.; Ma, L.; Shahroudy, A.; Shuai, B.; Liu, T.; Wang, X.; Wang, G.; Cai, J.; et al. Recent advances in convolutional neural networks. Pattern Recognit. 2018, 77, 354–377. [Google Scholar] [CrossRef]
- Wu, Z.; Pan, S.; Chen, F.; Long, G.; Zhang, C.; Yu, P.S. A Comprehensive Survey on Graph Neural Networks. IEEE Trans. Neural Networks Learn. Syst. 2021, 32, 4–24. [Google Scholar] [CrossRef]
- Vaswani, A.; Shazeer, N.; Parmar, N.; Uszkoreit, J.; Jones, L.; Gomez, A.N.; Kaiser, L.; Polosukhin, I. Attention Is All You Need. arXiv 2017, arXiv:1706.03762. [Google Scholar]
- Wang, Z.; Liu, M.; Luo, Y.; Xu, Z.; Xie, Y.; Wang, L.; Cai, L.; Qi, Q.; Yuan, Z.; Yang, T.; et al. Advanced graph and sequence neural networks for molecular property prediction and drug discovery. Bioinformatics 2022, 38, 2579–2586. [Google Scholar] [CrossRef]
- Fang, X.; Liu, L.; Lei, J.; He, D.; Zhang, S.; Zhou, J.; Wang, F.; Wu, H.; Wang, H. Geometry-enhanced molecular representation learning for property prediction. Nat. Mach. Intell. 2022, 4, 127–134. [Google Scholar] [CrossRef]
- Wang, Q.; Ma, Y.; Zhao, K.; Tian, Y. A Comprehensive Survey of Loss Functions in Machine Learning. Ann. Data Sci. 2022, 9, 187–212. [Google Scholar] [CrossRef]
- Ciampiconi, L.; Elwood, A.; Leonardi, M.; Mohamed, A.; Rozza, A. A survey and taxonomy of loss functions in machine learning. arXiv 2023, arXiv:2301.05579. [Google Scholar]
- Chauvin, Y.; Rumelhart, D.E. (Eds.) Backpropagation: Theory, Architectures, and Applications; Psychology Press: London, UK, 1995. [Google Scholar] [CrossRef]
- Lillicrap, T.P.; Santoro, A.; Marris, L.; Akerman, C.J.; Hinton, G. Backpropagation and the brain. Nat. Rev. Neurosci. 2020, 21, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Abdolrasol, M.G.M.; Hussain, S.M.S.; Ustun, T.S.; Sarker, M.R.; Hannan, M.A.; Mohamed, R.; Ali, J.A.; Mekhilef, S.; Milad, A. Artificial Neural Networks Based Optimization Techniques: A Review. Electronics 2021, 10, 2689. [Google Scholar] [CrossRef]
- AlQuraishi, M.; Sorger, P.K. Differentiable biology: Using deep learning for biophysics-based and data-driven modeling of molecular mechanisms. Nat. Methods 2021, 18, 1169–1180. [Google Scholar] [CrossRef]
- König, G.; Molnar, C.; Bischl, B.; Grosse-Wentrup, M. Relative Feature Importance. In Proceedings of the 25th International Conference on Pattern Recognition (ICPR), Milan, Italy, 10–15 January 2021; pp. 9318–9325. [Google Scholar] [CrossRef]
- Dhal, P.; Azad, C. A comprehensive survey on feature selection in the various fields of machine learning. Appl. Intell. 2022, 52, 4543–4581. [Google Scholar] [CrossRef]
- Bouchlaghem, Y.; Akhiat, Y.; Amjad, S. Feature Selection: A Review and Comparative Study. E3S Web Conf. 2022, 351, 01046. [Google Scholar] [CrossRef]
- Noé, F.; Tkatchenko, A.; Müller, K.R.; Clementi, C. Machine Learning for Molecular Simulation. Annu. Rev. Phys. Chem. 2020, 71, 361–390. [Google Scholar] [CrossRef]
- Haghighatlari, M.; Li, J.; Heidar-Zadeh, F.; Liu, Y.; Guan, X.; Head-Gordon, T. Learning to Make Chemical Predictions: The Interplay of Feature Representation, Data, and Machine Learning Methods. Chem 2020, 6, 1527–1542. [Google Scholar] [CrossRef]
