
Syntactic Pattern Recognition for the Prediction of L-Type Pseudoknots in RNA

 , and
, and
Abstract
:1. Introduction
2. Conceptual Framework
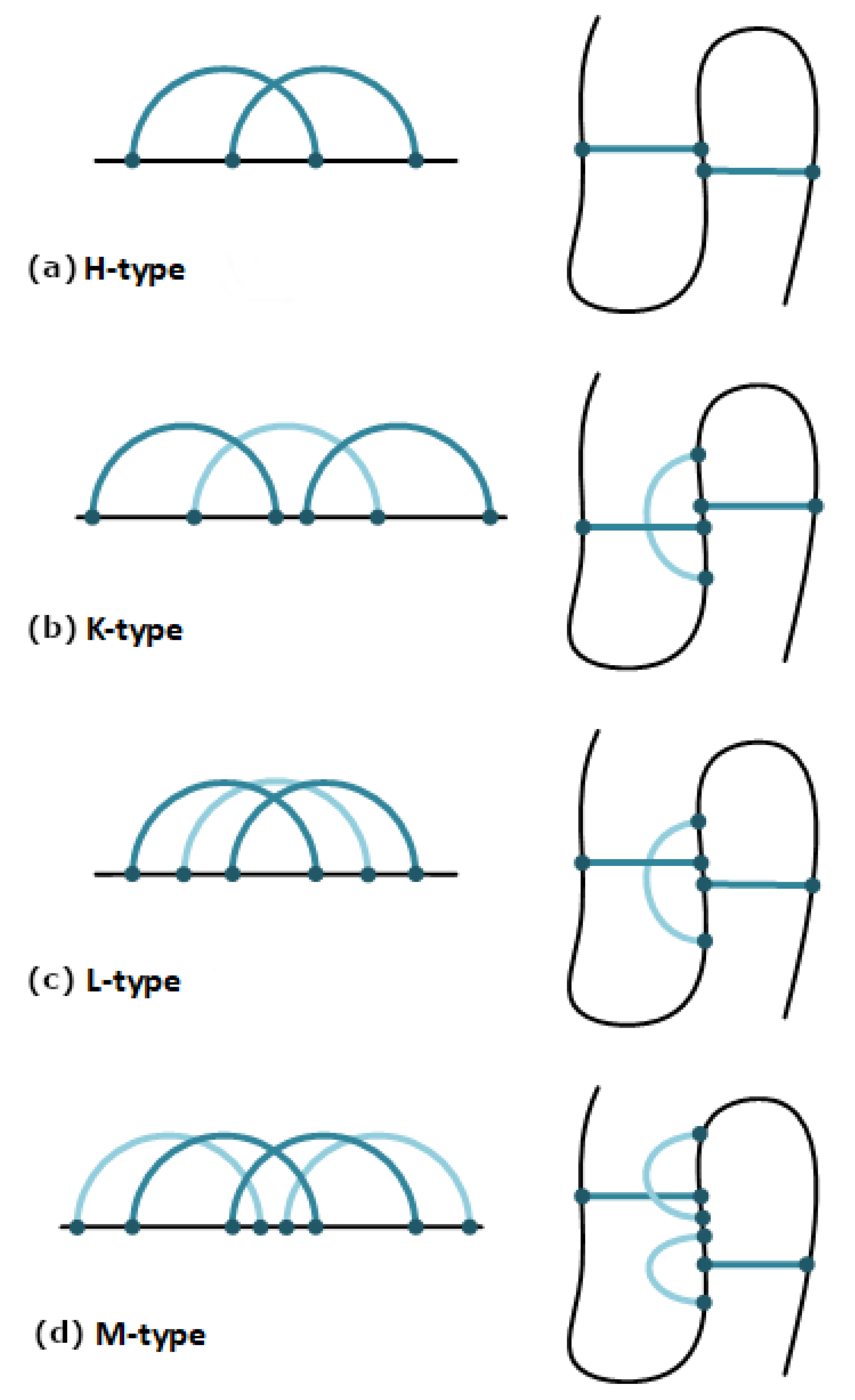
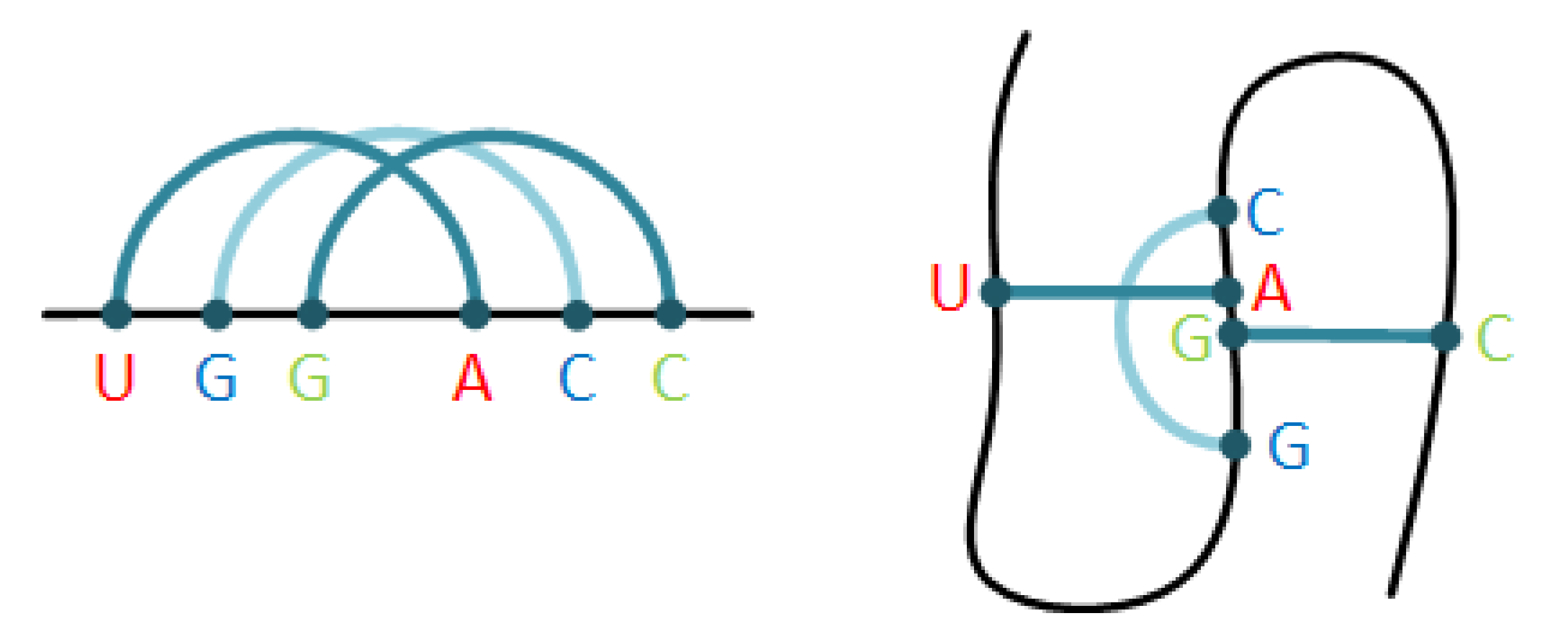
2.1. The Pseudoknot Motif
2.2. Syntactic Pattern Recognition—The Core Concept
Context-Free Grammars
3. Related Work
4. The Proposed Methodology—A Demonstration
4.1. Proposed CFG to Detect L-Type Pseudoknots
4.1.1. Decorate Core Stems
4.2. Choosing the Optimal Pseudoknot
5. Implementation Details
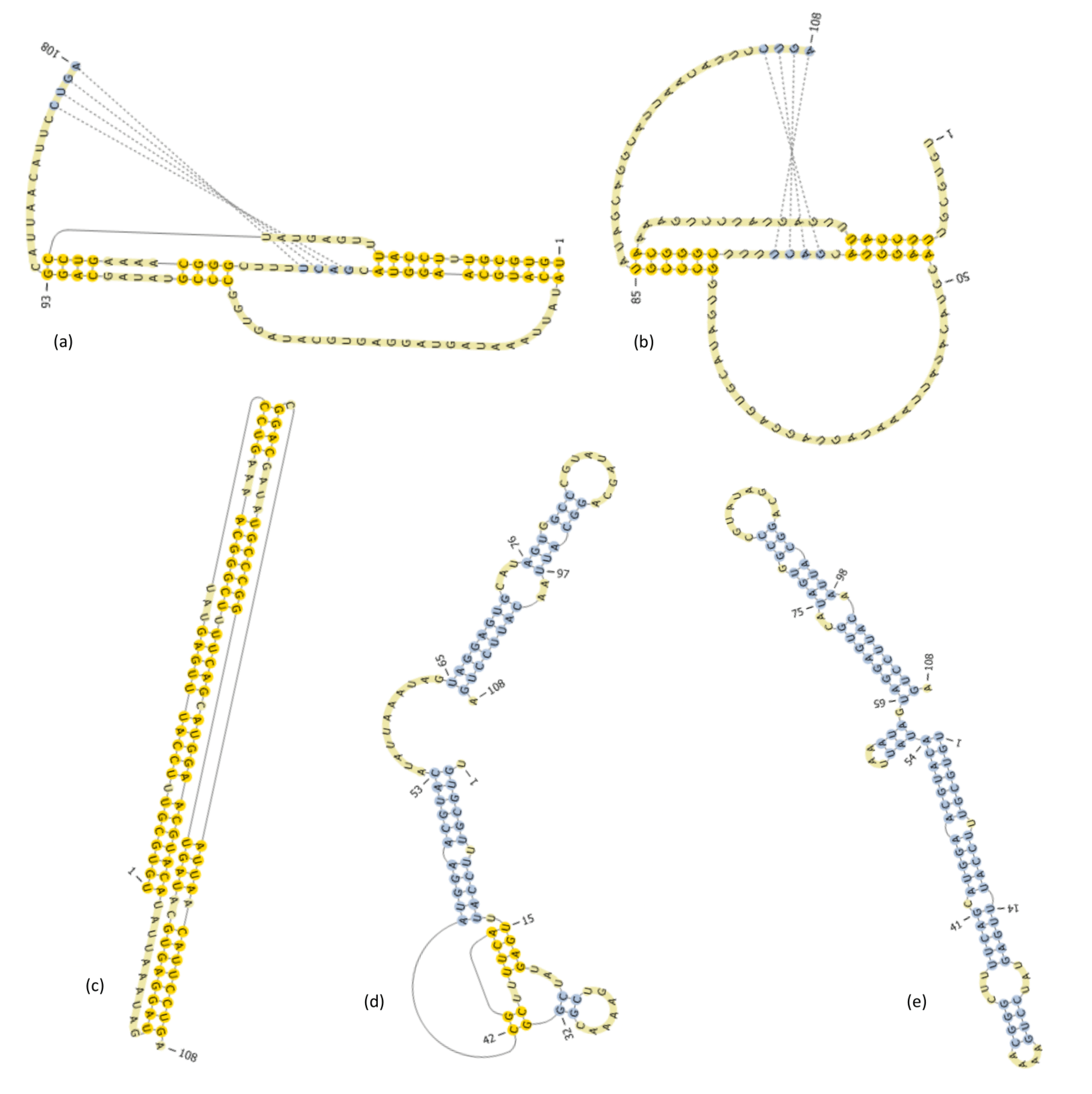
6. Predicting a Well-Known L-Type Pseudoknot
- Positive predictive value (PPV): PPV is a metric that measures the proportion of true positives among the positive predictions made by a classification model. It is calculated as the ratio of true positives (TPs) to the sum of true positives and false positives (FPs):
- Recall: Recall is a metric that measures the proportion of true positives that were correctly identified by a classification model. It is calculated as the ratio of TPs to the sum of true positives and false negatives (FNs):
- F1-score: The F1-score is a metric that balances precision (PPV) and recall. It is calculated as the harmonic mean of precision and recall:F1-score
