
Upper and Lower Motor Neuron Degenerations Are Somatotopically Related and Temporally Ordered in the Sod1 Mouse Model of Amyotrophic Lateral Sclerosis

 ,
,
Abstract
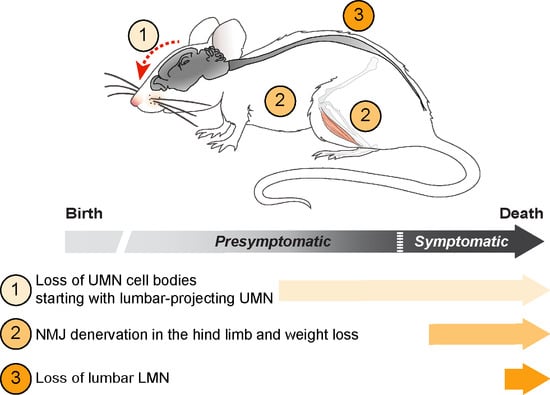
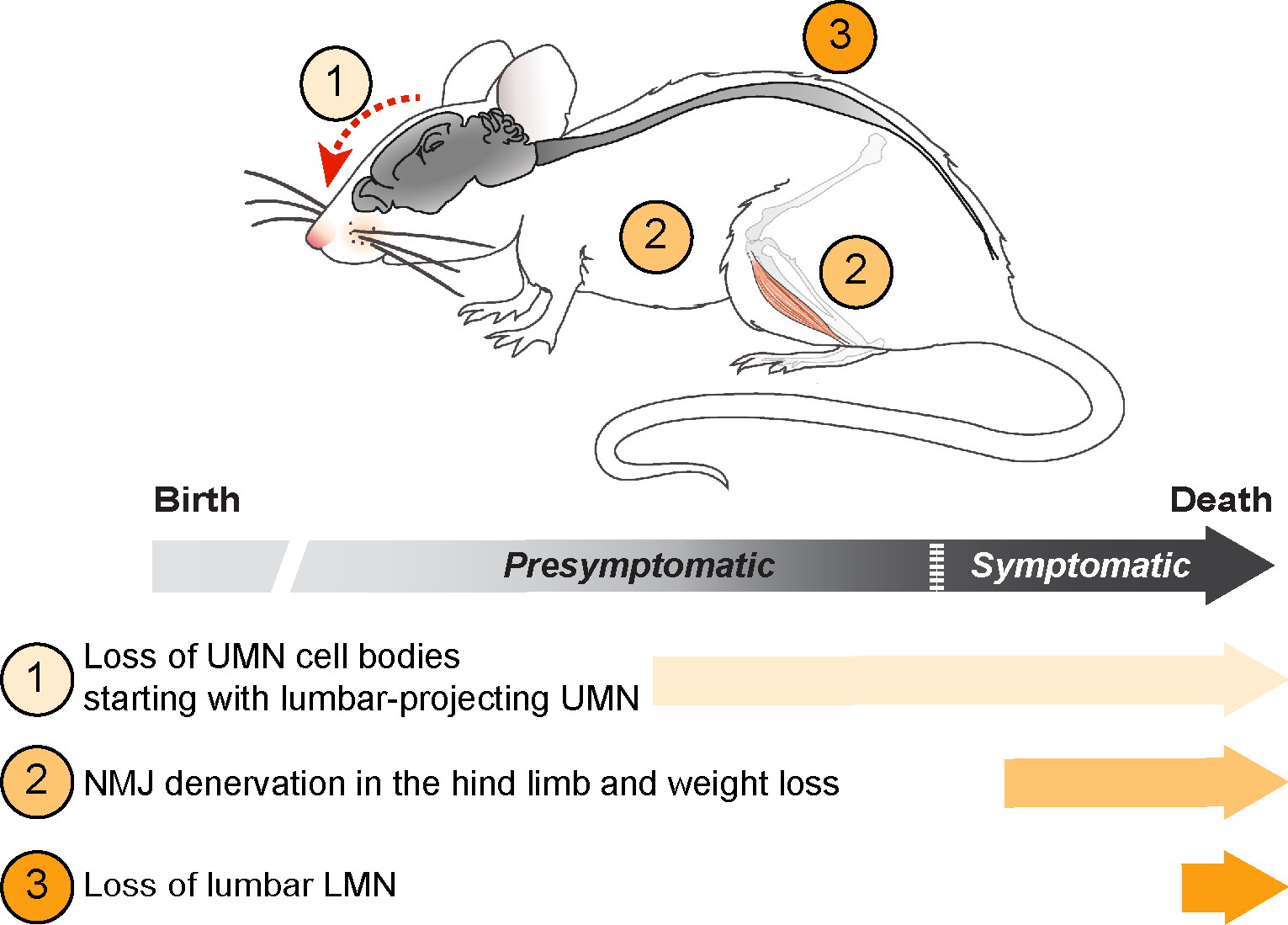
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
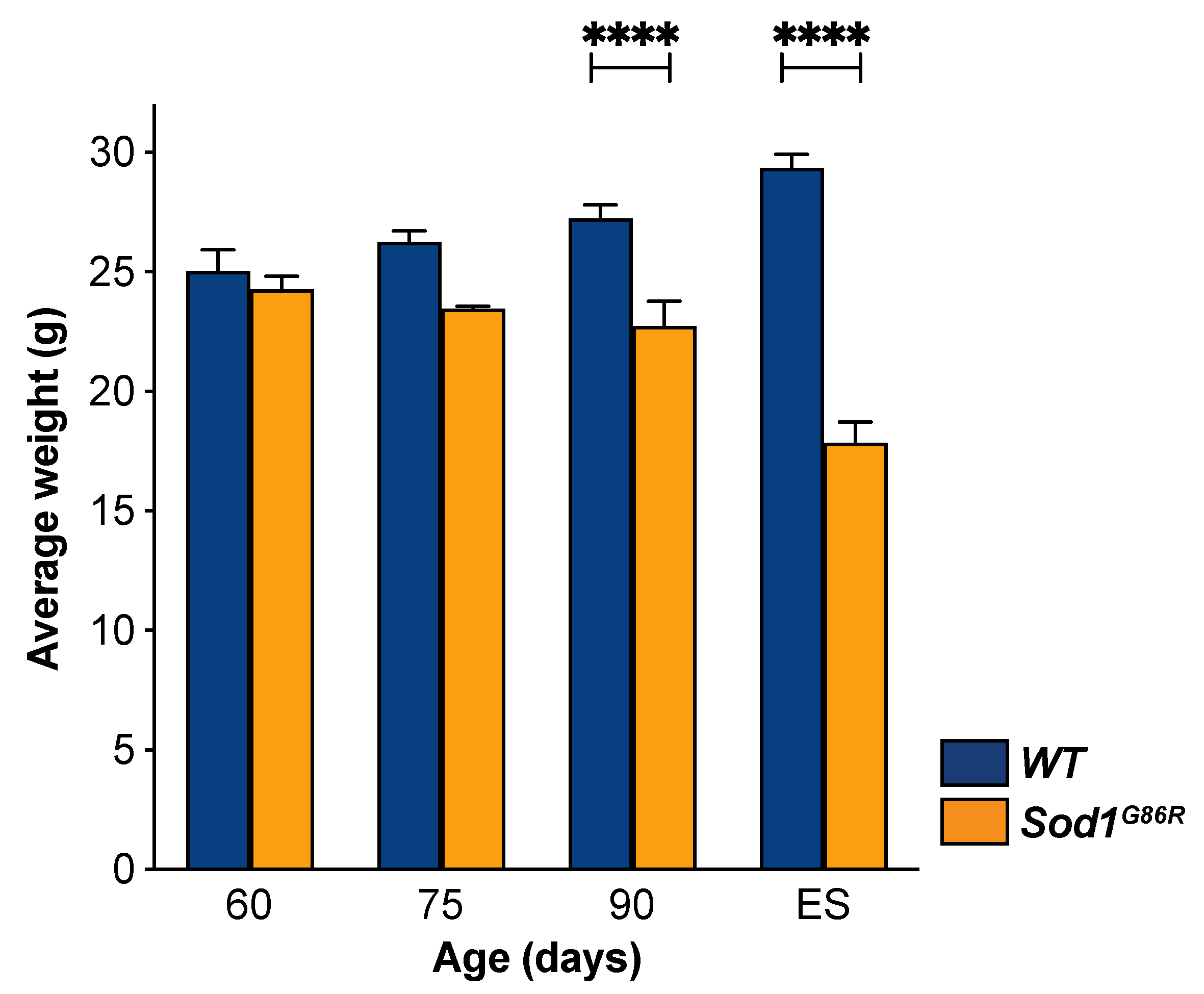{kind=link}
1. Introduction
2. Material and Methods
2.1. Animals
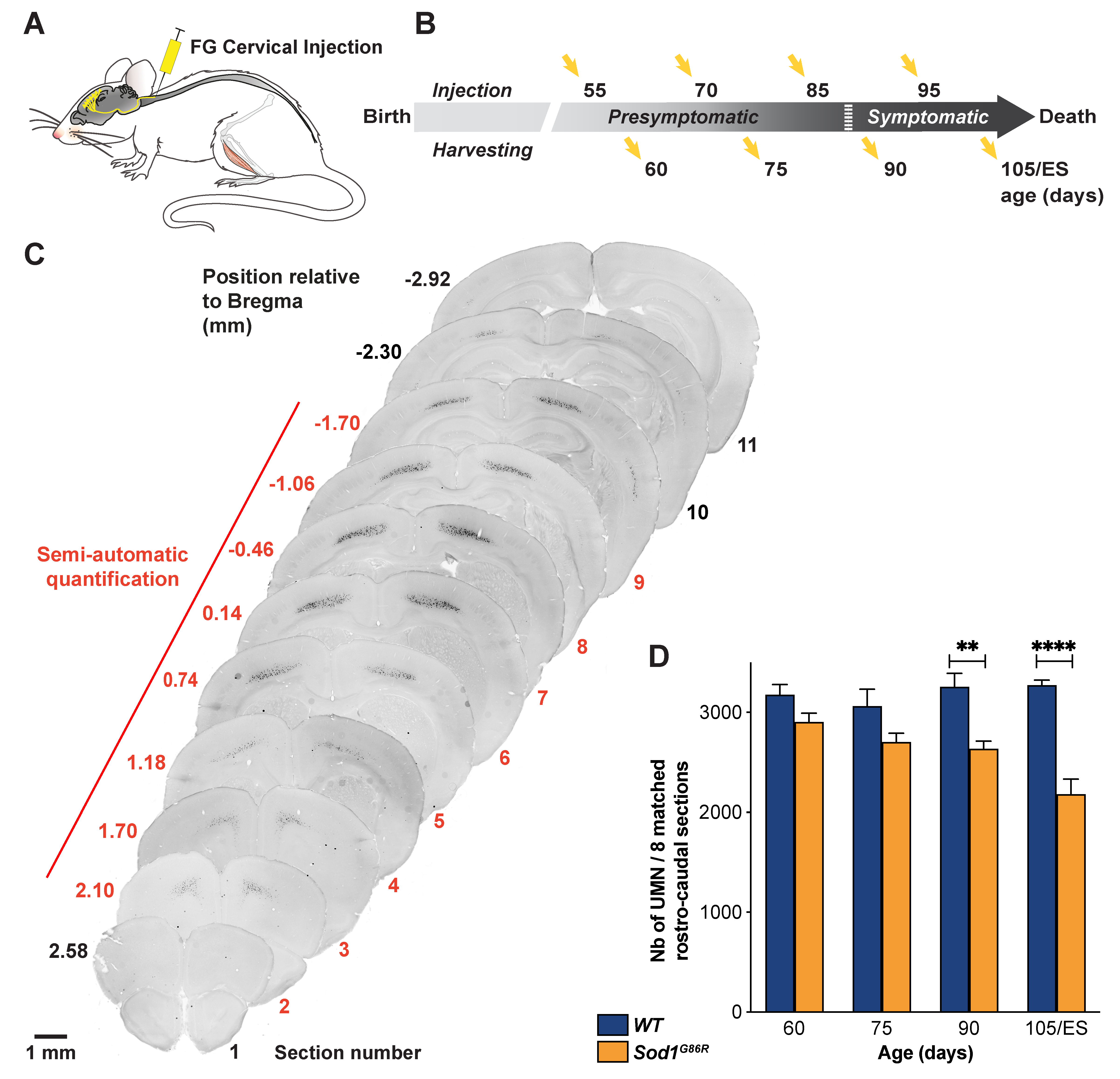
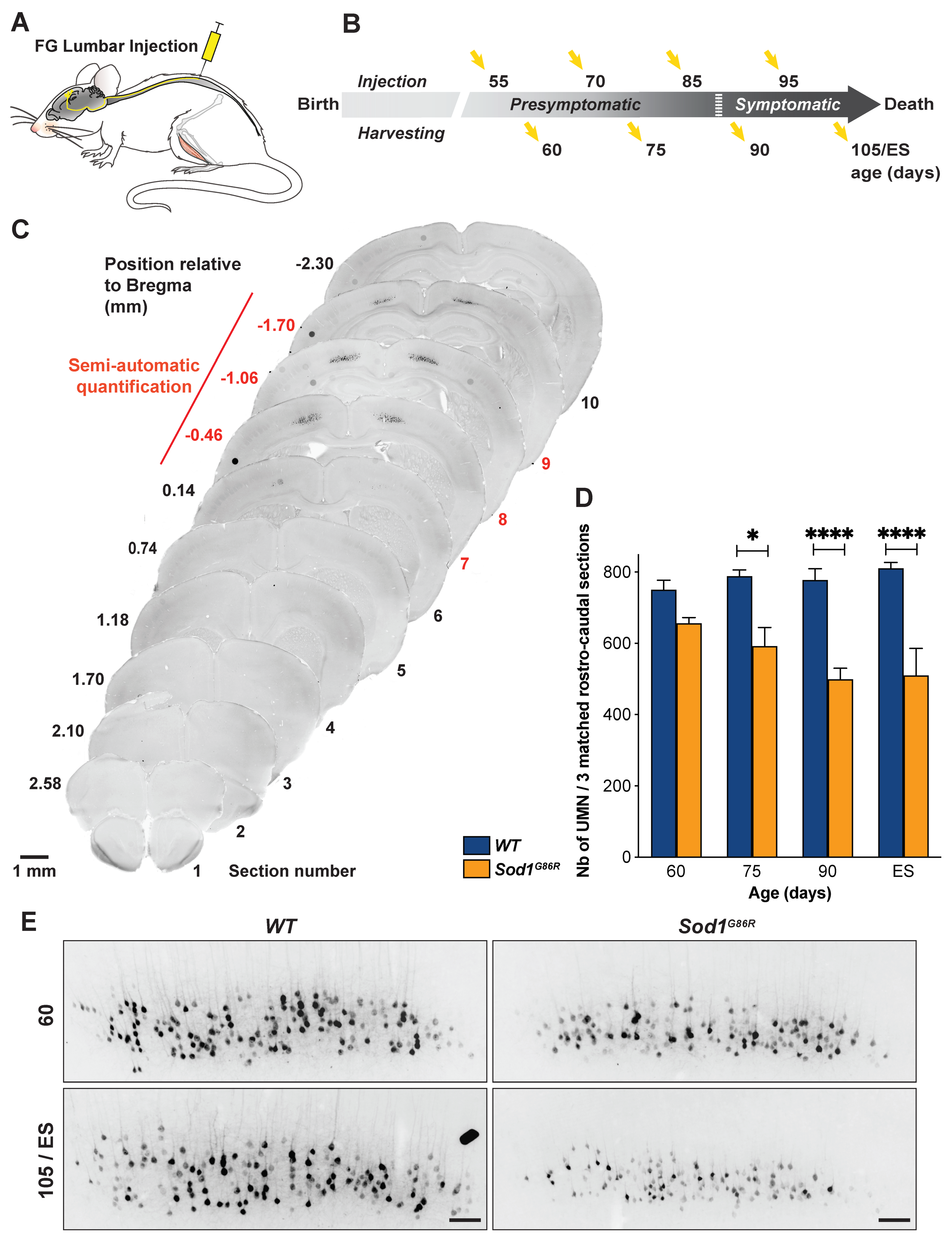
2.2. Retrograde Labelling of UMN
2.3. UMN Quantification
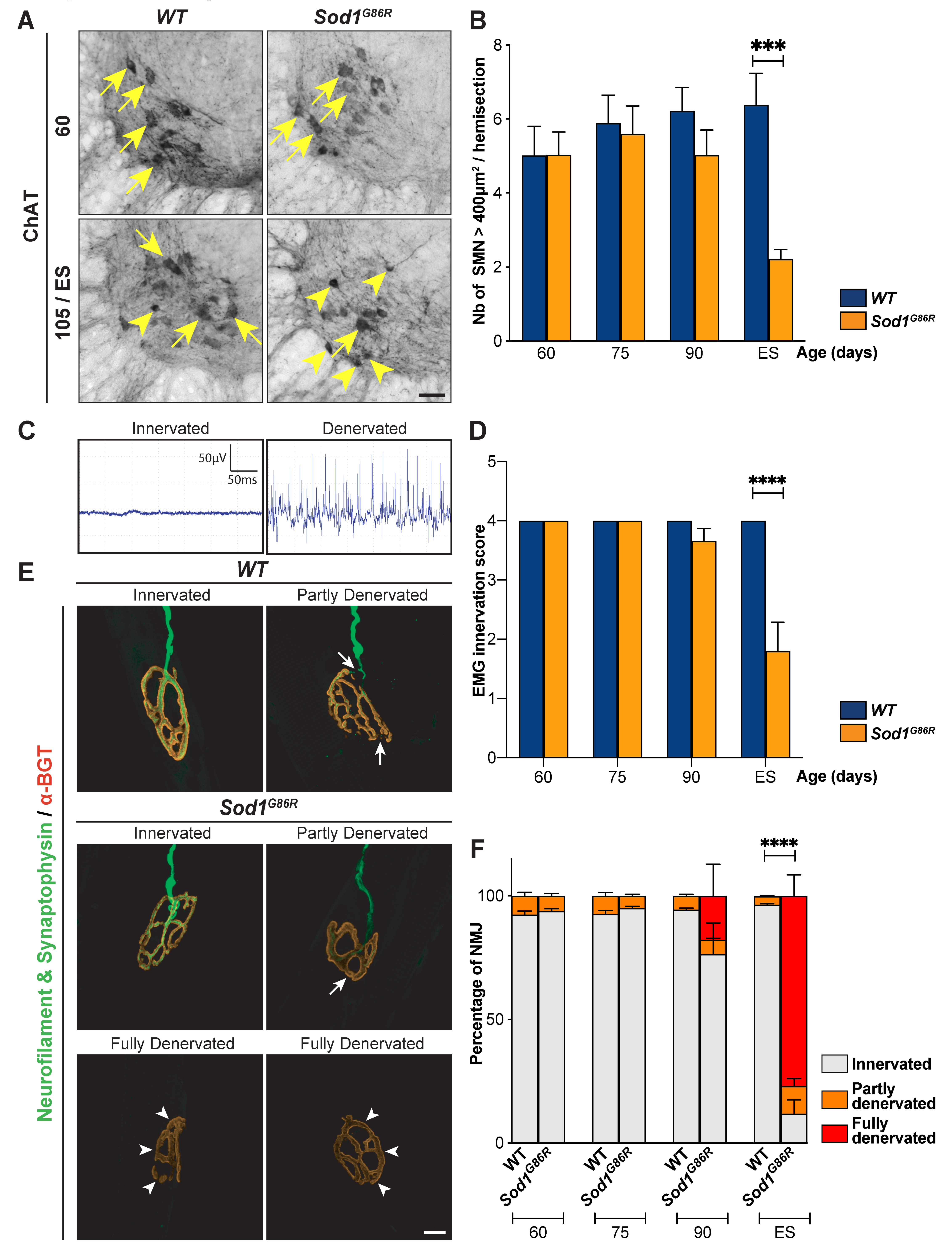
2.4. Lower Motor Neurons Quantification
2.5. Electromyography
2.6. Neuromuscular Junctions Staining and Morphological Analysis
2.7. Statistical Analyses
3. Results
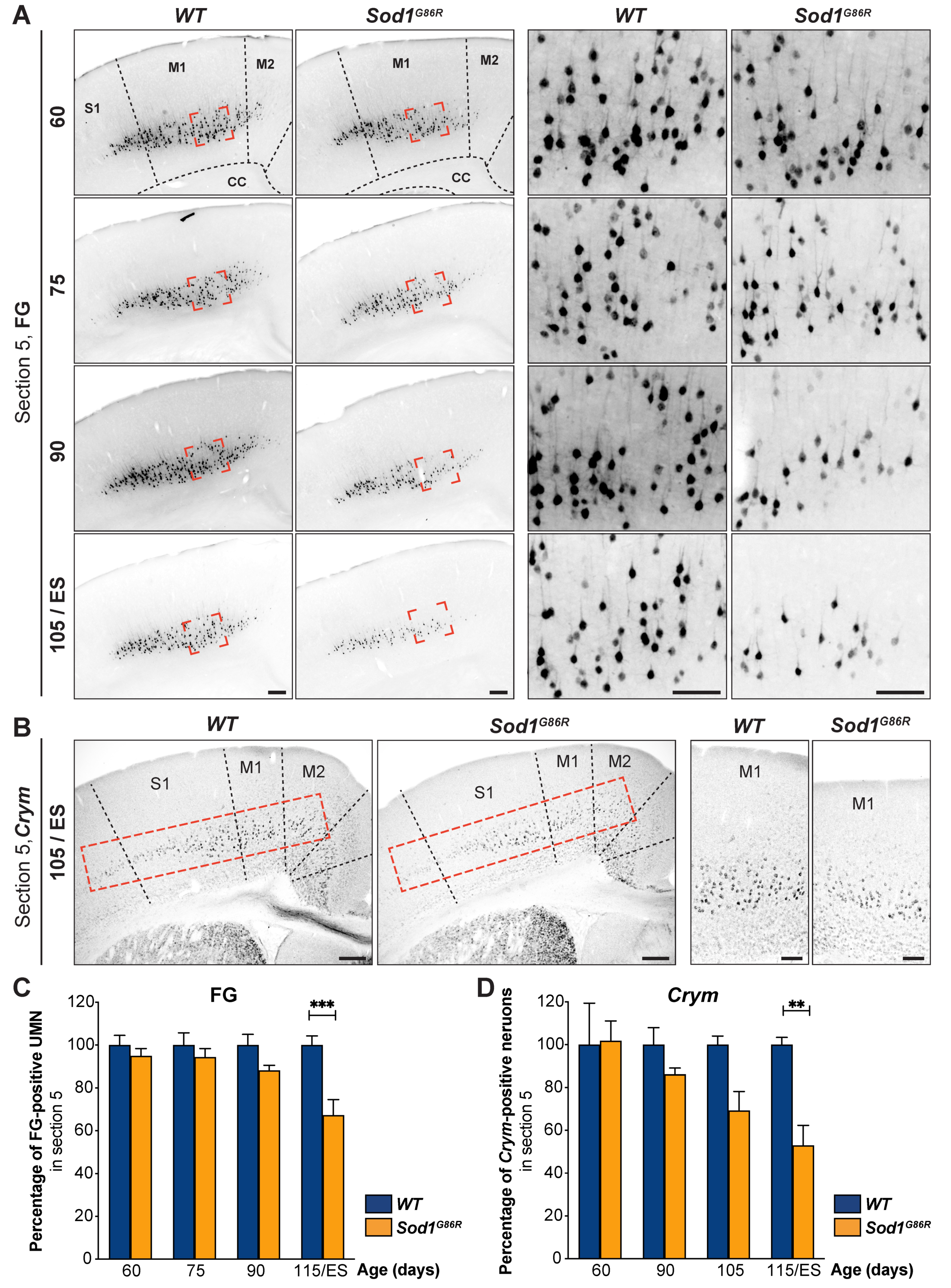
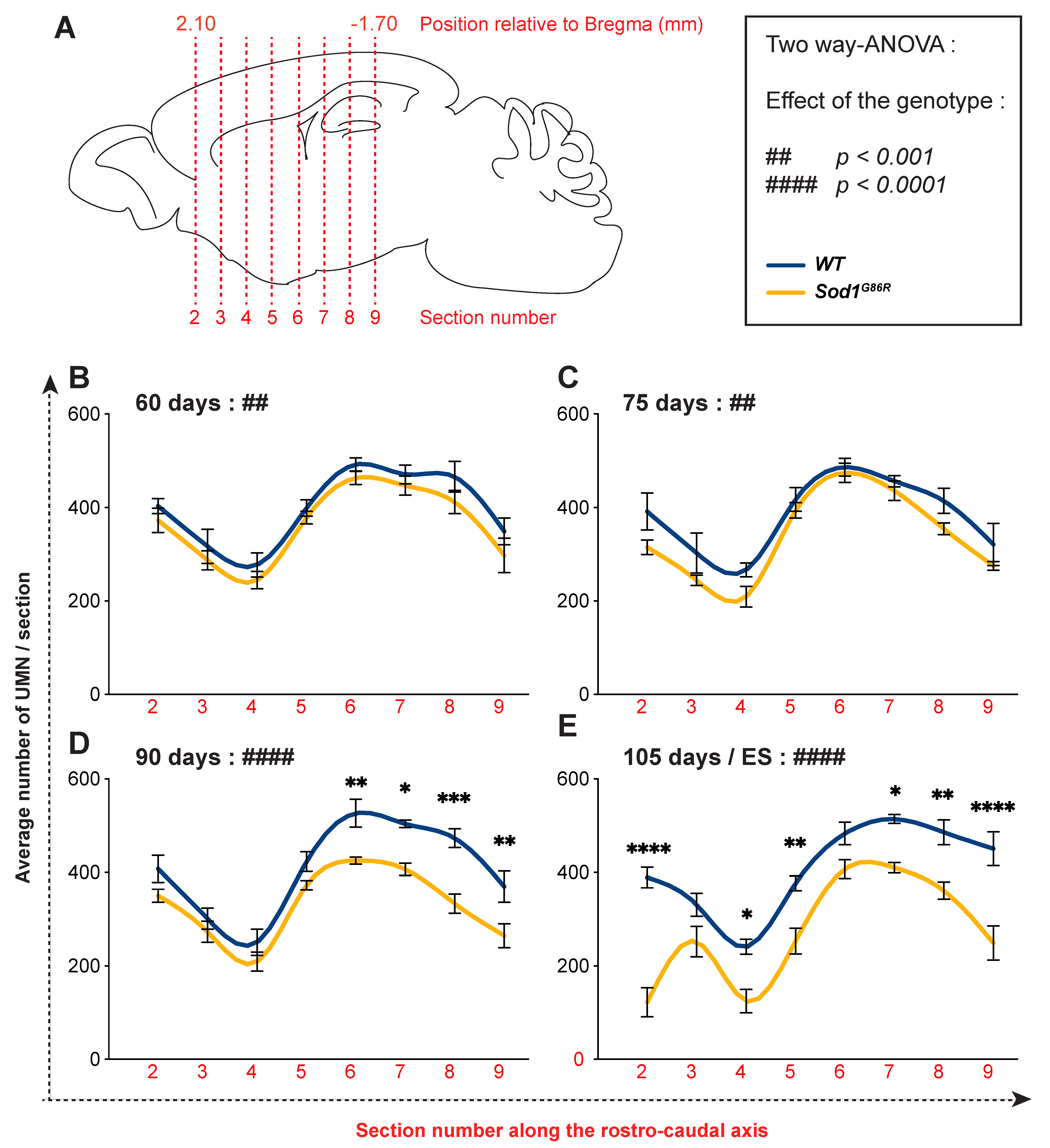
3.1. UMN Progressively Degenerate in Sod1G86R Mice
3.2. UMN Degeneration Propagates in a Caudo-Rostral Manner in Sod1G86R Mice
3.3. Lumbar-Projecting UMN Are Affected Earlier and to a Greater Extent Than the Rest of the UMN Population
3.4. UMN and LMN Degenerations Are Somatotopically Related and Temporally Ordered
3.5. UMN Degeneration Is a Particularly Early Event in the Disease Progression of Sod1G86R Mice
4. Discussion
4.1. Somatotopic Relationship between UMN and LMN Degenerations
4.2. Temporal Relationship between UMN and LMN Degenerations
4.3. Degeneration versus Disease Propagation in Mouse Models of ALS
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ALS | amyotrophic lateral sclerosis |
| ALS-FRS | amyotrophic lateral sclerosis functional rating scale |
| ChAT | choline acetyltransferase |
| CFuPN | corticofugal projection neurons |
