Thermodynamic Simulations for Determining the Recycling Path of a Spent Lead-Acid Battery Electrolyte Sample with Ca(OH)2


Abstract
:Featured Application
Abstract
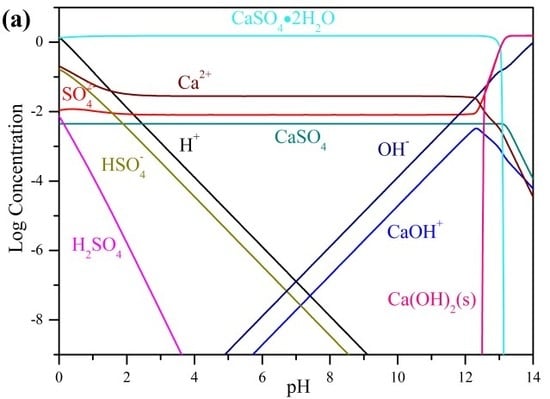
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
3. Results
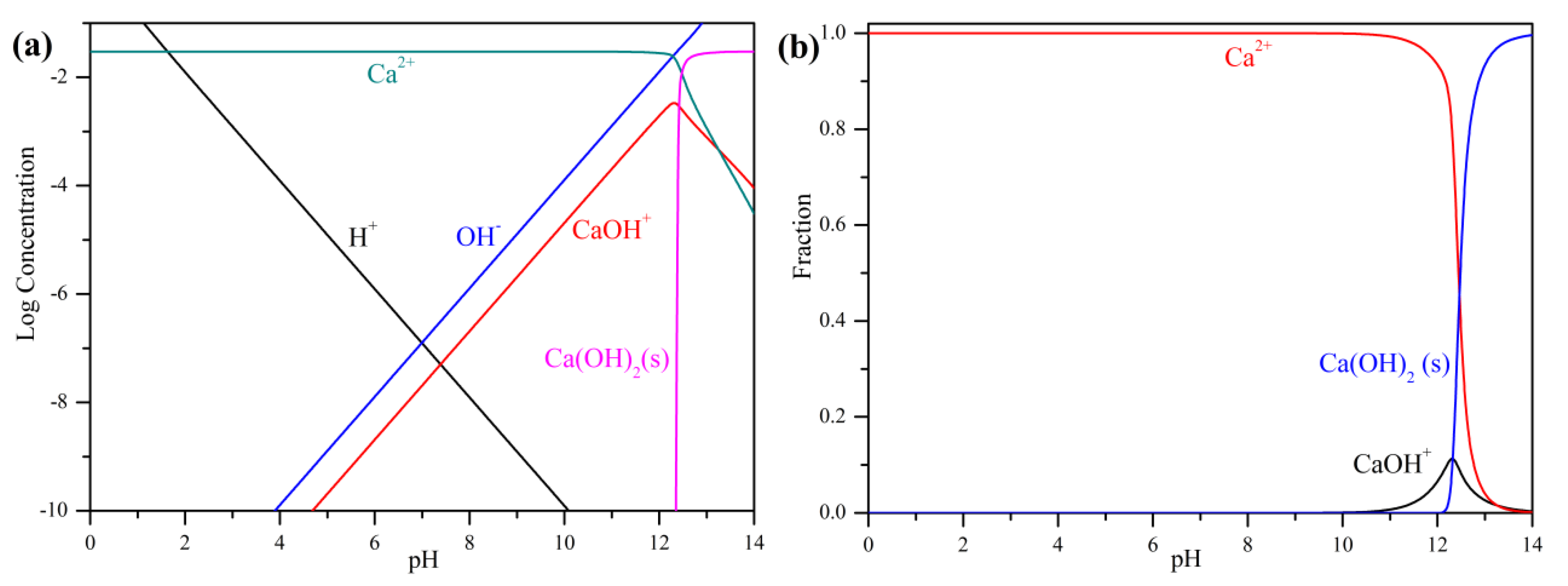
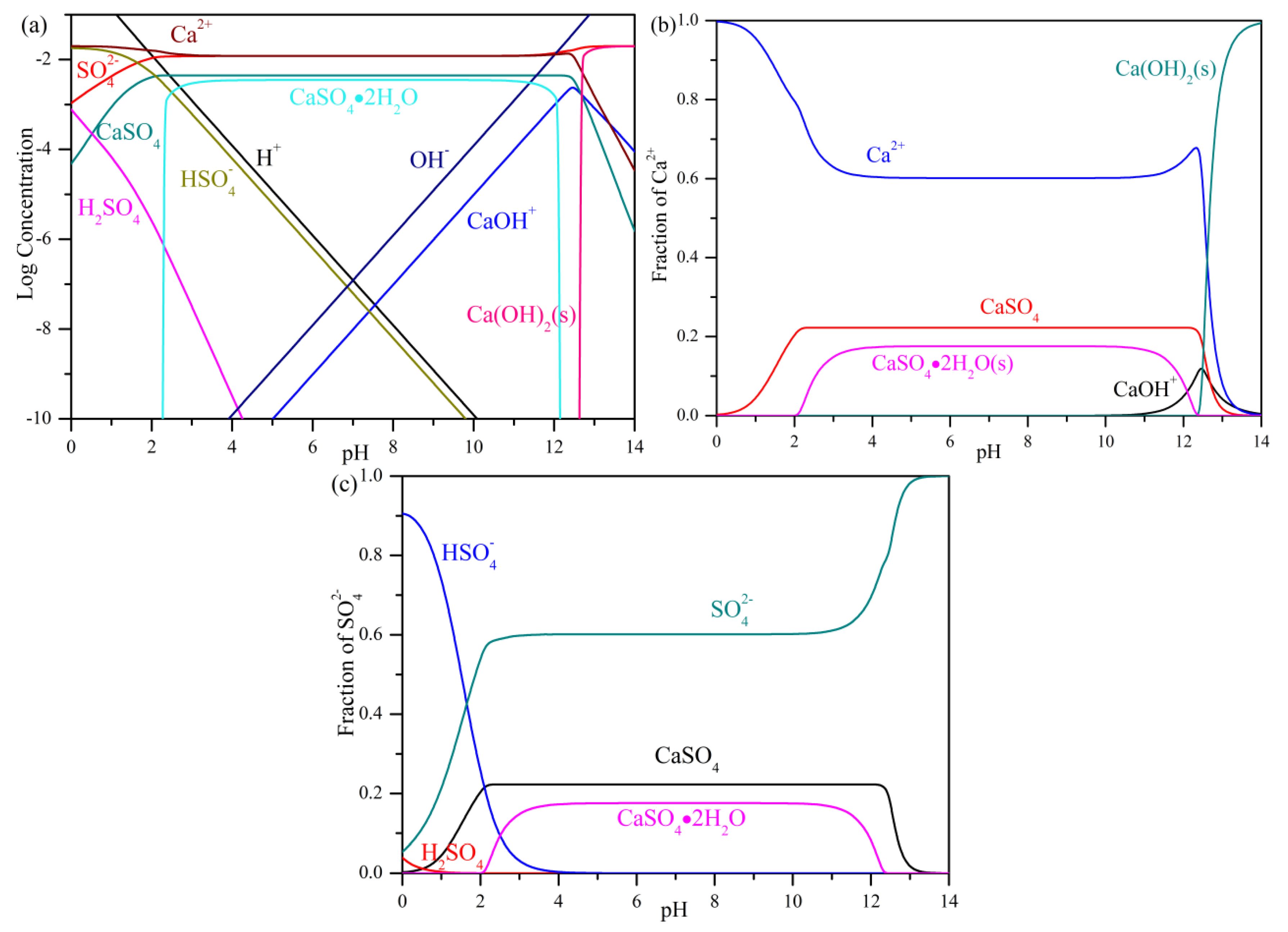