- Matthews correlation coefficient (MCC): MCC is a metric that takes into account all four outcomes of a binary classification model (true positives, false positives, true negatives, and false negatives). It is calculated as follows:
7. Discussion
8. Conclusions and Future Work
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CFG | Context-free grammar |
| CNN | Convolutional neural network |
| CPU | Central processing unit |
| CYK | Cocke–Younger–Kasami |
| DAG | Direct acyclic graph |
| FCN | Fully convolutional network |
| IBPMP | Improved base pair maximization principle |
| LSTM | Long short-term memory |
| MCC | Matthews correlation coefficient |
| MFE | Minimum free energy |
| NMR | Nuclear magnetic resonance |
| RNA | Ribonucleic acid |
| SCFG | Stochastic context-free grammar |
| YAEP | Yet Another Earley Parser |
References
- Andrikos, C.; Makris, E.; Kolaitis, A.; Rassias, G.; Pavlatos, C.; Tsanakas, P. Knotify: An Efficient Parallel Platform for RNA Pseudoknot Prediction Using Syntactic Pattern Recognition. Methods Protoc. 2022, 5, 14. [Google Scholar] [CrossRef]
- Makris, E.; Kolaitis, A.; Andrikos, C.; Moulos, V.; Tsanakas, P.; Pavlatos, C. Knotify+: Toward the Prediction of RNA H-Type Pseudoknots, Including Bulges and Internal Loops. Biomolecules 2023, 13, 308. [Google Scholar] [CrossRef]
