
Hereditary Spastic Paraplegia: From Genes, Cells and Networks to Novel Pathways for Drug Discovery


Abstract
:1. Introduction
2. Pathological Phenotypes in White Matter Tracts in Hereditary Spastic Paraplegias (HSP) Patients
3. Pathological Phenotypes in HSP Patient-Derived Cells
3.1. SPG4/SPAST
3.2. SPG7
3.3. Other Genotypes
4. Highly Connected Protein–Protein Interaction Networks of HSP Genes
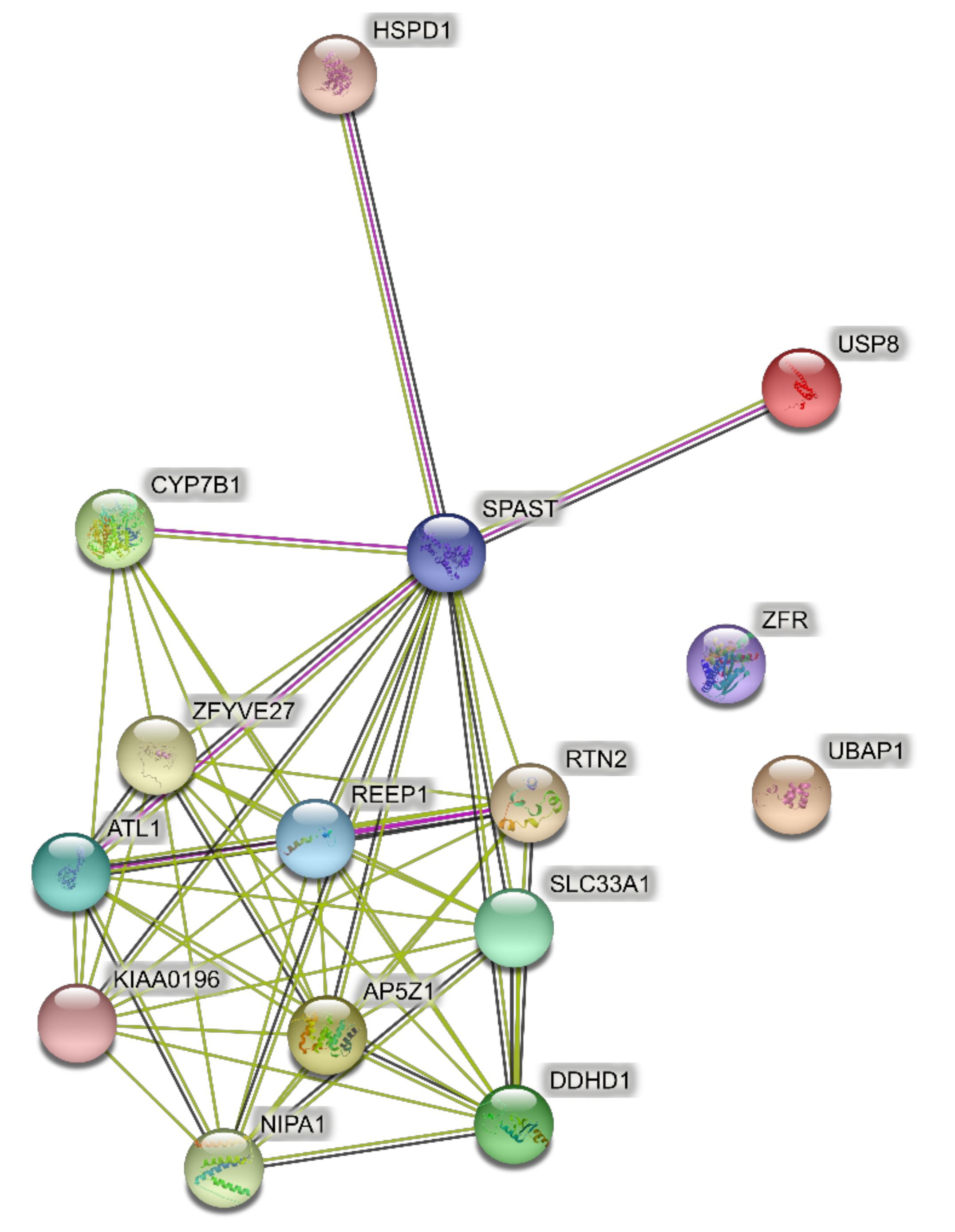
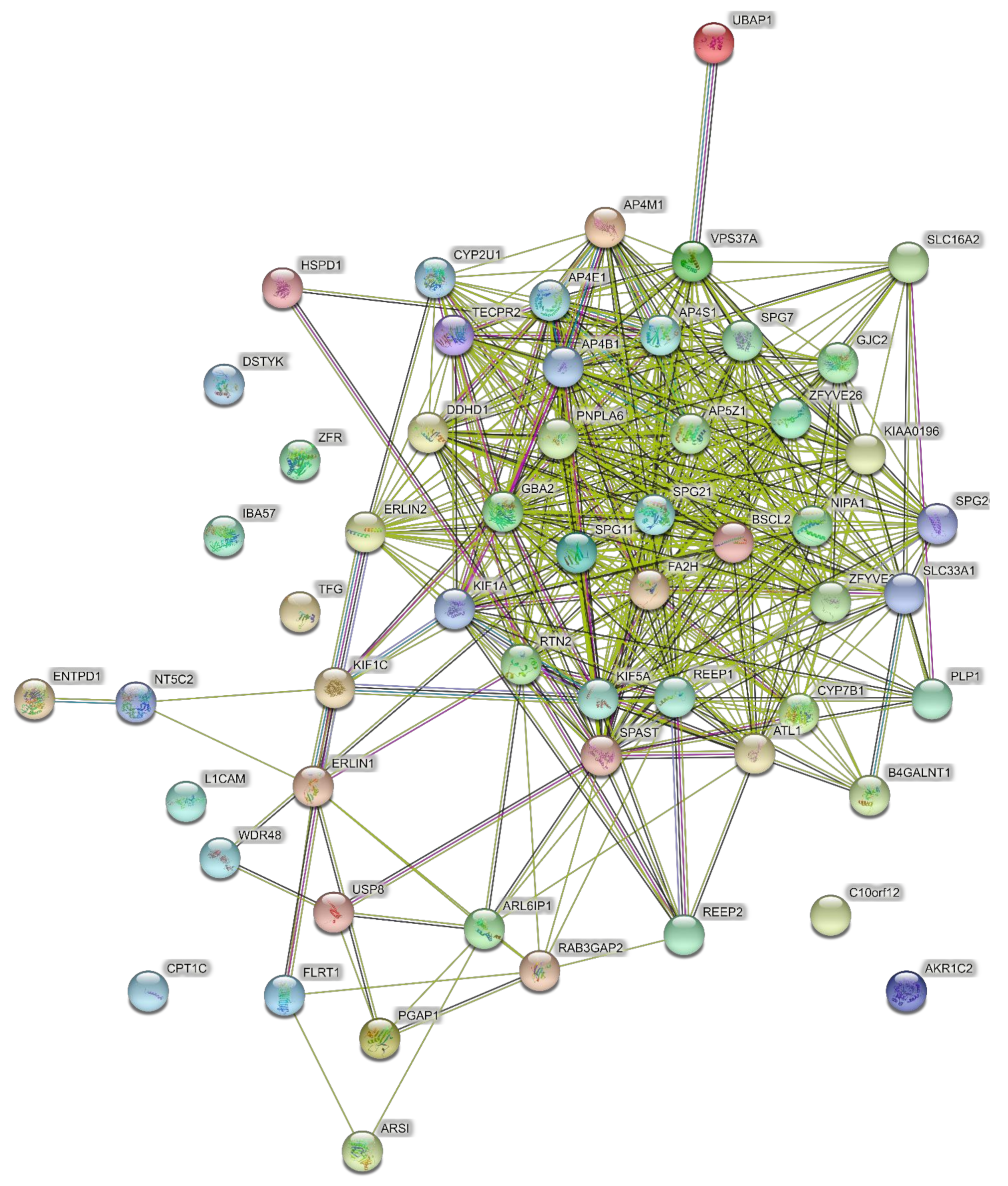
4.1. Networks of the Highest Prevalence Pure SPGs (Six Genes)
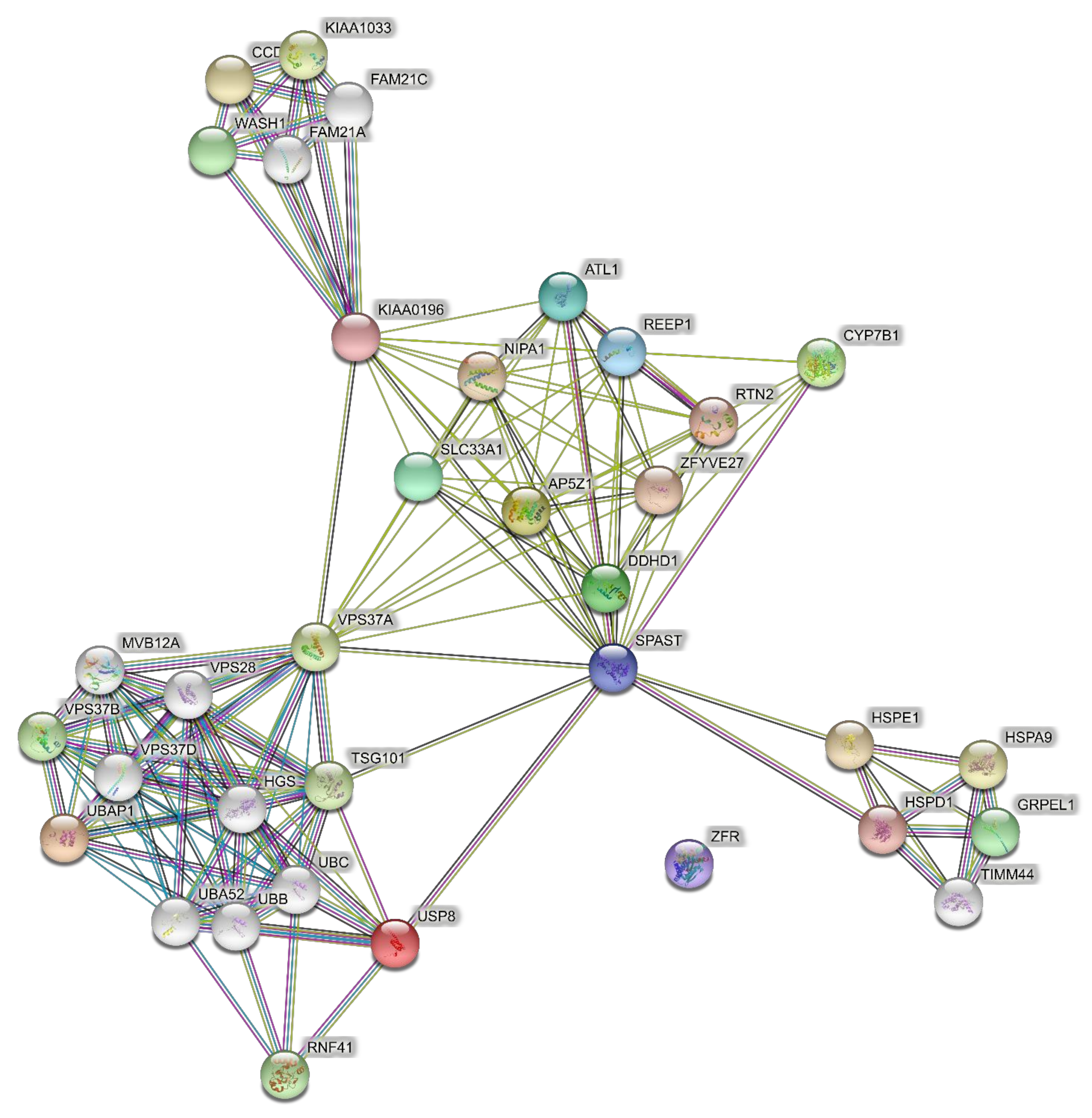
4.2. Networks of Pure SPGs (15 Genes)
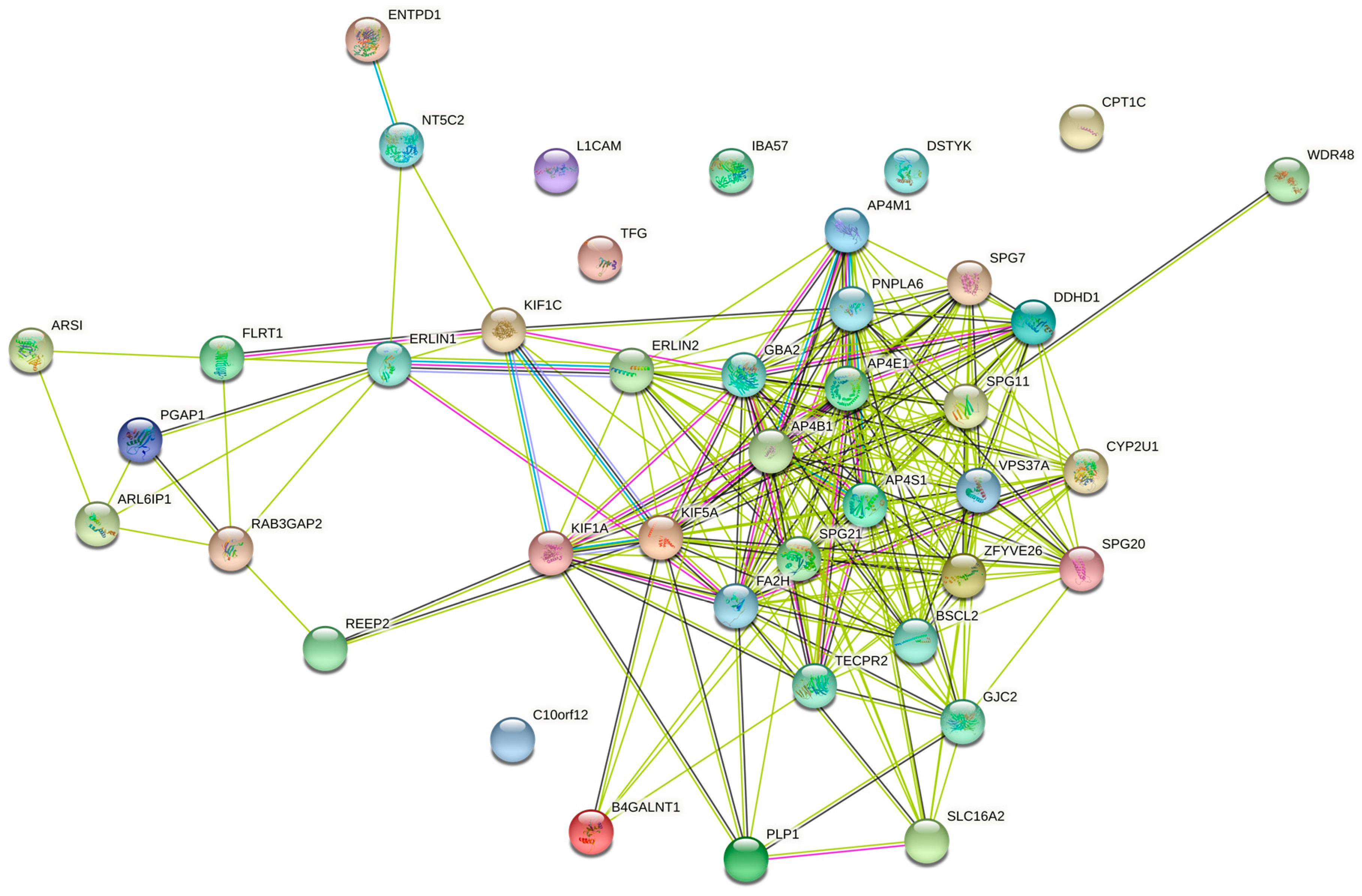
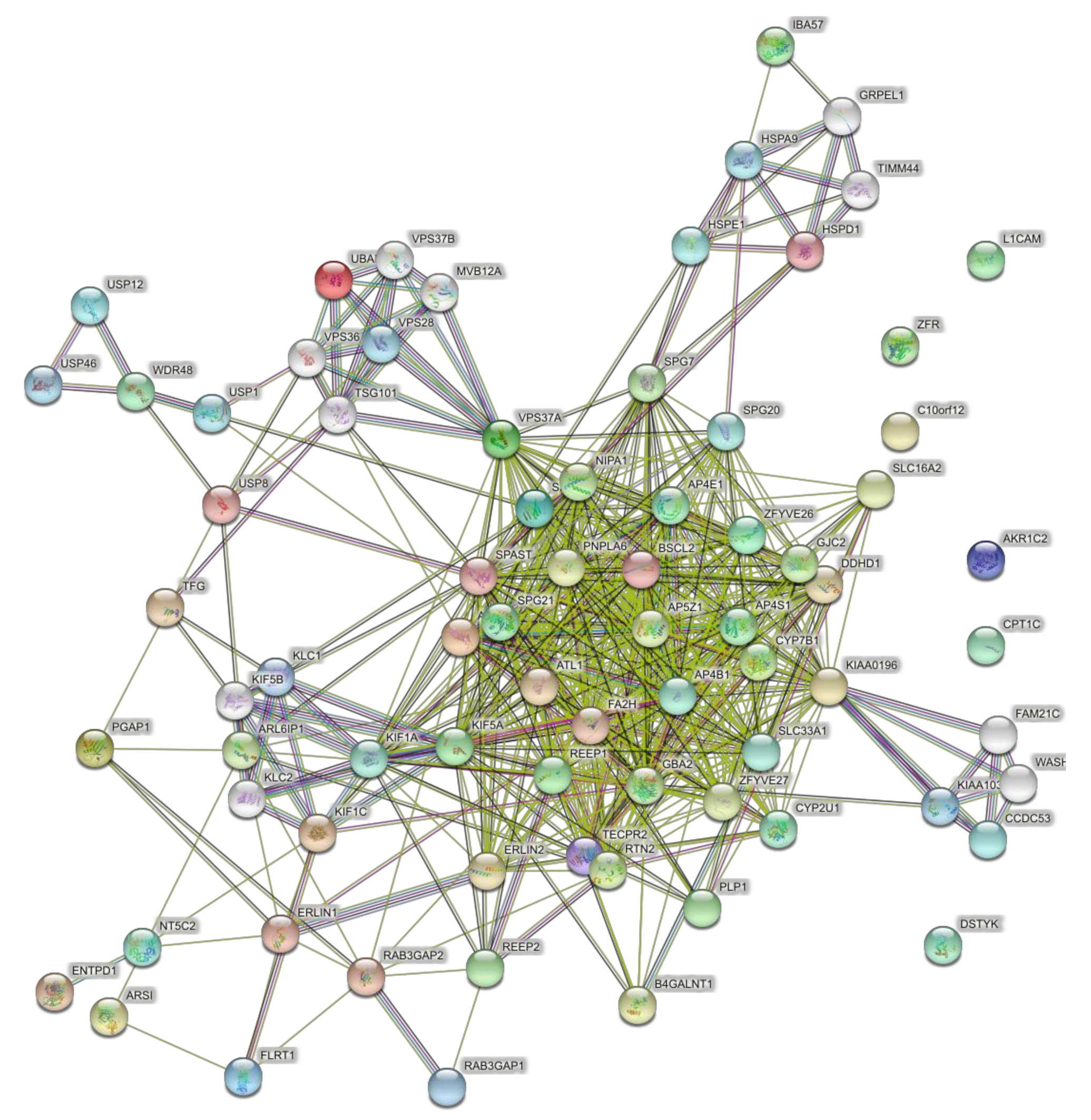
4.3. Networks of the Set of Complicated SPGs (42 Genes)
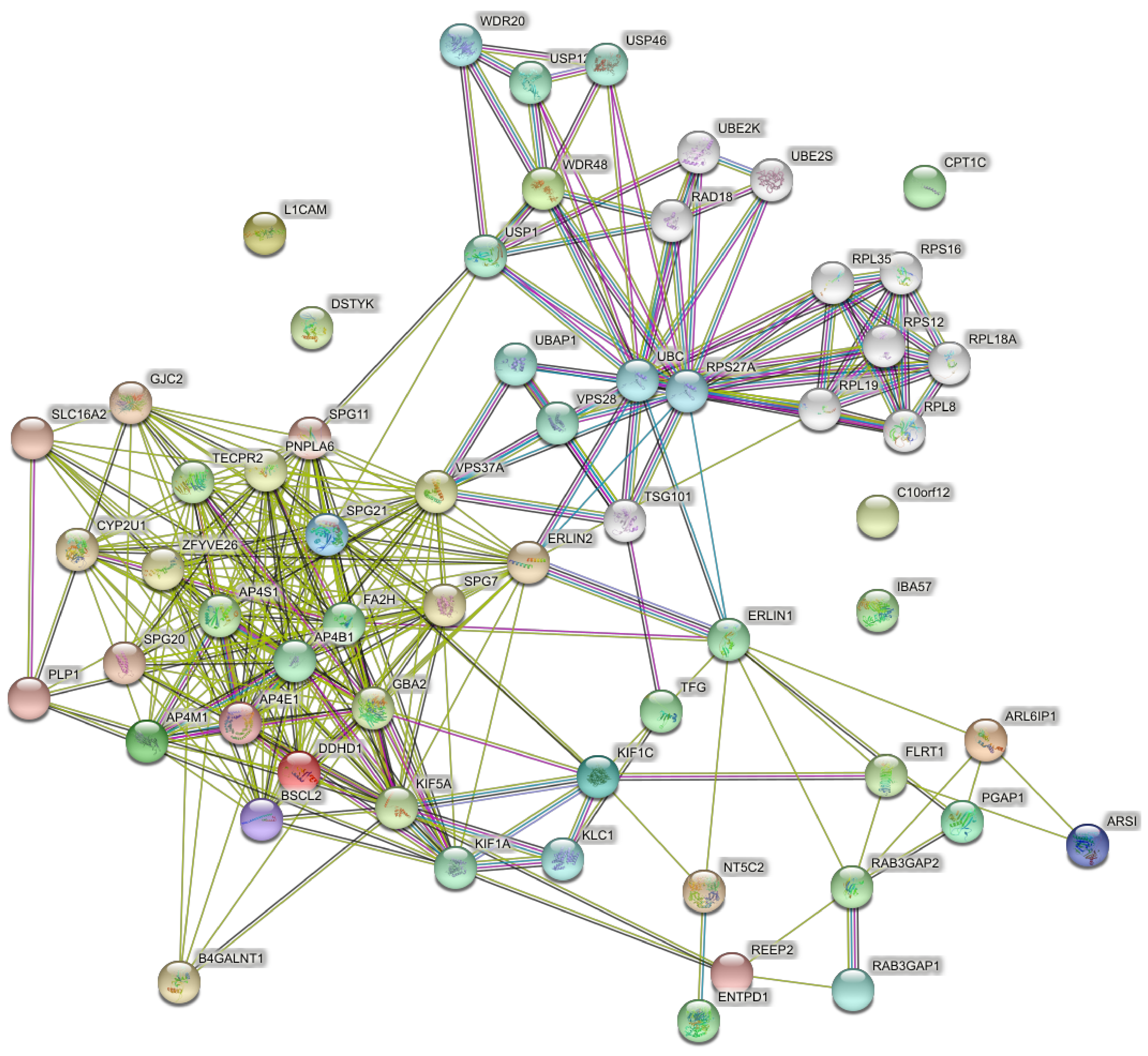