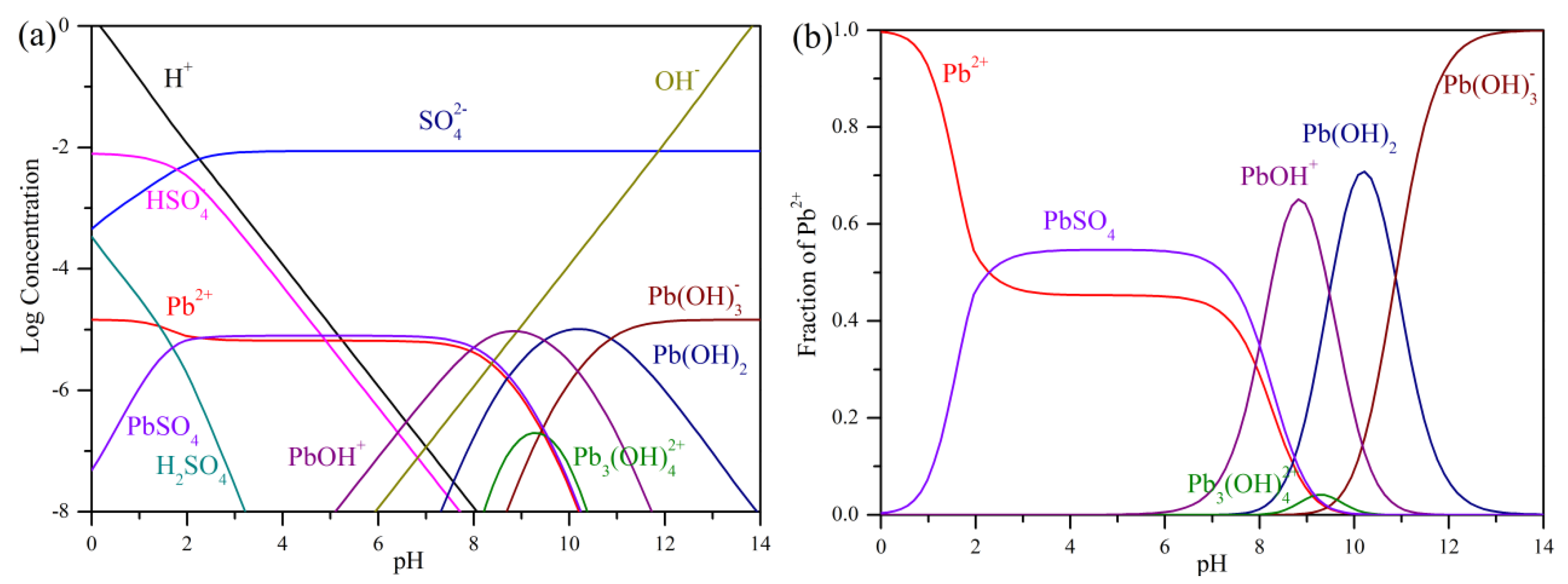
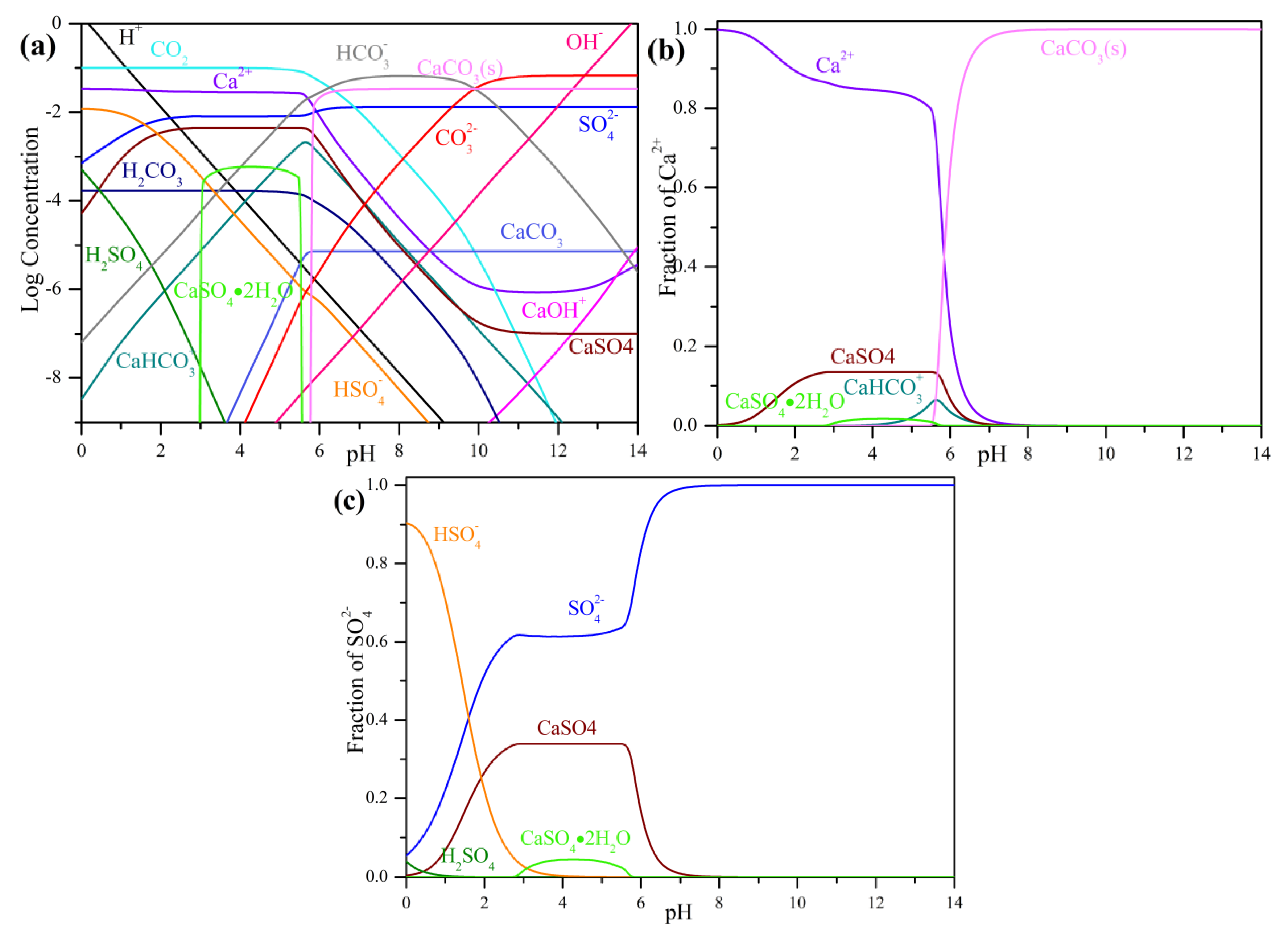
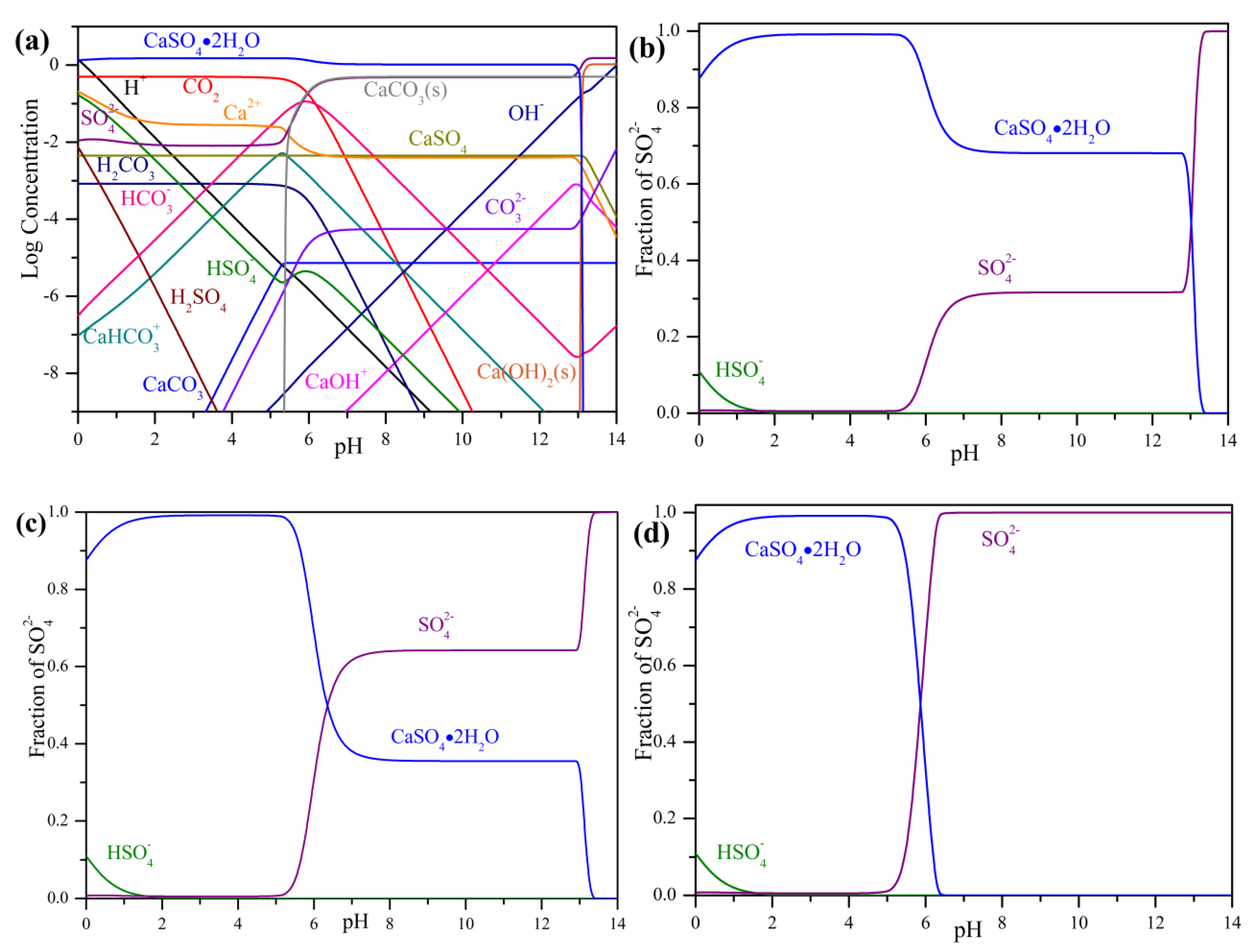
3.1. Precipitation
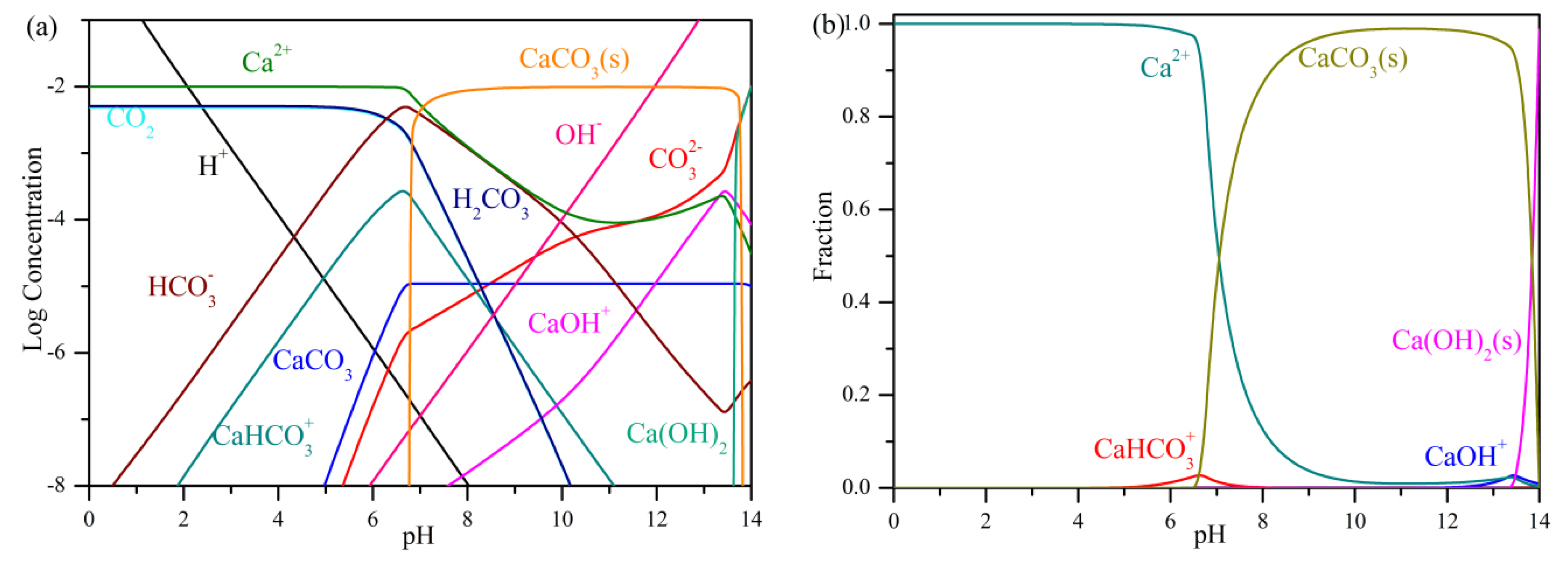
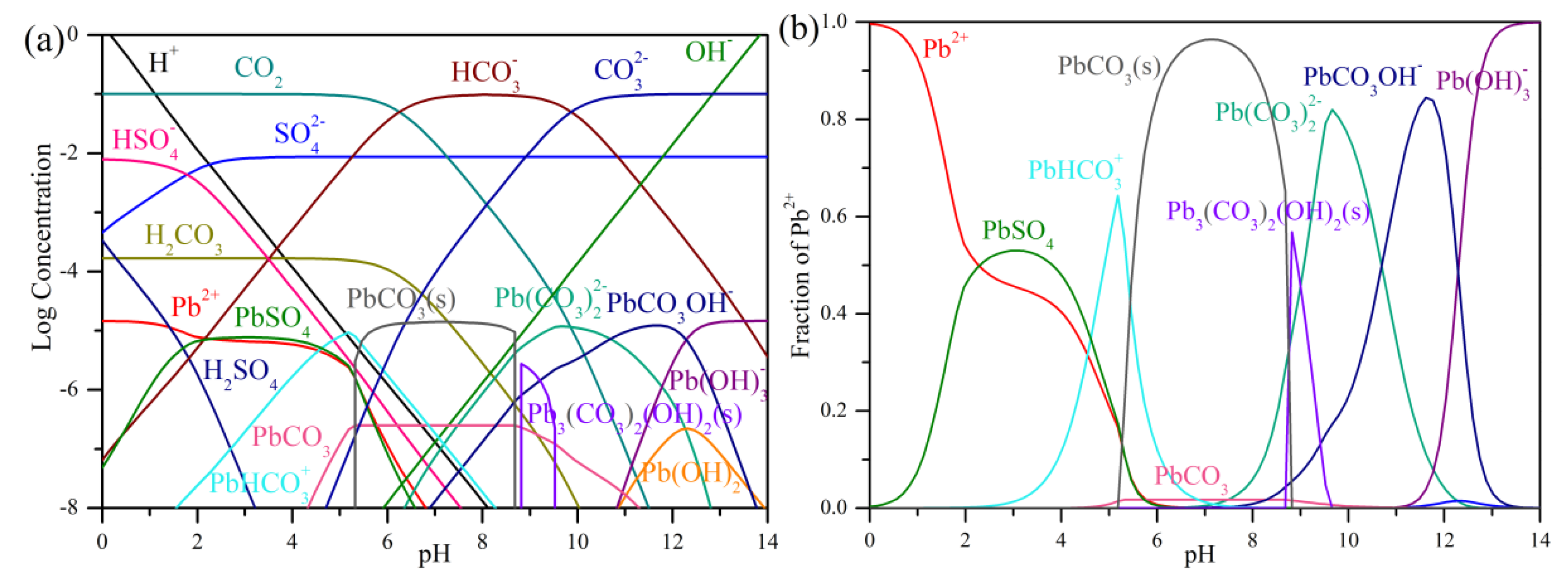
3.2. Carbonation
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bode, H. Lead-Acid Batteries; U.S. DOE: Washington, DC, USA, 1977; pp. 1–15.
