Loss of the Mitochondrial Fission GTPase Drp1 Contributes to Neurodegeneration in a Drosophila Model of Hereditary Spastic Paraplegia



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Drosophila Stocks and Conditions
2.2. Semi-Quantitative PCR
2.3. Immunoblot Analysis
2.4. Histology and Immunomicroscopy
2.5. Image Analysis
2.5.1. Drp1 Localisation Analysis
2.5.2. Mitochondrial Analysis
2.5.3. Autophagic Flux Analysis
2.5.4. Quantification of Ubiquitinylated Proteins
2.6. Live Mitochondrial Trafficking in Axons
2.7. Electron Microscopy
2.8. Ultrastructural Analysis
2.9. Behavioural Analysis
2.10. Statistical Analysis
3. Results
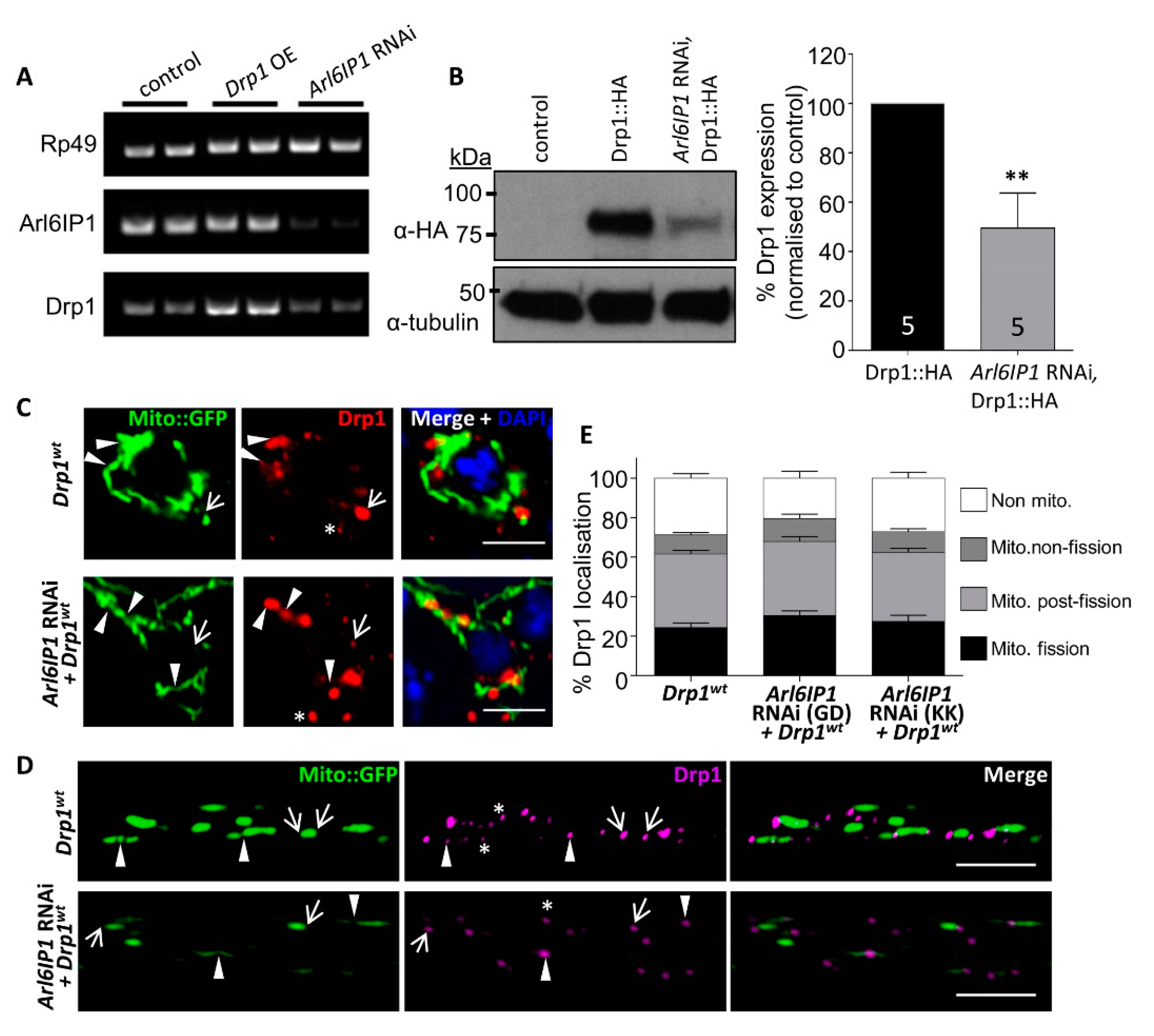
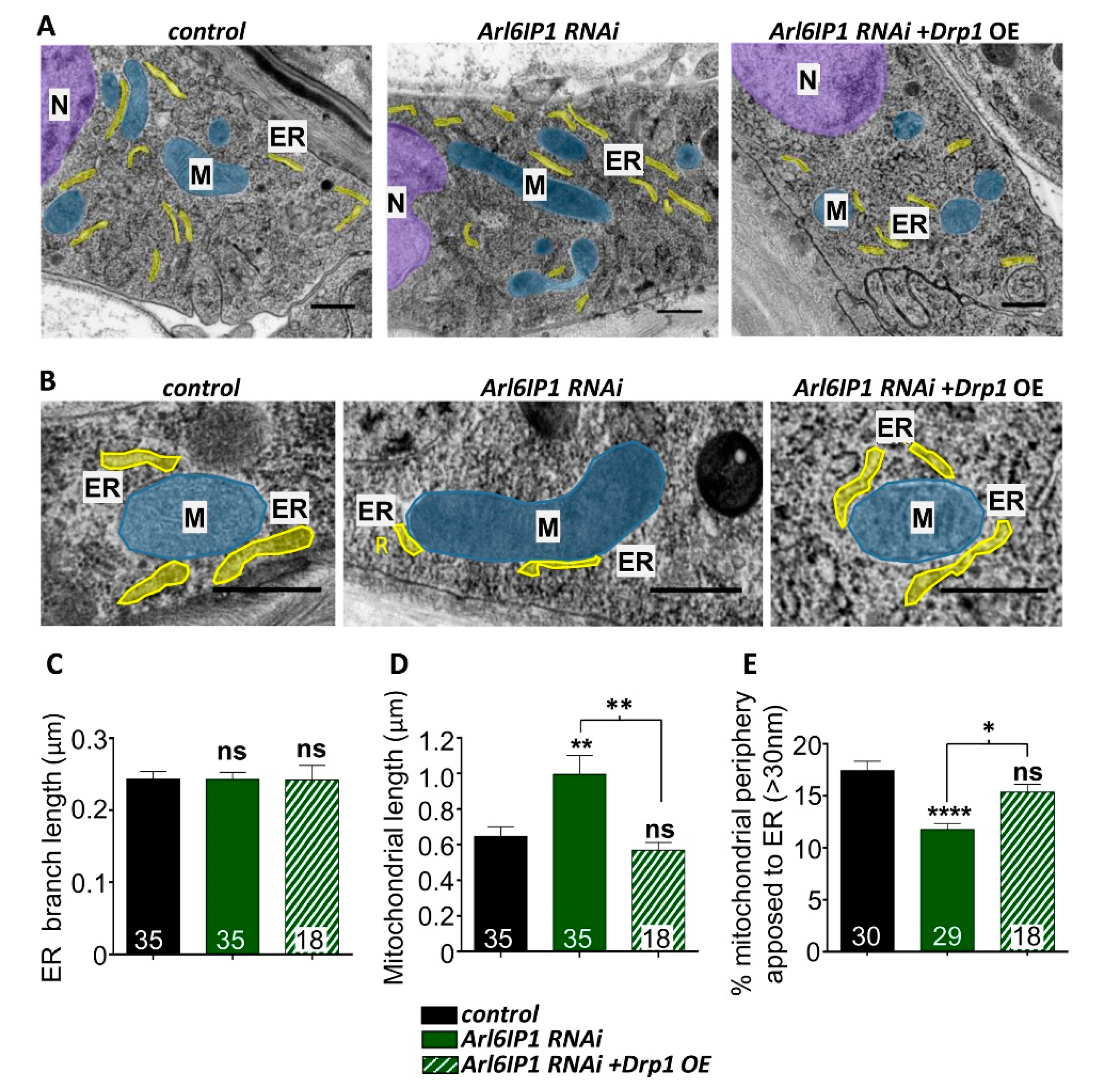
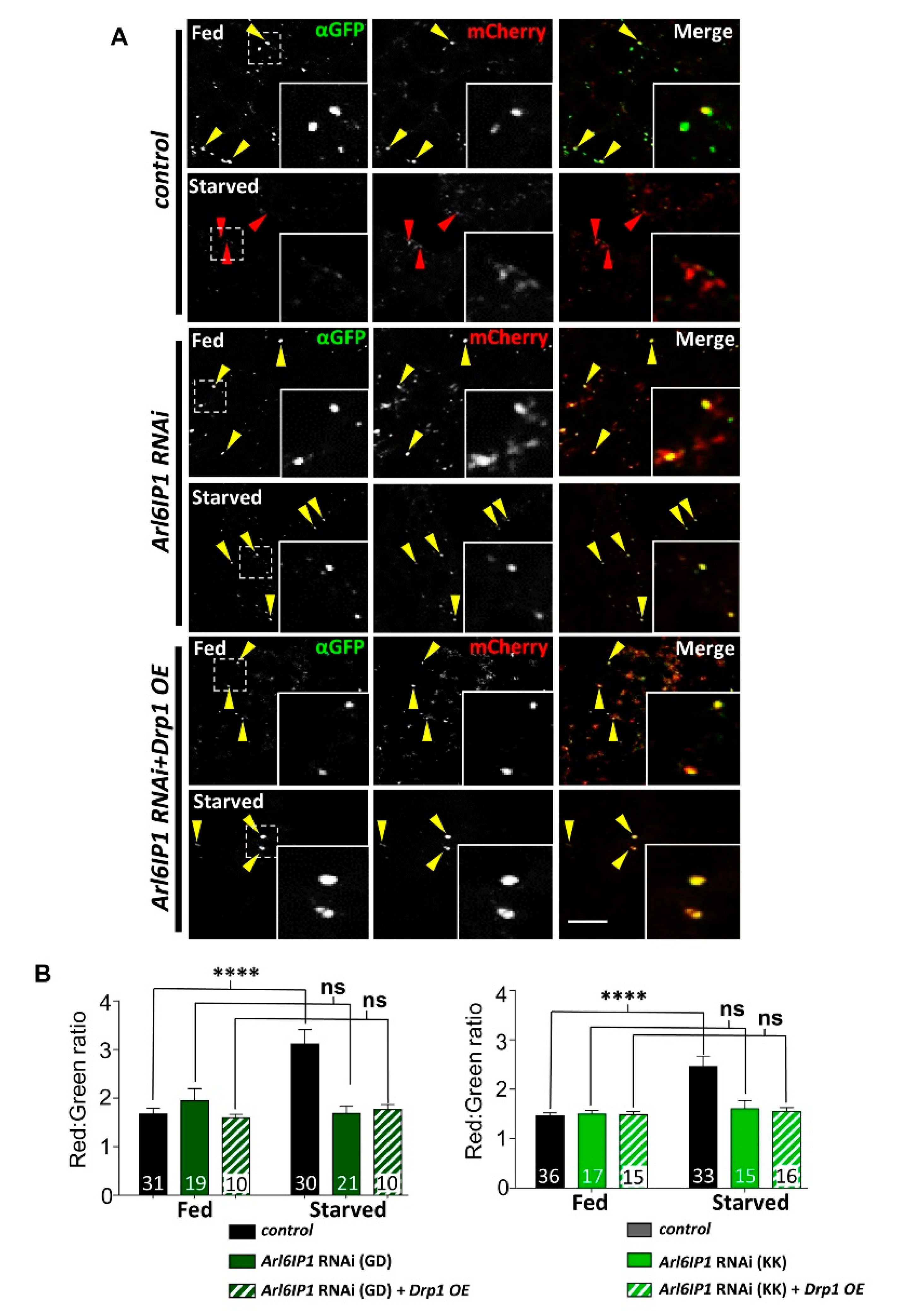
3.1. Mitochondrial Fission Is Disrupted in an Arl6IP1 Knockdown Model of HSP
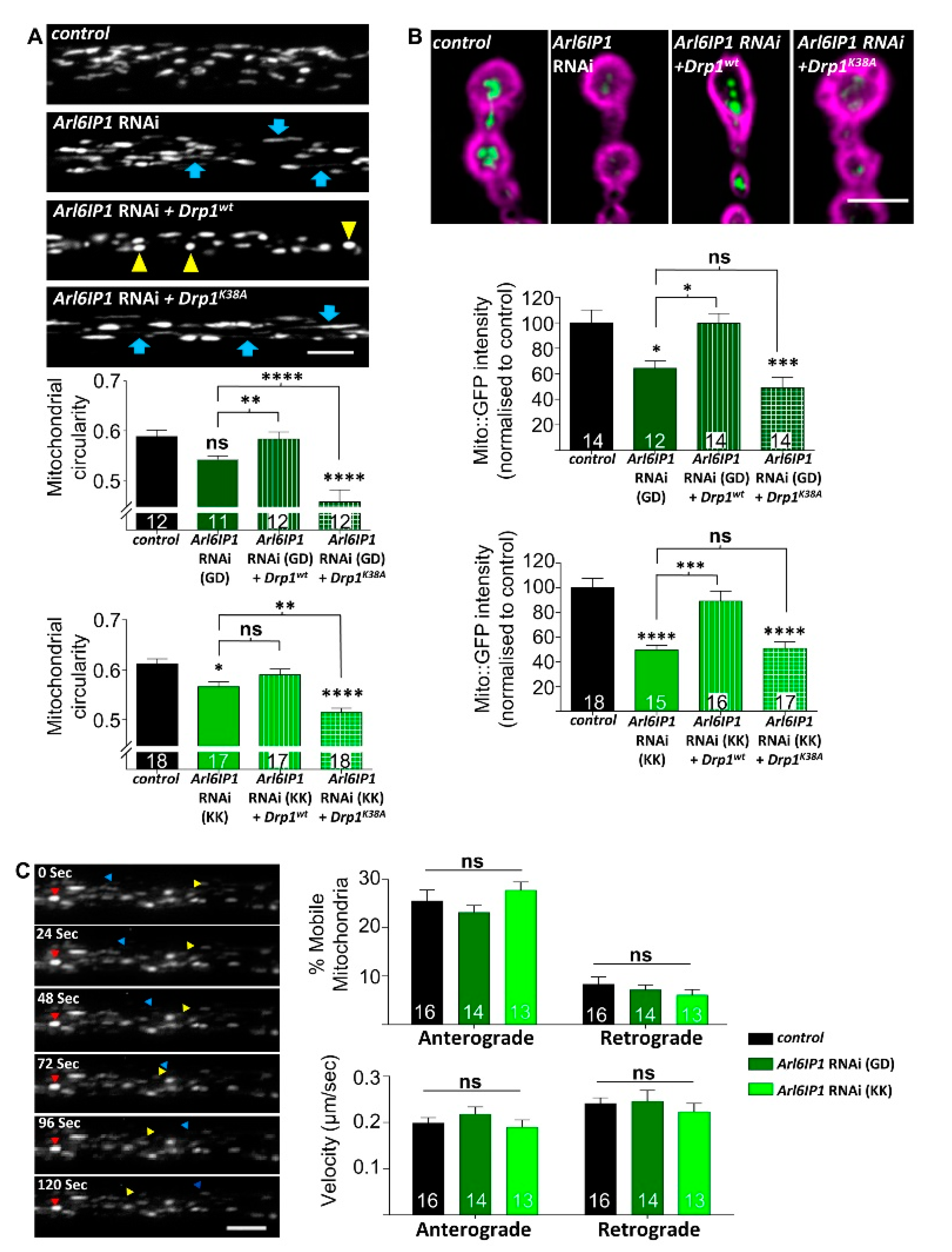
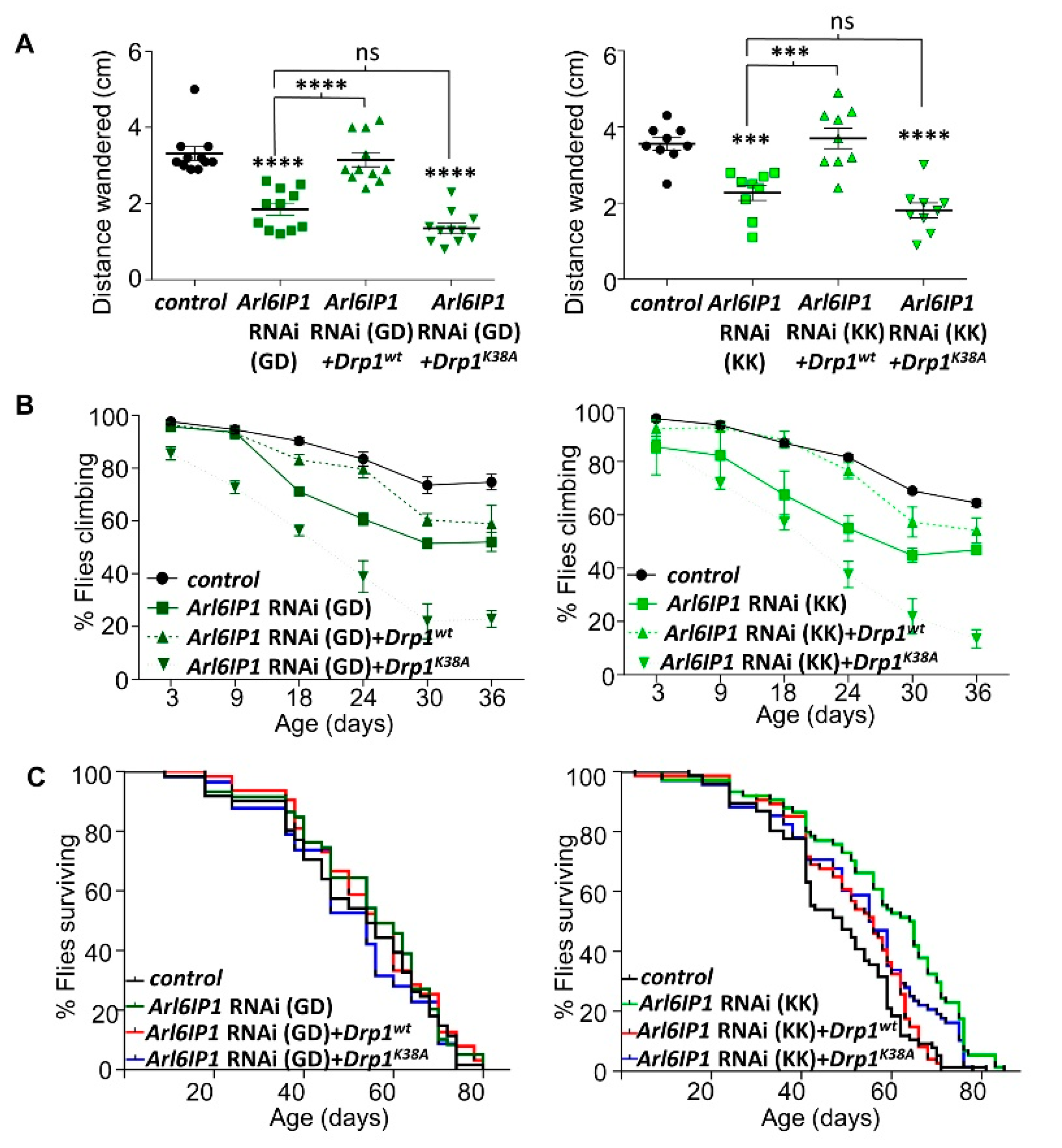
3.2. Increased Drp1 Activity Rescues Mitochondrial Morphology and Neurodegeneration in Arl6IP1 Knockdown Flies
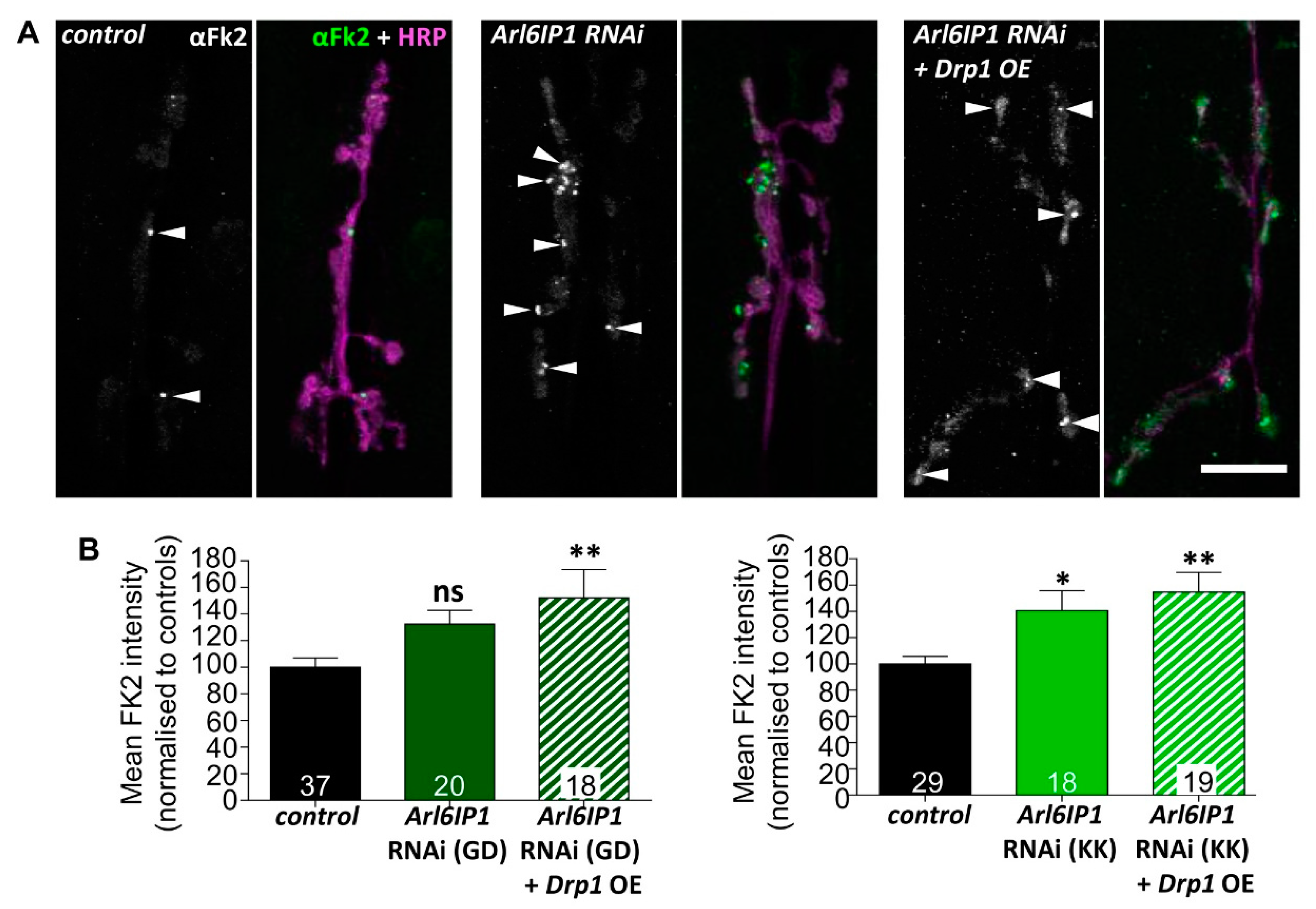
3.3. Protein Processing Pathways Are Impaired in Arl6IP1 Knockdown Flies Independent of Drp1 Expression
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tilokani, L.; Nagashima, S.; Paupe, V.; Prudent, J. Mitochondrial dynamics: Overview of molecular mechanisms. Essays Biochem. 2018, 62, 341–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smirnova, E.; Griparic, L.; Shurland, D.-L.; van der Bliek, A.M. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol. Biol. Cell 2001, 12, 2245–2256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pernas, L.; Scorrano, L. Mito-Morphosis: Mitochondrial fusion, fission, and cristae remodeling as key mediators of cellular function. Annu. Rev. Physiol. 2016, 78, 505–531. [Google Scholar] [CrossRef]
- Itoh, K.; Murata, D.; Kato, T.; Yamada, T.; Araki, Y.; Saito, A.; Adachi, Y.; Igarashi, A.; Li, S.; Pletnikov, M.; et al. Brain-specific Drp1 regulates postsynaptic endocytosis and dendrite formation independently of mitochondrial division. Elife 2019, 8, 11809–11822. [Google Scholar] [CrossRef]
- Vantaggiato, C.; Castelli, M.; Giovarelli, M.; Orso, G.; Bassi, M.T.; Clementi, E.; De Palma, C. The fine tuning of drp1-dependent mitochondrial remodeling and autophagy controls neuronal differentiation. Front. Cell. Neurosci. 2019, 13, 120. [Google Scholar] [CrossRef] [Green Version]