| Crym | mu-crystallin |
| CTIP2 | COUP TF1-interacting protein 2 (now designed as Bcl11b: BAF chromatin remodeling complex subunit BCL11B) |
| C9-BAC | C9ORF 72 BAC transgenic mice |
| DTI | diffusion tensor imaging |
| EMG | electromyography |
| ES | end stage |
| Fezf2 | FEZ Family Zinc Finger 2 |
| FG | fluorogold |
| hPFN1 | human Profilin 1 |
| ISH | in situ hybridization |
| LMN | lower motor neurons |
| MRI | magnetic resonance imaging |
| NMJ | neuromuscular junctions |
| SOD1 | superoxyde dismutase 1 |
| SubCerPN | subcerebtral projection neurons |
| TDP-43 | TAR DNA binding protein 43 |
| UMN | upper motor neurons |
| WT | wild type |
References
- Masrori, P.; Van Damme, P. Amyotrophic lateral sclerosis: A clinical review. Eur. J. Neurol. 2020, 27, 1918–1929. [Google Scholar] [CrossRef]
- Sorenson, E.J. The Electrophysiology of the Motor Neuron Diseases. Neurol. Clin. 2012, 30, 605–620. [Google Scholar] [CrossRef] [PubMed]
- Dash, R.P.; Babu, R.J.; Srinivas, N.R. Two Decades-Long Journey from Riluzole to Edaravone: Revisiting the Clinical Pharmacokinetics of the Only Two Amyotrophic Lateral Sclerosis Therapeutics. Clin. Pharmacokinet. 2018, 57, 1385–1398. [Google Scholar] [CrossRef]
- Bede, P.; Bodke, A.; Elamin, M.; Byrne, S.; McLaughlin, R.L.; Jordan, N.; Hampel, H.; Gallagher, L.; Lynch, C.; Fagan, A.J.; et al. Grey matter correlates of clinical variables in amyotrophic lateral sclerosis (ALS): A neuroimaging study of ALS motor phenotype heterogeneity and cortical focality. J. Neurol. Neurosurg. Psychiatry 2013, 84, 7166–7773. [Google Scholar] [CrossRef]