- Watson, J.; Crick, F. Molecular Structure of Nucleic Acids. Am. J. Psychiatry 2003, 160, 623–624. [Google Scholar] [CrossRef] [PubMed]
- Rietveld, K.; Van Poelgeest, R.; Pleij, C.W.; Van Boom, J.; Bosch, L. The tRNA-Uke structure at the 3’ terminus of turnip yellow mosaic virus RNA. Differences and similarities with canonical tRNA. Nucleic Acids Res. 1982, 10, 1929–1946. [Google Scholar] [CrossRef]
- Kucharík, M.; Hofacker, I.L.; Stadler, P.F.; Qin, J. Pseudoknots in RNA folding landscapes. Bioinformatics 2016, 32, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Staple, D.W.; Butcher, S.E. Pseudoknots: RNA structures with diverse functions. PLoS Biol. 2005, 3, e213. [Google Scholar] [CrossRef]
- Hopcroft, J.E.; Ullman, J.D. Formal Languages and Their Relation to Automata; Addison-Wesley Longman Publishing Co., Inc.: Boston, MA, USA, 1969. [Google Scholar]
- Chomsky, N. Three models for the description of language. IRE Trans. Inf. Theory 1956, 2, 113–124. [Google Scholar] [CrossRef]
- Sipser, M. Introduction to the Theory of Computation; Thomson Course Technology: Boston, MA, USA, 2006; Volume 2. [Google Scholar]