- Shields, L.Y.; Kim, H.; Zhu, L.; Haddad, D.; Berthet, A.; Pathak, D.; Lam, M.; Ponnusamy, R.; Diaz-Ramirez, L.G.; Gill, T.M.; et al. Dynamin-related protein 1 is required for normal mitochondrial bioenergetic and synaptic function in CA1 hippocampal neurons. Cell Death Dis. 2015, 6, e1725. [Google Scholar] [CrossRef] [Green Version]
- Manczak, M.; Calkins, M.J.; Reddy, P.H. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: Implications for neuronal damage. Hum. Mol. Genet. 2011, 20, 2495–2509. [Google Scholar] [CrossRef]
- Wang, W.; Yin, J.; Ma, X.; Zhao, F.; Siedlak, S.L.; Wang, Z.; Torres, S.; Fujioka, H.; Xu, Y.; Perry, G.; et al. Inhibition of mitochondrial fragmentation protects against Alzheimer’s disease in rodent model. Hum. Mol. Genet. 2017, 26, 4118–4131. [Google Scholar] [CrossRef] [Green Version]
- Filichia, E.; Hoffer, B.; Qi, X.; Luo, Y. Inhibition of Drp1 mitochondrial translocation provides neural protection in dopaminergic system in a Parkinson’s disease model induced by MPTP. Sci. Rep. 2016, 6, 32656. [Google Scholar] [CrossRef] [Green Version]
- Joshi, A.U.; Saw, N.L.; Vogel, H.; Cunnigham, A.D.; Shamloo, M.; Mochly-Rosen, D. Inhibition of Drp1/Fis1 interaction slows progression of amyotrophic lateral sclerosis. EMBO Mol. Med. 2018, 10, e8166. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.R.; Lackner, L.L.; West, M.; DiBenedetto, J.R.; Nunnari, J.; Voeltz, G.K. ER tubules mark sites of mitochondrial division. Science 2011, 334, 358–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korobova, F.; Ramabhadran, V.; Higgs, H.N. An actin-dependent step in mitochondrial fission mediated by the ER-associated Formin INF2. Science 2013, 339, 464–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, W.K.; Chakrabarti, R.; Fan, X.; Schoenfeld, L.; Strack, S.; Higgs, H.N. Receptor-mediated Drp1 oligomerization on endoplasmic reticulum. J. Cell Biol. 2017, 216, 4123–4139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cribbs, J.T.; Strack, S. Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. 2007, 8, 939–944. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.R.; Blackstone, C. Cyclic AMP-dependent protein kinase phosphorylation of Drp1 regulates its GTPase activity and mitochondrial morphology. J. Biol. Chem. 2007, 283, 21583–21587. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.; Kim, J.E. PDI-mediated S-nitrosylation of DRP1 facilitates DRP1-S616 phosphorylation and mitochondrial fission in CA1 neurons. Cell Death Dis. 2018, 9, 869. [Google Scholar] [CrossRef]
- Blackstone, C. Converging cellular themes for the hereditary spastic paraplegias. Curr. Opin. Neurobiol. 2018, 51, 139–146. [Google Scholar] [CrossRef]
- Fowler, P.C.; O’Sullivan, N.C. ER-shaping proteins are required for ER and mitochondrial network organization in motor neurons. Hum. Mol. Genet. 2016, 25, 2827–2837. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Guo, X.; Niu, L.; Li, X.; Sun, F.; Hu, J.; Wang, X.; Shen, K. Atlastin-1 regulates morphology and function of endoplasmic reticulum in dendrites. Nat. Commun. 2019, 10, 568. [Google Scholar] [CrossRef] [Green Version]
- Lavie, J.; Serrat, R.; Bellance, N.; Courtand, G.; Dupuy, J.W.; Tesson, C.; Coupry, I.; Brice, A.; Lacombe, D.; Durr, A.; et al. Mitochondrial morphology and cellular distribution are altered in SPG31 patients and are linked to DRP1 hyperphosphorylation. Hum. Mol. Genet. 2017, 26, 674–685. [Google Scholar] [CrossRef] [PubMed]
- Dietzl, G.; Chen, D.; Schnorrer, F.; Su, K.-C.; Barinova, Y.; Fellner, M.; Gasser, B.; Kinsey, K.; Oppel, S.; Scheiblauer, S.; et al. A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature 2007, 448, 151–156. [Google Scholar] [CrossRef] [PubMed]
- DuBoff, B.; Götz, J.; Feany, M.B. Tau promotes neurodegeneration via DRP1 mislocalization in vivo. Neuron 2012, 75, 618–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verstreken, P.; Ly, C.V.; Venken, K.J.T.; Koh, T.-W.; Zhou, Y.; Bellen, H.J. Synaptic mitochondria are critical for mobilization of reserve pool vesicles at Drosophila neuromuscular junctions. Neuron 2005, 47, 365–378. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Lee, G.; Chung, J. The PINK1–Parkin pathway is involved in the regulation of mitochondrial remodeling process. Biochem. Biophys. Res. Commun. 2009, 378, 518–523. [Google Scholar] [CrossRef]