- Dharmadasa, T.; Matamala, J.M.; Howells, J.; Vucic, S.; Kiernan, M.C. Clinical Neurophysiology. Clin. Neurophysiol. 2020, 131, 958–966. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Hu, F.; Zhang, Q.; Chen, Q.; Li, H.; Qin, X.; Jia, R.; Kang, L.; Dang, Y.; Dang, J. Dominant Heterogeneityof Upper and Lower Motorneuron Degeneration to MotorManifestation of involved Region inAmyotrophic Lateral Sclerosis. Sci. Rep. 2019, 9, 1–8. [Google Scholar] [CrossRef]
- Walhout, R.; Schmidt, R.; Westeneng, H.-J.; Verstraete, E.; Seelen, M.; van Rheenen, W.; de Reus, M.A.; van Es, M.A.; Hendrikse, J.; Veldink, J.H.; et al. Brain morphologic changes in asymptomatic C9orf72repeat expansion carriers. Neurology 2015, 85, 1780–1788. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Mao, C.; Jin, J.; Niu, C.; Bai, L.; Dang, J.; Zhang, M. Side of Limb-Onset Predicts Laterality of Gray Matter Loss in Amyotrophic Lateral Sclerosis. Biomed Res. Int 2014, 2014, 1–11. [Google Scholar] [CrossRef]
- Mochizuki, Y.; Mizutani, T.; Takasu, T. Amyotrophic lateral sclerosis with marked neurological asymmetry: Clinicopathological study. Acta Neuropathol. 1995, 90, 44–50. [Google Scholar] [CrossRef]
- Nihei, K.; McKee, A.C.; Kowall, N.W. Patterns of neuronal degeneration in the motor cortex of amyotrophic lateral sclerosis patients*. Acta Neuropathol. 1993, 86, 55–64. [Google Scholar] [CrossRef]
- Rivara, C.-B.N.D.; Sherwood, C.C.; Bouras, C.; Hof, P.R. Stereologic characterization and spatial distribution patterns of Betz cells in the human primary motor cortex. Anat. Rec. Adv. Integr. Anat. Evol. Biol. 2003, 270, 137–151. [Google Scholar] [CrossRef]