- Younger, D.H. Recognition and parsing of context-free languages in n3. Inf. Control. 1967, 10, 189–208. [Google Scholar] [CrossRef]
- Earley, J. An efficient context-free parsing algorithm. Commun. ACM 1970, 13, 94–102. [Google Scholar] [CrossRef]
- Graham, S.L.; Harrison, M.A.; Ruzzo, W.L. An improved context-free recognizer. ACM Trans. Program. Lang. Syst. 1980, 2, 415–462. [Google Scholar] [CrossRef]
- Ruzzo, W.L. General Context-Free Language Recognition. Ph.D. Thesis, University of California, Berkeley, CA, USA, 1978. [Google Scholar]
- Geng, T.; Xu, F.; Mei, H.; Meng, W.; Chen, Z.; Lai, C. A practical GLR parser generator for software reverse engineering. J. Netw. 2014, 9, 769–776. [Google Scholar] [CrossRef]
- Pavlatos, C.; Dimopoulos, A.C.; Koulouris, A.; Andronikos, T.; Panagopoulos, I.; Papakonstantinou, G. Efficient reconfigurable embedded parsers. Comput. Lang. Syst. Struct. 2009, 35, 196–215. [Google Scholar] [CrossRef]
- Chiang, Y.; Fu, K. Parallel parsing algorithms and VLSI implementations for syntactic pattern recognition. IEEE Trans. Pattern Anal. Mach. Intell. 1984, 6, 302–314. [Google Scholar] [CrossRef]
- Available online: https://github.com/vnmakarov/yaep (accessed on 25 March 2020).