- George, J.; Hautier, G. Chemist versus Machine: Traditional Knowledge versus Machine Learning Techniques. Trends Chem. 2021, 3, 86–95. [Google Scholar] [CrossRef]
- Kumar, V.; Minz, S. Feature Selection: A Literature Review. Smart Comput. Rev. 2014, 4, 211–229. [Google Scholar] [CrossRef]
- Liberati, A.; Altman, D.G.; Tetzlaff, J.; Mulrow, C.; Gøtzsche, P.C.; Ioannidis, J.P.A.; Clarke, M.; Devereaux, P.J.; Kleijnen, J.; Moher, D. The PRISMA statement for reporting systematic reviews and meta-analyses of studies that evaluate healthcare interventions: Explanation and elaboration. BMJ 2009, 339, b2700. [Google Scholar] [CrossRef] [PubMed]
- Demšar, J.; Curk, T.; Erjavec, A.; Črt Gorup; Hočevar, T.; Milutinovič, M.; Možina, M.; Polajnar, M.; Toplak, M.; Starič, A.; et al. Orange: Data Mining Toolbox in Python. J. Mach. Learn. Res. 2013, 14, 2349–2353. [Google Scholar]
- Hinton, G.E.; Roweis, S. Stochastic Neighbor Embedding. In Advances in Neural Information Processing Systems; MIT Press: Cambridge, MA, USA, 2002; Volume 15, pp. 857–864. [Google Scholar]
- Maaten, L.v.d.; Hinton, G. Visualizing Data using t-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2605. [Google Scholar]
- Miller, B.K.; Geiger, M.; Smidt, T.E.; Noé, F. Relevance of Rotationally Equivariant Convolutions for Predicting Molecular Properties. arXiv 2020, arXiv:2008.08461. [Google Scholar]
- Fuchs, F.B.; Worrall, D.E.; Fischer, V.; Welling, M. SE(3)-Transformers: 3D Roto-Translation Equivariant Attention Networks. In Proceedings of the 34th International Conference on Neural Information Processing Systems, Vancouver, BC, Canada, 6–12 December 2020; pp. 1970–1981. [Google Scholar]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Liu, Z.; Li, Y.; Han, L.; L, J.; Liu, J.; Zhao, Z.; Nie, W.; Yuchen, L.; Wang, R. PDB-wide Collection of Binding Data: Current Status of the PDBbind Database. Bioinformatics 2014, 31, 405–412. [Google Scholar] [CrossRef]
- Bourgeat, L.; Serghei, A.; Lesieur, C. Experimental Protein Molecular Dynamics: Broadband Dielectric Spectroscopy coupled with nanoconfinement. Sci. Rep. 2019, 9, 17988. [Google Scholar] [CrossRef]
- Pradeepkiran, J.; Reddy, P. Structure Based Design and Molecular Docking Studies for Phosphorylated Tau Inhibitors in Alzheimer’s Disease. Cells 2019, 8, 260. [Google Scholar] [CrossRef]
- Fout, A.; Byrd, J.; Shariat, B.; Ben-Hur, A. Protein Interface Prediction using Graph Convolutional Networks. In NIPS’17: Proceedings of the 31st International Conference on Neural Information Processing Systems; MIT Press: Cambridge, MA, USA, 2017; p. 10. [Google Scholar]
- Meng, Z.; Xia, K. Persistent spectral–based machine learning (PerSpect ML) for protein-ligand binding affinity prediction. Sci. Adv. 2021, 7, eabc5329. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Perilla, J.R.; Ning, J.; Lu, M.; Hou, G.; Ramalho, R.; Himes, B.A.; Zhao, G.; Bedwell, G.J.; Byeon, I.J.; et al. Cyclophilin A stabilizes the HIV-1 capsid through a novel non-canonical binding site. Nat. Commun. 2016, 7, 10714. [Google Scholar] [CrossRef]