- Mancinelli, S.; Lodato, S. Decoding neuronal diversity in the developing cerebral cortex: From single cells to functional networks. Curr. Opin. Neurobiol. 2018, 53, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Brunet, A.; Stuart-Lopez, G.; Burg, T.; Scekic-Zahirovic, J.; Rouaux, C. Cortical Circuit Dysfunction as a Potential Driver of Amyotrophic Lateral Sclerosis. Front. Neurosci. 2020, 14, 363. [Google Scholar] [CrossRef] [PubMed]
- Gunes, Z.I.; Kan, V.W.Y.; Ye, X.Q.; Liebscher, S. Exciting Complexity: The Role of Motor Circuit Elements in ALS Pathophysiology. Front. Neurosci. 2020, 14, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Fil, D.; DeLoach, A.; Yadav, S.; Alkam, D.; MacNicol, M.; Singh, A.; Compadre, C.M.; Goellner, J.J.; O’Brien, C.A.; Fahmi, T.; et al. Mutant Profilin1 transgenic mice recapitulate cardinal features of motor neuron disease. Hum. Mol. Genet. 2016, 364, 686–701. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Pattamatta, A.; Zu, T.; Reid, T.; Bardhi, O.; Borchelt, D.R.; Yachnis, A.T.; Ranum, L.P.W. C9orf72 BAC Mouse Model with Motor Deficits and Neurodegenerative Features of ALS/FTD. Neuron 2016, 90, 521–534. [Google Scholar] [CrossRef] [Green Version]
- Handley, E.E.; Pitman, K.A.; Dawkins, E.; Young, K.M.; Clark, R.M.; Jiang, T.C.; Turner, B.J.; Dickson, T.C.; Blizzard, C.A. Reduced Excitability and Increased Neurite Complexity of Cortical Interneurons in a Familial Mouse Model of Amyotrophic Lateral Sclerosis. Cereb. Cortex 2018, 27, 3630–3647. [Google Scholar]
- Zang, D.W.; Cheema, S.S. Degeneration of corticospinal and bulbospinal systems in the superoxide dismutase 1. Neurosci. Lett. 2002, 332, 99–102. [Google Scholar] [CrossRef]