- Perrin, L.; Bloyer, S.; Ferraz, C.; Agrawal, N.; Sinha, P.; Dura, J.M. The Leucine Zipper Motif of the Drosophila AF10 Homologue Can inhibit PRE-mediated repression: Implications for leukemogenic activity of human MLL-AF10 fusions. Mol. Cell. Biol. 2003, 23, 119–130. [Google Scholar] [CrossRef] [Green Version]
- Bushey, D.; Tononi, G.; Cirelli, C. The Drosophila fragile X mental retardation gene regulates sleep need. J. Neurosci. 2009, 29, 1948–1961. [Google Scholar] [CrossRef] [Green Version]
- Aberle, H.; Haghighi, A.P.; Fetter, R.D.; McCabe, B.D.; Magalhães, T.R.; Goodman, C.S. wishful thinking encodes a BMP type II receptor that regulates synaptic growth in Drosophila. Neuron 2002, 33, 545–558. [Google Scholar] [CrossRef] [Green Version]
- Asha, H.; Nagy, I.; Kovacs, G.; Stetson, D.; Ando, I.; Dearolf, C.R. Analysis of Ras-induced overproliferation in Drosophila hemocytes. Genetics 2003, 163, 203–215. [Google Scholar]
- Pilling, A.D.; Horiuchi, D.; Lively, C.M.; Saxton, W.M. Kinesin-1 and dynein are the primary motors for fast transport of mitochondria in Drosophila motor axons. Mol. Biol. Cell 2006, 17, 2057–2068. [Google Scholar] [CrossRef] [Green Version]
- Rusten, T.E.; Vaccari, T.; Lindmo, K.; Rodahl, L.M.W.; Nezis, I.P.; Sem-Jacobsen, C.; Wendler, F.; Vincent, J.-P.; Brech, A.; Bilder, D.; et al. ESCRTs and Fab1 regulate distinct steps of autophagy. Curr. Biol. 2007, 17, 1817–1825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Sullivan, N.C.; Dräger, N.; O’Kane, C.J. Characterization of the Drosophila Atlastin interactome reveals VCP as a functionally related interactor. J. Genet. Genomics 2013, 6, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Stewart, B.A.; Atwood, H.L.; Renger, J.J.; Wang, J.; Wu, C.F. Improved stability of Drosophila larval neuromuscular preparations in haemolymph-like physiological solutions. J. Comp. Physiol. A 1994, 175, 179–191. [Google Scholar] [CrossRef] [PubMed]
- Byrne, D.J.; Harmon, M.J.; Simpson, J.C.; Blackstone, C.; O’Sullivan, N.C. Roles for the VCP co-factors Npl4 and Ufd1 in neuronal function in Drosophila melanogaster. J. Genet. Genomics 2017, 44, 493–501. [Google Scholar] [CrossRef]
- Benard, G.; Karbowski, M. Mitochondrial fusion and division: Regulation and role in cell viability. Semin. Cell Dev. Biol. 2009, 20, 356–374. [Google Scholar] [CrossRef] [Green Version]