- Wicker, J.G.; Cooper, R.I. Beyond Rotatable Bond Counts: Capturing 3D Conformational Flexibility in a Single Descriptor. J. Chem. Inf. Model. 2016, 56, 2347–2352. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, J.; Wei, R.; Oeser, T.; Belisário-Ferrari, M.R.; Barth, M.; Then, J.; Zimmermann, W. Effect of Tris, MOPS, and phosphate buffers on the hydrolysis of polyethylene terephthalate films by polyester hydrolases. FEBS Open Bio 2016, 6, 919–927. [Google Scholar] [CrossRef] [PubMed]
- Dienes, Z.; Perner, J. A theory of implicit and explicit knowledge. Behav. Brain Sci. 1999, 22, 735–808. [Google Scholar] [CrossRef]
- Smith, J.D.; Berg, M.E.; Cook, R.G.; Murphy, M.S.; Crossley, M.J.; Boomer, J.; Spiering, B.; Beran, M.J.; Church, B.A.; Ashby, F.G.; et al. Implicit and explicit categorization: A tale of four species. Neurosci. Biobehav. Rev. 2012, 36, 2355–2369. [Google Scholar] [CrossRef] [PubMed]
- Lian, J.; Zhou, X.; Zhang, F.; Chen, Z.; Xie, X.; Sun, G. xDeepFM: Combining Explicit and Implicit Feature Interactions for Recommender Systems. In Proceedings of the 24th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining, London, UK, 19–23 August 2018; pp. 1754–1763. [Google Scholar] [CrossRef]
- Su, M.; Yang, Q.; Du, Y.; Feng, G.; Liu, Z.; Li, Y.; Wang, R. Comparative Assessment of Scoring Functions: The CASF-2016 Update. J. Chem. Inf. Model. 2019, 59, 895–913. [Google Scholar] [CrossRef]
- Duy Nguyen, D.; Cang, Z.; Wei, G.W. A review of mathematical representations of biomolecular data. Phys. Chem. Chem. Phys. 2020, 22, 4343–4367. [Google Scholar] [CrossRef]
- Wang, Y.; Lamim Ribeiro, J.M.; Tiwary, P. Machine learning approaches for analyzing and enhancing molecular dynamics simulations. Curr. Opin. Struct. Biol. 2020, 61, 139–145. [Google Scholar] [CrossRef]
- Doerr, S.; Majewski, M.; Pérez, A.; Krämer, A.; Clementi, C.; Noe, F.; Giorgino, T.; De Fabritiis, G. TorchMD: A Deep Learning Framework for Molecular Simulations. J. Chem. Theory Comput. 2021, 17, 2355–2363. [Google Scholar] [CrossRef]
- Pinheiro, M.; Ge, F.; Ferré, N.; O. Dral, P.; Barbatti, M. Choosing the right molecular machine learning potential. Chem. Sci. 2021, 12, 14396–14413. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.J.T.; Müller, R.; Toscano, M.D.; Kast, P.; Hellinga, H.W.; Hilvert, D.; Houk, K.N. Structural Reorganization and Preorganization in Enzyme Active Sites: Comparisons of Experimental and Theoretically Ideal Active Site Geometries in the Multistep Serine Esterase Reaction Cycle. J. Am. Chem. Soc. 2008, 130, 15361–15373. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, M.R.; Tlusty, T.; Leibler, S. Strain analysis of protein structures and low dimensionality of mechanical allosteric couplings. Proc. Natl. Acad. Sci. USA 2016, 113, E5847–E5855. [Google Scholar] [CrossRef]
- Eisenmesser, E.Z.; Millet, O.; Labeikovsky, W.; Korzhnev, D.M.; Wolf-Watz, M.; Bosco, D.A.; Skalicky, J.J.; Kay, L.E.; Kern, D. Intrinsic dynamics of an enzyme underlies catalysis. Nature 2005, 438, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Schramm, V.L.; Schwartz, S.D. Promoting Vibrations and the Function of Enzymes. Emerging Theoretical and Experimental Convergence. Biochemistry 2018, 57, 3299–3308. [Google Scholar] [CrossRef]
- Chalopin, Y.; Sparfel, J. Energy Bilocalization Effect and the Emergence of Molecular Functions in Proteins. Front. Mol. Biosci. 2021, 8, 736376. [Google Scholar] [CrossRef]