- Ozdinler, P.H.; Benn, S.; Yamamoto, T.H.; Guzel, M.; Brown, R.H.; Macklis, J.D. Corticospinal Motor Neurons and Related Subcerebral Projection Neurons Undergo Early and Specific Neurodegeneration in hSOD1G93A Transgenic ALS Mice. J. Neurosci. 2011, 31, 4166–4177. [Google Scholar] [CrossRef] [Green Version]
- Scekic-Zahirovic, J.; Fischer, M.; Stuart-Lopez, G.; Burg, T.; Gilet, J.; Dirrig-Grosch, S.; Marques, C.; Birling, M.-C.; Kessler, P.; Rouaux, C. Evidence that corticofugal propagation of ALS pathology is not mediated by prion-like mechanism. Prog. Neurobiol. 2020, 101972. [Google Scholar] [CrossRef]
- Ripps, M.E.; Huntley, G.W.; Hof, P.R.; Morrison, J.H.; Gordon, J.W. Transgenic mice expressing an altered murine superoxide dismutase gene provide an animal model of amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 1995, 92, 689–693. [Google Scholar] [CrossRef] [Green Version]
- Marques, C.; Fischer, M.; Keime, C.; Burg, T.; Brunet, A.; Scekic-Zahirovic, J.; Rouaux, C. Early alterations of RNA metabolism and splicing from adult corticospinal neurons in an ALS mouse model. bioRxiv 2019, 667733. [Google Scholar] [CrossRef]
- Rouaux, C.; Panteleeva, I.; Rene, F.; Gonzalez de Aguilar, J.-L.; Echaniz-Laguna, A.; Dupuis, L.; Menger, Y.; Boutillier, A.-L.; Loeffler, J.-P. Sodium valproate exerts neuroprotective effects in vivo through CREB-binding protein-dependent mechanisms but does not improve survival in an amyotrophic lateral sclerosis mouse model. J. Neurosci. 2007, 27, 5535–5545. [Google Scholar] [CrossRef] [PubMed]