- Rowland, A.A.; Voeltz, G.K. Endoplasmic reticulum–mitochondria contacts: Function of the junction. Nat. Rev. Mol. Cell Biol. 2012, 13, 607–615. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Zhang, M.; Zhang, C.; Zhou, X. Nuclear autophagy degrades a geminivirus nuclear protein to restrict viral infection in solanaceous plants. New Phytol. 2020, 225, 1746–1761. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.R.; Lingeman, E.; Ahmed, S.; Corn, J.E. Atlastins remodel the endoplasmic reticulum for selective autophagy. J. Cell Biol. 2018, 217, 3354–3367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, J.R.; Shen, W.L.; Yan, C.; Gao, P.J. Downregulation of dynamin-related protein 1 contributes to impaired autophagic flux and angiogenic function in senescent endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1413–1422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nezis, I.P.; Shravage, B.V.; Sagona, A.P.; Johansen, T.; Baehrecke, E.H.; Stenmark, H. Autophagy as a trigger for cell death: Autophagic degradation of inhibitor of apoptosis dBruce controls DNA fragmentation during late oogenesis in Drosophila. Autophagy 2010, 6, 1214–1215. [Google Scholar] [CrossRef] [Green Version]
- Mauvezin, C.; Ayala, C.; Braden, C.R.; Kim, J.; Neufeld, T.P. Assays to monitor autophagy in Drosophila. Methods 2014, 68, 134–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oz-Levi, D.; Ben-Zeev, B.; Ruzzo, E.K.; Hitomi, Y.; Gelman, A.; Pelak, K.; Anikster, Y.; Reznik-Wolf, H.; Bar-Joseph, I.; Olender, T.; et al. Mutation in TECPR2 reveals a role for autophagy in hereditary spastic paraparesis. Am. J. Hum. Genet. 2012, 91, 1065–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vantaggiato, C.; Crimella, C.; Airoldi, G.; Polishchuk, R.; Bonato, S.; Brighina, E.; Scarlato, M.; Musumeci, O.; Toscano, A.; Martinuzzi, A.; et al. Defective autophagy in spastizin mutated patients with hereditary spastic paraparesis type 15. Brain 2013, 136, 3119–3139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varga, R.-E.; Khundadze, M.; Damme, M.; Nietzsche, S.; Hoffmann, B.; Stauber, T.; Koch, N.; Hennings, J.C.; Franzka, P.; Huebner, A.K.; et al. In vivo evidence for lysosome depletion and impaired autophagic clearance in hereditary spastic paraplegia type SPG11. PLoS Genet. 2015, 11, e1005454. [Google Scholar] [CrossRef] [PubMed]