4.4. Networks of the Set of Pure and Complicated SPGs (57 Genes)
4.5. Gene Ontology (GO) Analysis Showed Similar Functions of the PPI Networks in Pure and Complicated HSPs
5. Future Directions: From Networks to Drug Discovery
Supplementary Materials
Funding
Acknowledgments
Conflicts of Interest
References
- Andersen, P.M. Amyotrophic lateral sclerosis associated with mutations in the CuZn superoxide dismutase gene. Curr. Neurol. Neurosci. Rep. 2006, 6, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Parodi, L.; Fenu, S.; Stevanin, G.; Durr, A. Hereditary spastic paraplegia: More than an upper motor neuron disease. Rev. Neurol. 2017, 173, 352–360. [Google Scholar] [CrossRef] [PubMed]
- Elsayed, L.E.O.; Eltazi, I.Z.M.; Ahmed, A.E.M.; Stevanin, G. Hereditary spastic paraplegias: Time for an objective case definition and a new nosology for neurogenetic disorders to facilitate biomarker/therapeutic studies. Expert Rev. Neurother. 2019, 19, 409–415. [Google Scholar] [CrossRef] [PubMed]
- De Souza, P.V.S.; de Rezende Pinto, W.B.V.; de Rezende Batistella, G.N.; Bortholin, T.; Oliveira, A.S.B. Hereditary Spastic Paraplegia: Clinical and Genetic Hallmarks. Cerebellum 2017, 16, 525–551. [Google Scholar] [CrossRef]
- Fink, J.K. Hereditary spastic paraplegia: Clinico-pathologic features and emerging molecular mechanisms. Acta Neuropathol. 2013, 126, 307–328. [Google Scholar] [CrossRef] [Green Version]
- Lallemant-Dudek, P.; Durr, A. Clinical and genetic update of hereditary spastic paraparesis. Rev. Neurol. 2020. [Google Scholar] [CrossRef]