- Bhangu, B.S.; Bentley, P.; Stone, D.A.; Bingham, C.M. Nonlinear observers for predicting state-of-charge and state-of-health of lead-acid batteries for hybrid-electric vehicles. IEEE Trans. Veh. Technol. 2005, 54, 783–794. [Google Scholar] [CrossRef]
- Parker, C.D. Lead–acid battery energy-storage systems for electricity supply networks. J. Power Sources 2001, 100, 18–28. [Google Scholar] [CrossRef]
- Zou, G.; Dou, Y.; Chang, X.; Chen, Y. Design and application of a standalone hybrid wind-solar system for automatic observation systems used in the polar region. Appl. Sci. 2018, 8, 2376. [Google Scholar]
- Chang, Y.; Mao, X.; Zhao, Y.; Feng, S.; Chen, H.; Finlow, D. Lead-acid battery use in the development of renewable energy systems in China. J. Power Sources 2009, 191, 176–183. [Google Scholar] [CrossRef]
- Pavlov, D. Lead-Acid Batteries: Science and Technology, 1st ed.; Elsevier Science: Oxford, UK, 2011; pp. 21–26. [Google Scholar]
- Recycling Used Lead-Acid Batteries: Health Considerations. Available online: https://apps.who.int/iris/bitstream/handle/10665/ (accessed on 31 May 2019).