- Pagano, P.; Guo, Q.; Ranasinghe, C.; Schroeder, E.; Robben, K.; Häse, F.; Ye, H.; Wickersham, K.; Aspuru-Guzik, A.; Major, D.T.; et al. Oscillatory Active-site Motions Correlate with Kinetic Isotope Effects in Formate Dehydrogenase. ACS Catal. 2019, 9, 11199. [Google Scholar] [CrossRef]
- Bonk, B.M.; Weis, J.W.; Tidor, B. Machine Learning Identifies Chemical Characteristics That Promote Enzyme Catalysis. J. Am. Chem. Soc. 2019, 141, 4108–4118. [Google Scholar] [CrossRef]
- An, Y.; Jessen, H.J.; Wang, H.; Shears, S.B.; Kireev, D. Dynamics of Substrate Processing by PPIP5K2, a Versatile Catalytic Machine. Structure 2019, 27, 1022–1028.e2. [Google Scholar] [CrossRef]
- Zhang, S.; Krieger, J.M.; Zhang, Y.; Kaya, C.; Kaynak, B.; Mikulska-Ruminska, K.; Doruker, P.; Li, H.; Bahar, I. ProDy 2.0: Increased Scale and Scope after 10 Years of Protein Dynamics Modelling with Python. Bioinformatics 2021, 37, 3657–3659. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, J.; Cao, Z.; Barati Farimani, A. Molecular contrastive learning of representations via graph neural networks. Nat. Mach. Intell. 2022, 4, 279–287. [Google Scholar] [CrossRef]
- Gallegos, L.C.; Luchini, G.; St. John, P.C.; Kim, S.; Paton, R.S. Importance of Engineered and Learned Molecular Representations in Predicting Organic Reactivity, Selectivity, and Chemical Properties. Acc. Chem. Res. 2021, 54, 827–836. [Google Scholar] [CrossRef] [PubMed]
- Ramsundar, B.; Eastman, P.; Walters, P.; Pande, V.; Leswing, K.; Wu, Z. Deep Learning for the Life Sciences; O’Reilly Media: Sebastopol, CA, USA; Beijing, China; Tokyo, Japan, 2019. [Google Scholar]
- Jamasb, A.R.; Lió, P.; Blundell, T.L. Graphein—a Python Library for Geometric Deep Learning and Network Analysis on Protein Structures. bioRxiv 2020. [Google Scholar] [CrossRef]
- Al-Tashi, Q.; Abdulkadir, S.J.; Rais, H.M.; Mirjalili, S.; Alhussian, H. Approaches to Multi-Objective Feature Selection: A Systematic Literature Review. IEEE Access 2020, 8, 125076–125096. [Google Scholar] [CrossRef]
- Abdollahzadeh, B.; Gharehchopogh, F.S. A multi-objective optimization algorithm for feature selection problems. Eng. Comput. 2022, 38, 1845–1863. [Google Scholar] [CrossRef]
- Chen, R.C.; Dewi, C.; Huang, S.W.; Caraka, R.E. Selecting critical features for data classification based on machine learning methods. J. Big Data 2020, 7, 52. [Google Scholar] [CrossRef]
- Zhu, G.; Xu, Z.; Guo, X.; Yuan, C.; Huang, Y. DIFER: Differentiable Automated Feature Engineering. arXiv 2021, arXiv:2010.08784. [Google Scholar]
- Gada, M.; Haria, Z.; Mankad, A.; Damania, K.; Sankhe, S. Automated Feature Engineering and Hyperparameter Optimization for Machine Learning. In Proceedings of the 2021 7th International Conference on Advanced Computing and Communication Systems (ICACCS), Coimbatore, India, 19–20 March 2021; Volume 1, pp. 981–986. [Google Scholar] [CrossRef]
- Chatzimparmpas, A.; Martins, R.M.; Kucher, K.; Kerren, A. FeatureEnVi: Visual Analytics for Feature Engineering Using Stepwise Selection and Semi-Automatic Extraction Approaches. IEEE Trans. Vis. Comput. Graph. 2022, 28, 1773–1791. [Google Scholar] [CrossRef] [PubMed]