- Arlotta, P.; Molyneaux, B.J.; Chen, J.; Inoue, J.; Kominami, R.; Macklis, J.D. Neuronal subtype-specific genes that control corticospinal motor neuron development in vivo. Neuron 2005, 45, 207–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burg, T.; Bichara, C.; Scekic-Zahirovic, J.; Fischer, M.; Stuart-Lopez, G.; Brunet, A.; Lefebvre, F.; Cordero-Erausquin, M.; Rouaux, C. Absence of Subcerebral Projection Neurons Is Beneficial in a Mouse Model of Amyotrophic Lateral Sclerosis. Ann. Neurol. 2020, 2014, 1–30. [Google Scholar] [CrossRef]
- Ebbesen, C.L.; Insanally, M.N.; Kopec, C.D.; Murakami, M.; Saiki, A.; Erlich, J.C. More than Just a “Motor”: Recent Surprises from the Frontal Cortex. J. Neurosci. 2018, 38, 9402–9413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, L.R.; Culver, D.G.; Tennant, P.; Davis, A.A.; Wang, M.; Castellano-Sanchez, A.; Khan, J.; Polak, M.A.; Glass, J.D. Amyotrophic lateral sclerosis is a distal axonopathy: Evidence in mice and man. Exp. Neurol. 2004, 185, 232–240. [Google Scholar] [CrossRef]
- de Carvalho, M.; Kiernan, M.C.; Swash, M. Fasciculation in amyotrophic lateral sclerosis: Origin and pathophysiological relevance. J. Neurol. Neurosurg. Psychiatry 2017, 88, 773–779. [Google Scholar] [CrossRef]
- Vandoorne, T.; De Bock, K.; Van Den Bosch, L. Energy metabolism in ALS: An underappreciated opportunity? Acta Neuropathol. 2018, 135, 489–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupuis, L.; Oudart, H.; Rene, F.; de Aguilar, J.-L.G.; Loeffler, J.-P. Evidence for defective energy homeostasis inamyotrophic lateral sclerosis: Benefit of ahigh-energy diet in a transgenic mouse model. Proc. Natl. Acad. Sci. USA 2004, 101, 1–6. [Google Scholar] [CrossRef] [Green Version]