- Sun, Z.; Cao, H.; Zhang, X.; Lin, X.; Zheng, W.; Cao, G.; Sun, Y.; Zhang, Y. Spent lead-acid battery recycling in China–A review and sustainable analyses on mass flow of lead. Waste Manag. 2017, 64, 190–201. [Google Scholar] [CrossRef] [PubMed]
- Ellis, T.W.; Mirza, A.H. The refining of secondary lead for use in advanced lead-acid batteries. J. Power Sources 2010, 195, 4525–4529. [Google Scholar] [CrossRef]
- Bernardes, A.M.; Espinosa, D.C.R.; Tenorio, J.A.S. Recycling of batteries: A review of current processes and technologies. J. Power Sources 2004, 130, 291–298. [Google Scholar] [CrossRef]
- Zhu, X.; Yang, J.; Gao, L.; Liu, J.; Yang, D.; Sun, X.; Zhang, W.; Wang, Q.; Li, L.; He, D.; et al. Preparation of lead carbonate from spent lead paste via chemical conversion. Hydrometallurgy 2013, 134–135, 47–53. [Google Scholar] [CrossRef]
- Andrews, D.; Raychaudhuri, A.; Frias, C. Environmentally sound technologies for recycling secondary lead. J. Power Sources 2000, 88, 124–129. [Google Scholar] [CrossRef]
- Liu, W.; Tian, J.; Chen, L.; Guo, Y. Temporal and spatial characteristics of lead emissions from the lead-acid battery manufacturing industry in China. Environ. Pollut. 2017, 220, 696–703. [Google Scholar] [CrossRef]
- Spanos, C.; Berlinger, S.A.; Jayan, A.; West, A.C. Inverse Charging Techniques for Sulfation Reversal in Flooded Lead-Acid Batteries. J. Electrochem. Soc. 2016, 163, A1612–A1618. [Google Scholar] [CrossRef]
- Jolly, R.; Rhin, C. The recycling of lead-acid batteries: Production of lead and polypropylene. Resour. Conserv. Recycl. 1994, 10, 137–143. [Google Scholar] [CrossRef]
- Rust, N.; Ferg, E.E.; Masalova, I. A degradation study of isotactic virgin and recycled polypropylene used in lead acid battery casings. Polym. Test. 2006, 25, 130–139. [Google Scholar] [CrossRef]
- Hong, S.; Cannon, F.S.; Hou, P.; Byrne, T.; Nieto-Delgado, C. Adsorptive removal of sulfate from acid mine drainage by polypyrrole modified activated carbons: Effects of polypyrrole deposition protocols and activated carbon source. Chemosphere 2017, 184, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Cuesta Mayorga, I.; Astilleros, J.M.; Fernández-Díaz, L. Precipitation of CaCO3 polymorphs from aqueous solutions: The role of pH and sulfate groups. Minerals 2019, 9, 178. [Google Scholar] [CrossRef]
- Gominsek, T.; Lubej, A.; Pohar, C. Continuous precipitation of calcium sulfate dehydrate from waste sulfuric acid and lime. J. Chem. Technol. Biotechnol. 2005, 80, 939–947. [Google Scholar] [CrossRef]
- Najib, T.; Solgi, M.; Farazmand, A.; Heydarian, S.M.; Nasernejad, B. Optimization of sulfate removal by sulfate reducing bacteria using response surface methodology and heavy metal removal in a sulfidogenic UASB reactor. J. Environ. Chem. Eng. 2017, 5, 3256–3265. [Google Scholar] [CrossRef]
- Arahman, N.; Mulyati, S.; Lubis, M.R.; Takagi, R.; Matsuyama, H. Removal profile of sulfate ion from mix ion solution with different type and configuration of anion exchange membrane in electrodialysis. J. Water Process Eng. 2017, 20, 173–179. [Google Scholar] [CrossRef]
- Kartic, D.N.; Narayana, B.C.H.A.; Arivazhagan, M. Removal of high concentration of sulfate from pigment industry effluent by chemical precipitation using barium chloride: RSM and ANN modeling approach. J. Environ. Manag. 2018, 206, 69–76. [Google Scholar] [CrossRef]
- Fang, P.; Tang, Z.; Chen, X.; Huang, J.; Tang, Z.; Cen, C. Removal of High-Concentration Sulfate Ions from the Sodium Alkali FGD Wastewater Using Ettringite Precipitation Method: Factor Assessment, Feasibility, and Prospect. J. Chem. 2018, 2018, 1265168. [Google Scholar] [CrossRef]