- McGibbon, R.; Beauchamp, K.; Harrigan, M.; Klein, C.; Swails, J.; Hernández, C.; Schwantes, C.; Wang, L.P.; Lane, T.; Pande, V. MDTraj: A Modern Open Library for the Analysis of Molecular Dynamics Trajectories. Biophys. J. 2015, 109, 1528–1532. [Google Scholar] [CrossRef]
- Beauchamp, K.A.; Bowman, G.R.; Lane, T.J.; Maibaum, L.; Haque, I.S.; Pande, V.S. MSMBuilder2: Modeling Conformational Dynamics on the Picosecond to Millisecond Scale. J. Chem. Theory Comput. 2011, 7, 3412–3419. [Google Scholar] [CrossRef]
- Scherer, M.K.; Trendelkamp-Schroer, B.; Paul, F.; Pérez-Hernández, G.; Hoffmann, M.; Plattner, N.; Wehmeyer, C.; Prinz, J.H.; Noé, F. PyEMMA 2: A Software Package for Estimation, Validation, and Analysis of Markov Models. J. Chem. Theory Comput. 2015, 11, 5525–5542. [Google Scholar] [CrossRef] [PubMed]
- Rombach, R.; Blattmann, A.; Lorenz, D.; Esser, P.; Ommer, B. High-Resolution Image Synthesis with Latent Diffusion Models. arXiv 2022, arXiv:2112.10752. [Google Scholar]
- OpenAI. ChatGPT: Optimizing Language Models for Dialogue. Available online: https://openai.com/blog/chatgpt/ (accessed on 4 February 2023).
- Chowdhury, R.; Bouatta, N.; Biswas, S.; Floristean, C.; Kharkar, A.; Roy, K.; Rochereau, C.; Ahdritz, G.; Zhang, J.; Church, G.M.; et al. Single-sequence protein structure prediction using a language model and deep learning. Nat. Biotechnol. 2022, 40, 1617–1623. [Google Scholar] [CrossRef]
- Leinonen, R.; Diez, F.G.; Binns, D.; Fleischmann, W.; Lopez, R.; Apweiler, R. UniProt archive. Bioinformatics 2004, 20, 3236–3237. [Google Scholar] [CrossRef]
- Zhong, E.D.; Bepler, T.; Berger, B.; Davis, J.H. CryoDRGN: Reconstruction of heterogeneous cryo-EM structures using neural networks. Nat. Methods 2021, 18, 176–185. [Google Scholar] [CrossRef]
- Iudin, A.; Korir, P.K.; Somasundharam, S.; Weyand, S.; Cattavitello, C.; Fonseca, N.; Salih, O.; Kleywegt, G.; Patwardhan, A. EMPIAR: The Electron Microscopy Public Image Archive. Nucleic Acids Res. 2023, 51, D1503–D1511. [Google Scholar] [CrossRef]
- Jamali, K.; Kimanius, D.; Scheres, S. ModelAngelo: Automated Model Building in Cryo-EM Maps. arXiv 2022, arXiv:2210.00006. [Google Scholar]
- Lawson, C.L.; Patwardhan, A.; Baker, M.L.; Hryc, C.; Garcia, E.S.; Hudson, B.P.; Lagerstedt, I.; Ludtke, S.J.; Pintilie, G.; Sala, R.; et al. EMDataBank unified data resource for 3DEM. Nucleic Acids Res. 2016, 44, D396–D403. [Google Scholar] [CrossRef]
- Ulrich, E.L.; Akutsu, H.; Doreleijers, J.F.; Harano, Y.; Ioannidis, Y.E.; Lin, J.; Livny, M.; Mading, S.; Maziuk, D.; Miller, Z.; et al. BioMagResBank. Nucleic Acids Res. 2007, 36, D402–D408. [Google Scholar] [CrossRef]
- Valentini, E.; Kikhney, A.G.; Previtali, G.; Jeffries, C.M.; Svergun, D.I. SASBDB, a repository for biological small-angle scattering data. Nucleic Acids Res. 2015, 43, D357–D363. [Google Scholar] [CrossRef]
- Ribeiro, A.; Holliday, G.L.; Furnham, N.; Tyzack, J.D.; Ferris, K.; Thornton, J.M. Mechanism and Catalytic Site Atlas (M-CSA): A database of enzyme reaction mechanisms and active sites. Nucleic Acids Res. 2018, 46, D618–D623. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Chang, P.M.; Ary, M.L.; Allen, B.D.; Chica, R.A.; Mayo, S.L.; Olafson, B.D. ProtaBank: A repository for protein design and engineering data. Protein Sci. 2019, 28, 672. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Property | Global % |