- Behrendt, L.; Kurth, I.; Kaether, C. A disease causing ATLASTIN 3 mutation affects multiple endoplasmic reticulum-related pathways. Cell. Mol. Life Sci. 2019, 76, 1433–1445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E.; et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006, 441, 880–884. [Google Scholar] [CrossRef] [PubMed]
- Novarino, G.; Fenstermaker, A.G.; Zaki, M.S.; Hofree, M.; Silhavy, J.L.; Heiberg, A.D.; Abdellateef, M.; Rosti, B.; Scott, E.; Mansour, L.; et al. Exome sequencing links corticospinal motor neuron disease to common neurodegenerative disorders. Science 2014, 343, 506–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gusdon, A.M.; Callio, J.; Distefano, G.; O’Doherty, R.M.; Goodpaster, B.H.; Coen, P.M.; Chu, C.T. Exercise increases mitochondrial complex I activity and DRP1 expression in the brains of aged mice. Exp. Gerontol. 2017, 90, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Klemm, R.W.; Norton, J.P.; Cole, R.A.; Li, C.S.; Park, S.H.; Crane, M.M.; Li, L.; Jin, D.; Boye-Doe, A.; Liu, T.Y.; et al. A conserved role for Atlastin GTPases in regulating lipid droplet size. Cell Rep. 2013, 3, 1465–1475. [Google Scholar] [CrossRef] [Green Version]
- Mancini, C.; Roncaglia, P.; Brussino, A.; Stevanin, G.; Lo Buono, N.; Krmac, H.; Maltecca, F.; Gazzano, E.; Bartoletti Stella, A.; Calvaruso, M.A.; et al. Genome-wide expression profiling and functional characterization of SCA28 lymphoblastoid cell lines reveal impairment in cell growth and activation of apoptotic pathways. BMC Med. Genom. 2013, 6, 22. [Google Scholar] [CrossRef] [PubMed]
- Rambold, A.S.; Kostelecky, B.; Elia, N.; Lippincott-Schwartz, J. Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc. Natl. Acad. Sci. USA 2011, 108, 10190–10195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomes, L.C.; Di Benedetto, G.; Scorrano, L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat. Cell Biol. 2011, 13, 589–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Vicente, M. Neuronal mitophagy in neurodegenerative diseases. Front. Mol. Neurosci. 2017, 10, 64. [Google Scholar] [CrossRef] [Green Version]