- vand den Bos, M.A.J.; Geevasinga, N.; Higashihara, M.; Menon, P.; Vucic, S. Pathophysiology and Diagnosis of ALS: Insights from Advances in Neurophysiological Techniques. Int. J. Mol. Sci. 2019, 20, 2818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charcot, J.M. Sclérose Latérale Amyotrophique: Oeuvres Complètes; Bureaux du Progrès Médical: Paris, France, 1874; Volume 2, pp. 249–266. [Google Scholar]
- Gowers, W.R. Manual of Diseases of the Nervous System; Churchill: London, UK, 1888. [Google Scholar]
- Devine, M.S.; Kiernan, M.C.; Heggie, S.; McCombe, P.A.; Henderson, R.D. Study of motor asymmetry in ALS indicates an effect of limb dominance on onset and spread of weakness, and an important role for upper motor neurons. Amyotroph. Lateral Scler. Front. Degener. 2014, 15, 481–487. [Google Scholar] [CrossRef]
- Takeda, T.; Kitagawa, K.; Arai, K. Phenotypic variability and its pathological basis in amyotrophic lateral sclerosis. Neuropathology 2020, 40, 40–56. [Google Scholar] [CrossRef]
- Yasvoina, M.V.; Genc, B.; Jara, J.H.; Sheets, P.L.; Quinlan, K.A.; Milosevic, A.; Shepherd, G.M.G.; Heckman, C.J.; Ozdinler, P.H. eGFP Expression under UCHL1 Promoter Genetically Labels Corticospinal Motor Neurons and a Subpopulation of Degeneration-Resistant Spinal Motor Neurons in an ALS Mouse Model. J. Neurosci. 2013, 33, 7890–7904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braak, H.; Brettschneider, J.; Ludolph, A.C.; Lee, V.M.; Trojanowski, J.Q.; Del Tredici, K. Amyotrophic lateral sclerosis-a model of corticofugal axonal spread. Nat. Rev. Neurol. 2013, 9, 708–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brettschneider, J.; Del Tredici, K.; Toledo, J.B.; Robinson, J.L.; Irwin, D.J.; Grossman, M.; Suh, E.; Van Deerlin, V.M.; Wood, E.M.; Baek, Y.; et al. Stages of pTDP-43 pathology in amyotrophic lateral sclerosis. Ann. Neurol. 2013, 74, 20–38. [Google Scholar] [CrossRef]