|---|---|---|
| Detail | Nonstructural | 39.1 |
| Structural | 65.1 | |
| Dynamic | 40.1 | |
| Interpretation | Energetic | 42.6 |
| Kinetic | 26.0 | |
| Thermo | 35.8 | |
| Quantum dynamic | 9.4 | |
| Topological | 59.1 | |
| Functional | 30.9 | |
| Complexation | 47.1 | |
| Structural interpretation | Sidechain | 50.9 |
| Folded | 31.2 | |
| Rotation invariant | 75.3 | |
| Translation invariant | 76.4 | |
| Biological interpretation | Evolutionary | 6.4 |
| Toxological | 2.8 | |
| Metabolic | 8.7 | |
| Forces | Electrostatic | 23.4 |
| Hydrophobic | 23.7 | |
| Van der Waals | 23.7 | |
| Bonds | Hydrogen bond | 29.3 |
| Salt bridge | 27.0 | |
| Pi | 16.0 | |
| Metal | 20.9 | |
| Environment | Solvent | 50.2 |
| Solute | 47.8 | |
| Trapped water | 37.7 | |
| Pocket/void | 46.7 | |
| Mathematical | Fixed size | 42.3 |
| Hierarchical | 44.5 | |
| Unique | 25.5 | |
| Subset unique | 3.7 | |
| Differentiable | 33.1 | |
| Reversible | 18.7 | |
| Guaranteed valid | 73.7 | |
| Probabilistic | 32.3 | |
| Graph/network | 25.8 |
| Index | SCC | Property 1 | Property 2 | Group |
|---|---|---|---|---|
| 1 | 0.977 | Rotation invariant | Translation invariant | Mathematical |
| 2 | 0.830 | Metal | Salt bridge | Bonds |
| 3 | 0.813 | Metal | Pi | Bonds |
| 4 | 0.806 | Hydrophobic | van der Waals | Forces |
| 5 | 0.776 | Hydrogen bond | Salt bridge | Bonds |
| 6 | 0.760 | Hydrogen bond | Metal | Bonds |
| 7 | 0.748 | Pi | Salt bridge | Bonds |
| 8 | 0.724 | Metal | van der Waals | Interactions |
| 9 | 0.721 | Solute | Solvent | Environment |
| 10 | 0.697 | Hydrogen bond | Pi | Bonds |
| 11 | 0.697 | Pi | van der Waals | Interactions |
| 12 | 0.644 | Salt bridge | van der Waals | Interactions |
| 13 | 0.634 | Reversible | Subset unique | Mathematical |
| 14 | 0.626 | Hydrogen bond | van der Waals | Interactions |
| 15 | 0.615 | Electrostatic | Hydrophobic | Forces |
| 16 | 0.601 | Kinetic | Thermodynamic | Energy |
| 17 | 0.601 | Hydrophobic | Pi | Interactions |
| 18 | 0.598 | Energetic | Thermodynamic | Energy |
| 19 | 0.592 | Hydrophobic | Pi | Interactions |
| 20 | 0.589 | Linearly combinable | Metabolic | Math/Bio |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
North, J.L.; Hsu, V.L. PREFMoDeL: A Systematic Review and Proposed Taxonomy of Biomolecular Features for Deep Learning. Appl. Sci. 2023, 13, 4356. https://doi.org/10.3390/app13074356
North JL, Hsu VL. PREFMoDeL: A Systematic Review and Proposed Taxonomy of Biomolecular Features for Deep Learning. Applied Sciences. 2023; 13(7):4356. https://doi.org/10.3390/app13074356
Chicago/Turabian StyleNorth, Jacob L., and Victor L. Hsu. 2023. "PREFMoDeL: A Systematic Review and Proposed Taxonomy of Biomolecular Features for Deep Learning" Applied Sciences 13, no. 7: 4356. https://doi.org/10.3390/app13074356
APA StyleNorth, J. L., & Hsu, V. L. (2023). PREFMoDeL: A Systematic Review and Proposed Taxonomy of Biomolecular Features for Deep Learning. Applied Sciences, 13(7), 4356. https://doi.org/10.3390/app13074356