- Garrels, R.M.; Thompson, M.E. A chemical model for sea water at 25 degrees C and one atmosphere total pressure. Am. J. Sci. 1962, 260, 57–66. [Google Scholar] [CrossRef]
- Helgeson, H.C.; Kirkham, D.H.; Flowers, G.C. Theoretical prediction of the thermodynamic behavior of aqueous electrolytes by high pressures and temperatures; IV, Calculation of activity coefficients, osmotic coefficients, and apparent molal and standard and relative partial molal properties to 600 degrees C and 5kb. Am. J. Sci. 1981, 281, 1249–1516. [Google Scholar]
- Oelkers, E.H.; Helgeson, H.C. Triple-ion anions and polynuclear complexing in supercritical electrolyte solutions. Geochim. Cosmochim. Acta 1990, 54, 727–738. [Google Scholar] [CrossRef]
- Shock, E.L.; Oelkers, E.H.; Johnson, J.W.; Sverjensky, D.A.; Helgeson, H.C. Calculation of the thermodynamic properties of aqueous species at high pressures and temperatures. Effective electrostatic radii, dissociation constants and standard partial molal properties to 1000 °C and 5 kbar. J. Chem. Soc. Faraday Trans. 1992, 88, 803–826. [Google Scholar] [CrossRef]
- Karimi, H. Effect of pH and initial Pb(II) concentration on the lead removal efficiency from industrial wastewater using Ca(OH)2. Int. J. Water Wastewater Treat. 2017, 3, 1–4. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulas | Possible Chemical Reactions | Log10(K) 1 | |
|---|---|---|---|
| H+/OH- | H2O H+ + OH− | (1) | −13.99 |
| Ca2+ | 2H+ + Ca(OH)2(s) Ca2+ + 2H2O | (2) | 22.62 |
| CaOH+ | H+ + Ca(OH)2(s) CaOH+ + H2O | (3) | 9.785 |
| Ca(OH)2(s) | CaCO3(s) + 2H2O 2H+ + 2CO32− + Ca(OH)2(s) | (4) | −30.88 |
| CaO(s) | CaCO3(s) + H2O 2H+ + 2CO32− + CaO(s) | (5) | −41.0 |
| CaCO3 | CaCO3(s) CaCO3 | (6) | −4.961 |
| CaHCO3+ | CaCO3(s) + H+ CaHCO3+ | (7) | 3.08 |
| CaOH+ | CaCO3(s) + H2O H+ + 2CO32− + CaOH+ | (8) | −21.11 |
| CO2 | 2H+ + CO32− H2O + CO2 | (9) | 16.68 |
| CO2(g) | 2H+ + CO32− H2O + CO2(g) | (10) | 18.15 |
| H2CO3 | 2H+ + CO32− H2CO3 | (11) | 16.70 |
| HCO3− | H+ + CO32− HCO3− | (12) | 10.33 |
| Ca2+ | CaCO3(s) Ca2+ + CO32− | (13) | −8.30 |
| CaOH+ | CaSO4(s) + H2O CaOH+ + H+ + SO42− | (14) | −17.21 |
| CaSO4 | CaSO4(s) CaSO4 | (15) | −2.190 |
| Ca2+/SO42− | CaSO4(s) Ca2+ + SO42− | (16) | −4.378 |
| Ca(OH)2 (s) | CaSO4(s) +2H2O Ca(OH)2(s) + 2H+ + SO42− | (17) | −26.99 |
| CaO(s) | CaSO4(s) +H2O CaO + 2H+ + SO42− | (18) | −37.08 |
| CaSO4·2H2O (s) | CaSO4(s) +2H2O CaSO4·2H2O(s) | (19) | 0.161 |
| H2SO4 | 2H+ + SO42− H2SO4 | (20) | 0.780 |
| HSO4− | H+ + SO42− HSO4− | (21) | 1.982 |
| CaOH+ | Ca(OH)2(s) + H+ H2O + CaOH+ | (22) | 9.785 |
| CaSO4 | CaSO4·2H2O(s) CaSO4 +2H2O | (23) | −2.351 |
| Ca2+ | Ca(OH)2(s) + 2H+ Ca2+ + 2H2O | (24) | 22.62 |
| CaO(s) | Ca(OH)2(s) CaO(s) + H2O | (25) | −10.09 |
| CaSO4(s) | CaSO4·2H2O(s) CaSO4(s) +2H2O | (26) | −0.161 |
| H2SO4 | CaSO4·2H2O(s) H2SO4 + Ca(OH)2(s) | (27) | −26.37 |
| HSO4− | CaSO4·2H2O(s) HSO4− + Ca(OH)2(s) + H+ | (28) | −25.17 |
| SO42− | CaSO4·2H2O(s) SO42− + Ca(OH)2(s) + 2H+ | (29) | −13.99 |
| CaCO3 | Ca2+ + CO2 + H2O CaCO3 + 2H+ | (30) | 22.62 |
| CaHCO3+ | Ca2+ + CO2 + H2O CaHCO3+ + H+ | (31) | 9.785 |
| CaOH+ | Ca2+ + H2O CaOH+ + H+ | (32) | −30.88 |
| CaSO4 | Ca2+ + SO42− CaSO4 | (33) | −41.0 |
| CO2(g) | CO2 CO2(g) | (34) | −4.961 |
| H2CO3 | CO2 + H2O H2CO3 | (35) | 3.080 |
| H2SO4 | 2H+ + SO42− H2SO4 | (36) | −21.11 |
| HCO3− | CO2 + H2O H+ + HCO3− | (37) | 16.68 |
| HSO4− | H+ + SO42− HSO4− | (38) | 18.15 |
| CO32− | CO2 + H2O 2H+ + CO32− | (39) | 16.7 |
| Ca(OH)2(s) | Ca2+ + 2H2O Ca(OH)2(s) + 2H+ | (40) | 10.33 |
| CaCO3(s) | Ca2+ + 2H2O + CO2 CaCO3(s) | (41) | −8.30 |
| CaSO4(s) | Ca2+ + SO42− CaSO4(s) | (42) | −17.21 |