- Schule, R.; Wiethoff, S.; Martus, P.; Karle, K.N.; Otto, S.; Klebe, S.; Klimpe, S.; Gallenmuller, C.; Kurzwelly, D.; Henkel, D.; et al. Hereditary spastic paraplegia: Clinicogenetic lessons from 608 patients. Ann. Neurol. 2016, 79, 646–658. [Google Scholar] [CrossRef]
- Ruano, L.; Melo, C.; Silva, M.C.; Coutinho, P. The global epidemiology of hereditary ataxia and spastic paraplegia: A systematic review of prevalence studies. Neuroepidemiology 2014, 42, 174–183. [Google Scholar] [CrossRef]
- Blackstone, C. Hereditary spastic paraplegia. Handb Clin. Neurol. 2018, 148, 633–652. [Google Scholar] [CrossRef]
- Hedera, P. Hereditary Spastic Paraplegia Overview. In GeneReviews((R)); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2018. [Google Scholar]
- Das Bhowmik, A.; Patil, S.J.; Deshpande, D.V.; Bhat, V.; Dalal, A. Novel splice-site variant of UCHL1 in an Indian family with autosomal recessive spastic paraplegia-79. J. Hum. Genet. 2018, 63, 927–933. [Google Scholar] [CrossRef]
- Lin, X.; Su, H.Z.; Dong, E.L.; Lin, X.H.; Zhao, M.; Yang, C.; Wang, C.; Wang, J.; Chen, Y.J.; Yu, H.; et al. Stop-gain mutations in UBAP1 cause pure autosomal-dominant spastic paraplegia. Brain 2019, 142, 2238–2252. [Google Scholar] [CrossRef] [Green Version]
- Nan, H.; Ichinose, Y.; Tanaka, M.; Koh, K.; Ishiura, H.; Mitsui, J.; Mizukami, H.; Morimoto, M.; Hamada, S.; Ohtsuka, T.; et al. UBAP1 mutations cause juvenile-onset hereditary spastic paraplegias (SPG80) and impair UBAP1 targeting to endosomes. J. Hum. Genet. 2019, 64, 1055–1065. [Google Scholar] [CrossRef]
- Aghakhanyan, G.; Martinuzzi, A.; Frijia, F.; Vavla, M.; Hlavata, H.; Baratto, A.; Martino, N.; Paparella, G.; Montanaro, D. Brain white matter involvement in hereditary spastic paraplegias: Analysis with multiple diffusion tensor indices. AJNR Am. J. Neuroradiol. 