- Drummond, M.J.; Addison, O.; Brunker, L.; Hopkins, P.N.; McClain, D.A.; Lastayo, P.C.; Marcus, R.L. Downregulation of E3 ubiquitin ligases and mitophagy-related genes in skeletal muscle of physically inactive, frail older women: A cross-sectional comparison. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2014, 69, 1040–1048. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Yun, J.; Liu, J.; Malide, D.; Liu, C.; Rovira, I.I.; Holmström, K.M.; Fergusson, M.M.; Yoo, Y.H.; Combs, C.A.; et al. Measuring in vivo mitophagy. Mol. Cell 2015, 60, 685–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rana, A.; Oliveira, M.P.; Khamoui, A.V.; Aparicio, R.; Rera, M.; Rossiter, H.B.; Walker, D.W. Promoting Drp1-mediated mitochondrial fission in midlife prolongs healthy lifespan of Drosophila melanogaster. Nat. Commun. 2017, 8, 448. [Google Scholar] [CrossRef]
- Kageyama, Y.; Zhang, Z.; Roda, R.; Fukaya, M.; Wakabayashi, J.; Wakabayashi, N.; Kensler, T.W.; Hemachandra Reddy, P.; Iijima, M.; Sesaki, H. Mitochondrial division ensures the survival of postmitotic neurons by suppressing oxidative damage. J. Cell Biol. 2012, 197, 535–551. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Hu, C.; Huang, J.; Liu, W.; Lai, W.; Leng, F.; Tang, Q.; Liu, Y.; Wang, Q.; Zhou, M.; et al. ROCK1 induces dopaminergic nerve cell apoptosis via the activation of Drp1-mediated aberrant mitochondrial fission in Parkinson’s disease. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef]
- Farmer, B.C.; Walsh, A.E.; Kluemper, J.C.; Johnson, L.A. Lipid droplets in neurodegenerative disorders. Front. Neurosci. 2020, 14, 742. [Google Scholar] [CrossRef]
- Rambold, A.S.; Cohen, S.; Lippincott-Schwartz, J. Fatty acid trafficking in starved cells: Regulation by lipid droplet lipolysis, autophagy, and mitochondrial fusion dynamics. Dev. Cell 2015, 32, 678–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larrea, D.; Pera, M.; Gonnelli, A.; Quintana-Cabrera, R.; Akman, H.O.; Guardia-Laguarta, C.; Velasco, K.R.; Area-Gomez, E.; Dal Bello, F.; De Stefani, D.; et al. MFN2 mutations in charcot-marie-Tooth disease alter mitochondria-Associated er membrane function but do not impair bioenergetics. Hum. Mol. Genet. 2019, 28, 1782–1800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandoval, H.; Yao, C.K.; Chen, K.; Jaiswal, M.; Donti, T.; Lin, Y.Q.; Bayat, V.; Xiong, B.; Zhang, K.; David, G.; et al. Mitochondrial fusion but not fission regulates larval growth and synaptic development through steroid hormone production. Elife 2014, 3, 1–23. [Google Scholar] [CrossRef]