- Cao, S.; Chen, S. Predicting structures and stabilities for H-type pseudoknots with interhelix loops. RNA 2009, 4, 696–706. [Google Scholar] [CrossRef]
- Akutsu, T. Dynamic programming algorithms for RNA secondary structure prediction with pseudoknots. Discret. Appl. Math. 2000, 104, 45–62. [Google Scholar] [CrossRef]
- Meyer, I.M.; Miklos, I. SimulFold: Simultaneously inferring RNA structures including pseudoknots, alignments, and trees using a Bayesian MCMC framework. PLoS Comput. Biol. 2007, 3, e149. [Google Scholar] [CrossRef]
- Van Batenburg, F.; Gultyaev, A.P.; Pleij, C.W. An APL-programmed genetic algorithm for the prediction of RNA secondary structure. J. Theor. Biol. 1995, 174, 269–280. [Google Scholar] [CrossRef]
- Isambert, H.; Siggia, E.D. Modeling RNA folding paths with pseudoknots: Application to hepatitis delta virus ribozyme. Proc. Natl. Acad. Sci. USA 2000, 97, 6515–6520. [Google Scholar] [CrossRef] [PubMed]
- Jabbari, H.; Wark, I.; Montemagno, C.; Will, S. Knotty: Efficient and accurate prediction of complex RNA pseudoknot structures. Bioinformatics 2018, 34, 3849–3856. [Google Scholar] [CrossRef]
- Chen, H.L.; Condon, A.; Jabbari, H. An O(n5) algorithm for MFE prediction of kissing hairpins and 4-chains in nucleic acids. J. Comput. Biol. 2009, 16, 803–815. [Google Scholar] [CrossRef]
- Bellaousov, S.; Mathews, D.H. ProbKnot: Fast prediction of RNA secondary structure including pseudoknots. RNA 2010, 16, 1870–1880. [Google Scholar] [CrossRef]
- Sato, K.; Kato, Y.; Hamada, M.; Akutsu, T.; Asai, K. IPknot: Fast and accurate prediction of RNA secondary structures with pseudoknots using integer programming. Bioinformatics 2011, 27, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Kato, Y. Prediction of RNA secondary structure including pseudoknots for long sequences. Briefings Bioinform. 2021, 23, bbab395. [Google Scholar] [CrossRef]
- Singh, J.; Hanson, J.; Paliwal, K.; Zhou, Y. RNA secondary structure prediction using an ensemble of two-dimensional deep neural networks and transfer learning. Nat. Commun. 2019, 10, 5407. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, Y.; Zhong, X.; Liu, H.; Lu, C.; Li, C.; Zhang, H. DMfold: A novel method to predict RNA secondary structure with pseudoknots based on deep learning and improved base pair Maximization Principle. Front. Genet. 2019, 10, 143. [Google Scholar] [CrossRef]
- Kangkun, M.; Jun, W.; Yi, X. Prediction of RNA secondary structure with pseudoknots using coupled deep neural networks. Biophys. Rep. 2020, 6, 146–154. [Google Scholar]
- Wang, Y.; Liu, Y.; Wang, S.; Liu, Z.; Gao, Y.; Zhang, H.; Dong, L. ATTfold: RNA secondary structure prediction with pseudoknots based on attention mechanism. Front. Genet. 2020, 11, 1564. [Google Scholar] [CrossRef]
- Fu, L.; Cao, Y.; Wu, J.; Peng, Q.; Nie, Q.; Xie, X. UFold: Fast and accurate RNA secondary structure prediction with deep learning. Nucleic Acids Res. 2021, 50, e14. [Google Scholar] [CrossRef]
- Knudsen, B.; Hein, J. RNA secondary structure prediction using stochastic context-free grammars and evolutionary history. Bioinformatics 1999, 15, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, B.; Hein, J. Pfold: RNA Secondary Structure Prediction Using Stochastic Context-Free Grammars. Nucleic Acids Res. 2003, 31, 3423–3428. [Google Scholar] [CrossRef] [PubMed]