- Kassubek, J.; Muller, H.P.; Del Tredici, K.; Brettschneider, J.; Pinkhardt, E.H.; Lule, D.; Bohm, S.; Braak, H.; Ludolph, A.C. Diffusion tensor imaging analysis of sequential spreading of disease in amyotrophic lateral sclerosis confirms patterns of TDP-43 pathology. Brain A J. Neurol. 2014, 137, 1733–1740. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marques, C.; Burg, T.; Scekic-Zahirovic, J.; Fischer, M.; Rouaux, C. Upper and Lower Motor Neuron Degenerations Are Somatotopically Related and Temporally Ordered in the Sod1 Mouse Model of Amyotrophic Lateral Sclerosis. Brain Sci. 2021, 11, 369. https://doi.org/10.3390/brainsci11030369
Marques C, Burg T, Scekic-Zahirovic J, Fischer M, Rouaux C. Upper and Lower Motor Neuron Degenerations Are Somatotopically Related and Temporally Ordered in the Sod1 Mouse Model of Amyotrophic Lateral Sclerosis. Brain Sciences. 2021; 11(3):369. https://doi.org/10.3390/brainsci11030369
Chicago/Turabian StyleMarques, Christine, Thibaut Burg, Jelena Scekic-Zahirovic, Mathieu Fischer, and Caroline Rouaux. 2021. "Upper and Lower Motor Neuron Degenerations Are Somatotopically Related and Temporally Ordered in the Sod1 Mouse Model of Amyotrophic Lateral Sclerosis" Brain Sciences 11, no. 3: 369. https://doi.org/10.3390/brainsci11030369
APA StyleMarques, C., Burg, T., Scekic-Zahirovic, J., Fischer, M., & Rouaux, C. (2021). Upper and Lower Motor Neuron Degenerations Are Somatotopically Related and Temporally Ordered in the Sod1 Mouse Model of Amyotrophic Lateral Sclerosis. Brain Sciences, 11(3), 369. https://doi.org/10.3390/brainsci11030369