- Darios, F.; Mochel, F.; Stevanin, G. Lipids in the physiopathology of hereditary spastic paraplegias. Front. Neurosci. 2020, 14, 74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, H.; Dodson, M.W.; Huang, H.; Guo, M. The Parkinson’s disease genes pink1 and parkin promote mitochondrial fission and/or inhibit fusion in Drosophila. Proc. Natl. Acad. Sci. USA 2008, 105, 14503–14508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Touvier, T.; De Palma, C.; Rigamonti, E.; Scagliola, A.; Incerti, E.; Mazelin, L.; Thomas, J.L.; D’Antonio, M.; Politi, L.; Schaeffer, L.; et al. Muscle-specific Drp1 overexpression impairs skeletal muscle growth via translational attenuation. Cell Death Dis. 2015, 6, e1663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denton, K.; Mou, Y.; Xu, C.-C.; Shah, D.; Chang, J.; Blackstone, C.; Li, X.-J. Impaired mitochondrial dynamics underlie axonal defects in hereditary spastic paraplegias. Hum. Mol. Genet. 2018, 27, 2517–2530. [Google Scholar] [CrossRef] [PubMed] [Green Version]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fowler, P.C.; Byrne, D.J.; Blackstone, C.; O'Sullivan, N.C. Loss of the Mitochondrial Fission GTPase Drp1 Contributes to Neurodegeneration in a Drosophila Model of Hereditary Spastic Paraplegia. Brain Sci. 2020, 10, 646. https://doi.org/10.3390/brainsci10090646
Fowler PC, Byrne DJ, Blackstone C, O'Sullivan NC. Loss of the Mitochondrial Fission GTPase Drp1 Contributes to Neurodegeneration in a Drosophila Model of Hereditary Spastic Paraplegia. Brain Sciences. 2020; 10(9):646. https://doi.org/10.3390/brainsci10090646
Chicago/Turabian StyleFowler, Philippa C., Dwayne J. Byrne, Craig Blackstone, and Niamh C. O'Sullivan. 2020. "Loss of the Mitochondrial Fission GTPase Drp1 Contributes to Neurodegeneration in a Drosophila Model of Hereditary Spastic Paraplegia" Brain Sciences 10, no. 9: 646. https://doi.org/10.3390/brainsci10090646
APA StyleFowler, P. C., Byrne, D. J., Blackstone, C., & O'Sullivan, N. C. (2020). Loss of the Mitochondrial Fission GTPase Drp1 Contributes to Neurodegeneration in a Drosophila Model of Hereditary Spastic Paraplegia. Brain Sciences, 10(9), 646. https://doi.org/10.3390/brainsci10090646