- Sukosd, Z.; Knudsen, B.; Vaerum, M.; Kjems, J.; Andersen, E.S. Multithreaded comparative RNA secondary structure prediction using stochastic context-free grammars. BMC Bioinform. 2011, 12, 103. [Google Scholar] [CrossRef]
- Pedersen, J.S.; Meyer, I.M.; Forsberg, R.; Simmonds, P.; Hein, J. A comparative method for finding and folding RNA secondary structures within protein-coding regions. Nucleic Acids Res. 2004, 32, 4925–4936. [Google Scholar] [CrossRef] [PubMed]
- Do, C.B.; Woods, D.A.; Batzoglou, S. CONTRAfold: RNA secondary structure prediction without physics-based models. Bioinformatics 2006, 22, e90–e98. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, J.S.; Bejerano, G.; Siepel, A.; Rosenbloom, K.; Lindblad-Toh, K.; Lander, E.S.; Kent, J.; Miller, W.; Haussler, D. Identification and classification of conserved RNA secondary structures in the human genome. PLoS Comput. Biol. 2006, 2, e33. [Google Scholar] [CrossRef]
- Nawrocki, E.P.; Kolbe, D.L.; Eddy, S.R. Infernal 1.0: Inference of RNA alignments. Bioinformatics 2009, 25, 1335–1337. [Google Scholar] [CrossRef]
- Anderson, J.W.; Haas, P.A.; Mathieson, L.A.; Volynkin, V.; Lyngsø, R.; Tataru, P.; Hein, J. Oxfold: Kinetic folding of RNA using stochastic context-free grammars and evolutionary information. Bioinformatics 2013, 29, 704–710. [Google Scholar] [CrossRef]
- Bradley, R.K.; Pachter, L.; Holmes, I. Specific alignment of structured RNA: Stochastic grammars and sequence annealing. Bioinformatics 2008, 24, 2677–2683. [Google Scholar] [CrossRef]
- Makris, E.; Kolaitis, A.; Andrikos, C.; Moulos, V.; Tsanakas, P.; Pavlatos, C. An intelligent grammar-based platform for RNA H-type pseudoknot prediction. In Artificial Intelligence Applications and Innovations, Proceedings of the AIAI 2022 IFIP WG 12.5 International Workshops: IFIP Advances in Information and Communication Technology, Crete, Greece, 17–20 June 2022; Springer: Berlin/Heidelberg, Germany, 2022; Volume 652. [Google Scholar]
- Trotta, E. On the normalization of the minimum free energy of RNAs by sequence length. PLoS ONE 2014, 9, e113380. [Google Scholar] [CrossRef]
- Nussinov, R.; Jacobson, A.B. Fast algorithm for predicting the secondary structure of single-stranded RNA. Proc. Natl. Acad. Sci. USA 1980, 77, 6309–6313. [Google Scholar] [CrossRef] [PubMed]
- Mathews, D.H. Using an RNA secondary structure partition function to determine confidence in base pairs predicted by free energy minimization. RNA 2004, 10, 1178–1190. [Google Scholar] [CrossRef]
- Rivas, E.; Eddy, S.R. Noncoding RNA gene detection using comparative sequence analysis. BMC Bioinform. 2001, 2, 8. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.; Corey, D.R. RNA Sequencing: Platform Selection, Experimental Design, and Data Interpretation. Nucleic Acid Ther. 2012, 22, 271–274. [Google Scholar] [CrossRef]
- Ren, J.; Rastegari, B.; Condon, A.; Hoos, H.H. HotKnots: Heuristic prediction of RNA secondary structures including pseudoknots. RNA 2005, 11, 1494–1504. [Google Scholar] [CrossRef] [PubMed]
- Mathews, D.; Sabina, J.; Zuker, M.; Turner, D. Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure1. J. Mol. Biol. 1999, 288, 911–940. [Google Scholar] [CrossRef]
- Dirks, R.; Pierce, N. Introduction A Partition Function Algorithm for Nucleic Acid Secondary Structure Including Pseudoknots. J. Comput. Chem. 2003, 24, 1664–1677. [Google Scholar] [CrossRef]
- Available online: https://github.com/chriskor1 (accessed on 9 March 2023).