| CaSO4·2H2O(s) | Ca2+ + SO42−+ 2H2O CaSO4·2H2O (s) | (43) | −2.19 |
| CaCO3 | Ca(OH)2(s) + CO2 CaCO3 + H2O | (44) | −4.378 |
| CaHCO3+ | Ca(OH)2(s) + CO2 + H+ CaHCO3+ + H2O | (45) | −26.99 |
| CaOH+ | Ca(OH)2(s) + H+ CaOH++ H2O | (46) | −37.08 |
| CaCO3(s) | Ca(OH)2(s) + CO2 CaCO3(s) + H2O | (47) | 0.161 |
| Pb(OH)2 | Pb2+ + 2H2O Pb(OH)2 + 2H+ | (48) | −17.12 |
| Pb(OH)3− | Pb2+ + 3H2O Pb(OH)3− + 3H+ | (49) | −28.06 |
| Pb3(OH)42+ | 2Pb2+ + 4H2O Pb3(OH)42+ + 4 H+ | (50) | −23.88 |
| PbOH+ | Pb2+ + H2O PbOH+ + H+ | (51) | −7.71 |
| PbSO4 | Pb2+ + SO42− PbSO4 | (52) | 2.69 |
| Pb(OH)2(s) | Pb2+ + 2H2O Pb(OH)2(s) + 2H+ | (53) | −13.6 |
| PbO(s) | Pb2+ + 2H2O PbO(s) + 2H+ | (54) | −12.9 |
| PbSO4(s) | Pb2+ + SO42− PbSO4(s) | (55) | 7.79 |
| Pb(CO3)22− | Pb2+ + 2H2O + 2CO2 Pb(CO3)22− + 4H+ | (56) | −23.26 |
| PbCO3 | Pb2+ + H2O + CO2 PbCO3 + 2H+ | (57) | −10.08 |
| PbCO3OH− | Pb2+ + 2H2O + 2CO2 PbCO3OH− + 3H+ | (58) | −18.48 |
| PbHCO3+ | Pb2+ + H2O + CO2 PbHCO3+ + H+ | (59) | −3.331 |
| Pb3(CO3)2(OH)2(s) | 3Pb2+ + 3H2O + 2CO2 Pb3(CO3)2(OH)2(s) + 6H+ | (60) | −13.96 |
| PbCO3(s) | Pb2+ + H2O + CO2 PbCO3(s) + 2H+ | (61) | −3.48 |
| Contents | Species | Concentrations (mM) |
|---|---|---|
| Saturated Ca(OH)2 (30 mM) | Ca(OH)2 (s) | 7.203 |
| Ca2+ | 19.48 | |
| OH− | 28.44 | |
| CaOH+ | 3.318 | |
| Saturated CaCO3 (10 mM) | CaCO3(s) | 9.847 |
| CaCO3 | 1.094 × 10−2 | |
| CaHCO3+ | 1.415 × 10−4 | |
| OH− | 8.886 × 10−2 | |
| Ca2+ | 0.1415 | |
| CO32− | 4.318 × 10−2 | |
| HCO3− | 9.840 × 10−2 | |
| H2CO3 | 2.560 × 10−5 | |
| CO2 | 2.468 × 10−5 | |
| Saturated CaSO4 (20 mM) | OH− | 1.212 × 10−4 |
| SO42− | 12.03 | |
| CaOH+ | 9.681 × 10−6 | |
| CaSO4 | 4.457 | |
| H2SO4 | 3.236 × 10−13 | |
| HSO4− | 6.279 × 10−5 | |
| Ca2+ | 12.03 | |
| CaSO4·2H2O | 3.516 |
| Contents | Species | Concentrations (mM) | Removal Efficiency of SO42−/Pb2+ |
|---|---|---|---|
| 1.55 M Ca(OH)2 + 1.53 M SO42− | OH− | 26.53 | 99.15% |
| CaOH+ | 3.605 | ||
| CaSO4 | 4.457 | ||
| Ca2+ | 25.06 | ||
| SO42− | 8.668 | ||
| CaSO4·2H2O | 1517 | ||
| Pb(OH)2 | 3.447 × 10−4 | 0 | |
| Pb(OH)3− | 1.416 × 10−2 | ||
| Carbonation after filtration at pH 7 (0.1 M CO2) | Ca2+ | 0.4509 | 99.15% |
| SO42− | 13.01 | ||
| CaSO4 | 0.1118 | ||
| HCO3− | 57.32 | ||
| CaCO3(s) | 32.44 | ||
| PbCO3(s) | 1.397 × 10−2 | 96.35% | |
| PbCO3 | 2.512 × 10−4 | ||
| PbHCO3+ | 1.834 × 10−4 | ||
| Pb(CO3)22− | 7.332 × 10−5 | ||
| Carbonation without filtration at pH 7 (0.1 M CO2) | CaSO4 | 4.457 | 69.61% |
| HCO3− | 20.48 | ||
| Ca2+ | 4.098 | ||
| SO42− | 460.43 | ||
| CaCO3(s) | 476.2 | ||
| CaSO4·2H2O | 1065 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gu, S.; Fu, B.; Fujita, T.; Ahn, J.W. Thermodynamic Simulations for Determining the Recycling Path of a Spent Lead-Acid Battery Electrolyte Sample with Ca(OH)2. Appl. Sci. 2019, 9, 2262. https://doi.org/10.3390/app9112262
Gu S, Fu B, Fujita T, Ahn JW. Thermodynamic Simulations for Determining the Recycling Path of a Spent Lead-Acid Battery Electrolyte Sample with Ca(OH)2. Applied Sciences. 2019; 9(11):2262. https://doi.org/10.3390/app9112262
Chicago/Turabian StyleGu, Shuai, Bitian Fu, Toyohisa Fujita, and Ji Whan Ahn. 2019. "Thermodynamic Simulations for Determining the Recycling Path of a Spent Lead-Acid Battery Electrolyte Sample with Ca(OH)2" Applied Sciences 9, no. 11: 2262. https://doi.org/10.3390/app9112262
APA StyleGu, S., Fu, B., Fujita, T., & Ahn, J. W. (2019). Thermodynamic Simulations for Determining the Recycling Path of a Spent Lead-Acid Battery Electrolyte Sample with Ca(OH)2. Applied Sciences, 9(11), 2262. https://doi.org/10.3390/app9112262