- Bon, M.; Vernizzi, G.; Orland, H.; Zee, A. Topological classification of RNA structures. J. Mol. Biol. 2008, 379, 900–911. [Google Scholar] [CrossRef]
- Byun, Y.; Han, K. PseudoViewer3: Generating planar drawings of large-scale RNA structures with pseudoknots. Bioinformatics 2009, 25, 1435–1437. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enumeration | Syntactic Rules |
|---|---|
| 0 | S → “A” L “A” L “A” D “U” L “U” L “U” |
| 1 | S → “A” L “A” L “U” D “U” L “U” L “A” |
| 2 | S → “A” L “A” L “G” D “U” L “U” L “C” |
| 3 | S → “A” L “A” L “C” D “U” L “U” L “G” |
| 4 | S → “A” L “U” L “A” D “U” L “A” L “U” |
| 5 | S → “A” L “U” L “U” D “U” L “A” L “A” |
| 6 | S → “A” L “U” L “G” D “U” L “A” L “C” |
| 7 | S → “A” L “U” L “C” D “U” L “A” L “G” |
| 8 | S → “A” L “G” L “A” D “U” L “C” L “U” |
| 9 | S → “A” L “G” L “U” D “U” L “C” L “A” |
| 10 | S → “A” L “G” L “G” D “U” L “C” L “C” |
| 11 | S → “A” L “G” L “C” D “U” L “C” L “G” |
| . | |
| . | |
| . | |
| 60 | S → “C” L “A” L “C” D “G” L “G” L “U” |
| 61 | S → “C” L “U” L “C” D “G” L “G” L “A” |
| 62 | S → “C” L “G” L “C” D “G” L “G” L “C” |
| 63 | S → “C” L “C” L “C” D “G” L “G” L “G” |
| 64 | L → “A” L |
| 65 | L → “U” L |
| 66 | L → “C” L |
| 67 | L → “G” L |
| 68 | L → “A” |
| 69 | L → “U” |
| 70 | L → “C” |
| 71 | L → “G” |
| 72 | D → K K |
| 73 | K → “A” |
| 74 | K → “U” |
| 75 | K → “C” |
| 76 | K → “G” |
| 77 | K |
| String Enumeration | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | 19 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| String | U | U | U | A | C | G | G | C | C | G | A | A | U | U | C | G | C | G | G | A |
| Parser output: | . | . | ( | . | . | [ | . | . | . | { | ) | . | . | . | ] | . | } | . | . | . |
| Step 1 | . | ( | ( | . | . | [ | . | . | . | { | ) | ) | . | . | ] | . | } | . | . | . |
| Step 2 | . | ( | ( | . | . | [ | . | { | { | { | ) | ) | . | . | ] | . | } | } | } | . |
| Step 3 | . | ( | ( | . | [ | [ | . | { | { | { | ) | ) | . | . | ] | ] | } | } | } | . |
| Platform | RNA/Dot Bracket |
|---|---|
| UGUGCGUUUCCAUUUGAGUAUCCUGAAAACGGGCUUUUCAGCAUGGAACGUACAUAUUAAAUAGUAGGAGUGCAUAGUGGCCCGUAUAGCAGGCAUUAACAUUCCUGA | |
| Ground truth | (((((((.(((((........[[[[....[[[[....{{{{.))))))))))))..........................]]]].....]]]]...........}}}} |
| Knotify | .......((((((...............[[[[[[...{{{{.))))))...............................]]]]]]...................}}}} |
| IPknot | .((((((.(((((.[[[[...((.......))((..]]]])))))))))))))...........((((((((...(((.(((.........))))))..)))))))). |
| Knotty | (((((((.((((((((((...[[[[...[[[[[[[.))))).))))))))))))..........((((((((..((((]]]]]]]....]]]].)))).)))))))). |
| ProbKnot | (((((((.((((((((((...((((....))))...))))).))))))))))))(((...))).((((((((..((((.(((.........))))))).)))))))). |
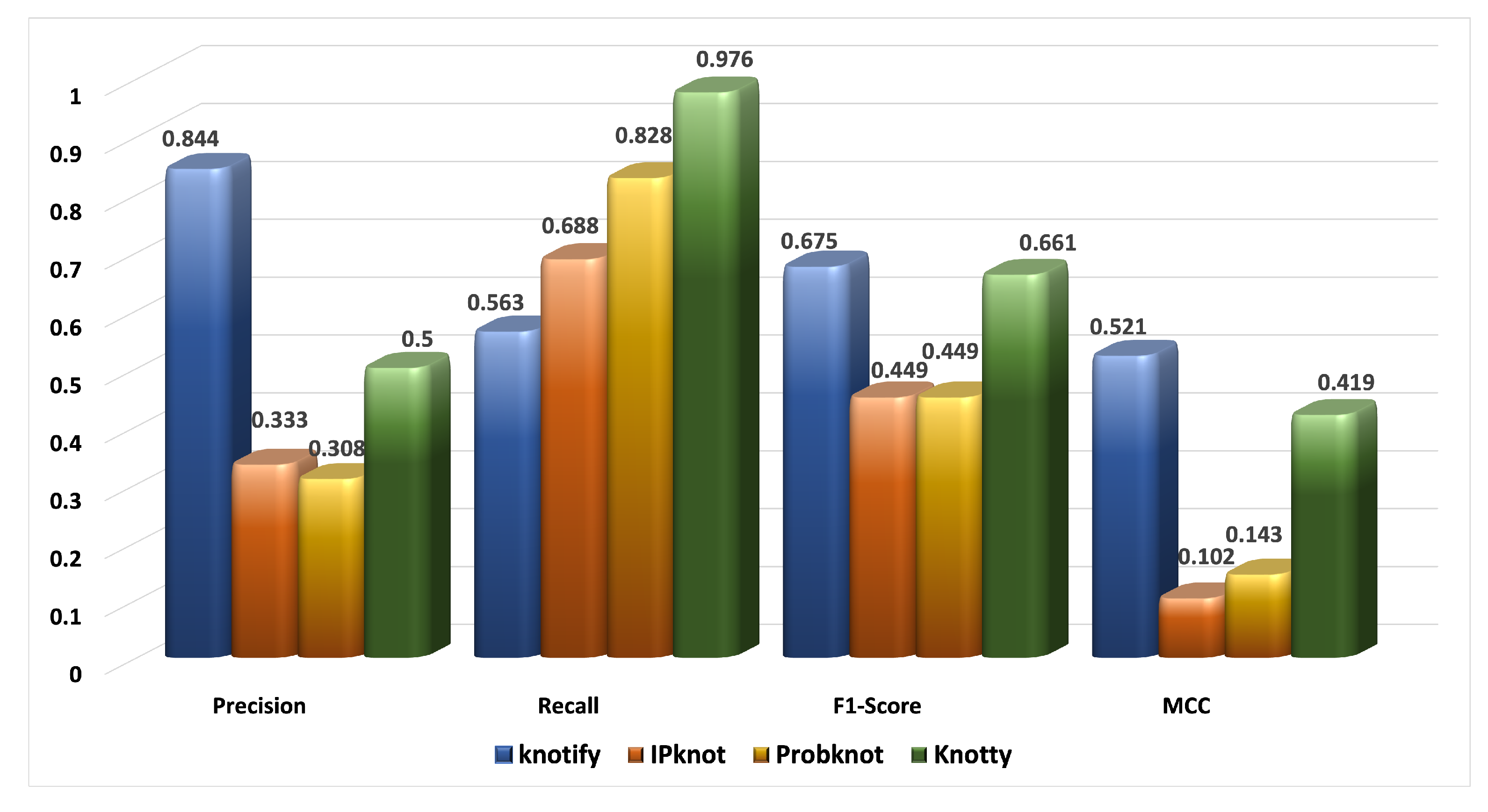
| Platform | tp | tn | fp | fn | Precision | Recall | F1-Score | MCC |
|---|---|---|---|---|---|---|---|---|
| Knotify | 27 | 55 | 5 | 21 | 0.844 | 0.563 | 0.675 | 0.521 |
| IPknot | 22 | 32 | 44 | 10 | 0.333 | 0.688 | 0.449 | 0.102 |
| ProbKnot | 24 | 25 | 54 | 5 | 0.308 | 0.828 | 0.449 | 0.143 |
| Knotty | 40 | 27 | 40 | 1 | 0.500 | 0.976 | 0.661 | 0.419 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koroulis, C.; Makris, E.; Kolaitis, A.; Tsanakas, P.; Pavlatos, C. Syntactic Pattern Recognition for the Prediction of L-Type Pseudoknots in RNA. Appl. Sci. 2023, 13, 5168. https://doi.org/10.3390/app13085168
Koroulis C, Makris E, Kolaitis A, Tsanakas P, Pavlatos C. Syntactic Pattern Recognition for the Prediction of L-Type Pseudoknots in RNA. Applied Sciences. 2023; 13(8):5168. https://doi.org/10.3390/app13085168
Chicago/Turabian StyleKoroulis, Christos, Evangelos Makris, Angelos Kolaitis, Panayiotis Tsanakas, and Christos Pavlatos. 2023. "Syntactic Pattern Recognition for the Prediction of L-Type Pseudoknots in RNA" Applied Sciences 13, no. 8: 5168. https://doi.org/10.3390/app13085168
APA StyleKoroulis, C., Makris, E., Kolaitis, A., Tsanakas, P., & Pavlatos, C. (2023). Syntactic Pattern Recognition for the Prediction of L-Type Pseudoknots in RNA. Applied Sciences, 13(8), 5168. https://doi.org/10.3390/app13085168