2014, 35, 1533–1538. [Google Scholar] [CrossRef] [Green Version]
- Agosta, F.; Scarlato, M.; Spinelli, E.G.; Canu, E.; Benedetti, S.; Bassi, M.T.; Casali, C.; Sessa, M.; Copetti, M.; Pagani, E.; et al. Hereditary Spastic Paraplegia: Beyond Clinical Phenotypes toward a Unified Pattern of Central Nervous System Damage. Radiology 2015, 276, 207–218. [Google Scholar] [CrossRef] [Green Version]
- Duning, T.; Warnecke, T.; Schirmacher, A.; Schiffbauer, H.; Lohmann, H.; Mohammadi, S.; Young, P.; Deppe, M. Specific pattern of early white-matter changes in pure hereditary spastic paraplegia. Mov. Disord. 2010, 25, 1986–1992. [Google Scholar] [CrossRef]
- Franca, M.C., Jr.; Yasuda, C.L.; Pereira, F.R.; D’Abreu, A.; Lopes-Ramos, C.M.; Rosa, M.V.; Cendes, F.; Lopes-Cendes, I. White and grey matter abnormalities in patients with SPG11 mutations. J. Neurol. Neurosurg. Psychiatry 2012, 83, 828–833. [Google Scholar] [CrossRef]
- Garaci, F.; Toschi, N.; Lanzafame, S.; Meschini, A.; Bertini, E.; Simonetti, G.; Santorelli, F.M.; Guerrisi, M.; Floris, R. Diffusion tensor imaging in SPG11- and SPG4-linked hereditary spastic paraplegia. Int. J. Neurosci. 2014, 124, 261–270. [Google Scholar] [CrossRef]
- Lindig, T.; Bender, B.; Hauser, T.K.; Mang, S.; Schweikardt, D.; Klose, U.; Karle, K.N.; Schule, R.; Schols, L.; Rattay, T.W. Gray and white matter alterations in hereditary spastic paraplegia type SPG4 and clinical correlations. J. Neurol. 2015, 262, 1961–1971. [Google Scholar] [CrossRef]
- Oguz, K.K.; Sanverdi, E.; Has, A.; Temucin, C.; Turk, S.; Doerschner, K. Tract-based spatial statistics of diffusion tensor imaging in hereditary spastic paraplegia with thin corpus callosum reveals widespread white matter changes. Diagn Interv. Radiol. 2013, 19, 181–186. [Google Scholar] [CrossRef] [Green Version]
- Rezende, T.J.; de Albuquerque, M.; Lamas, G.M.; Martinez, A.R.; Campos, B.M.; Casseb, R.F.; Silva, C.B.; Branco, L.M.; D’Abreu, A.; Lopes-Cendes, I.; et al. Multimodal MRI-based study in patients with SPG4 mutations. PLoS ONE 2015, 10, e0117666. [Google Scholar] [CrossRef] [Green Version]
- Unrath, A.; Muller, H.P.; Riecker, A.; Ludolph, A.C.; Sperfeld, A.D.; Kassubek, J. Whole brain-based analysis of regional white matter tract alterations in rare motor neuron diseases by diffusion tensor imaging. Hum. Brain Mapp. 2010, 31, 1727–1740. [Google Scholar] [CrossRef]
- Martinuzzi, A.; Montanaro, D.; Vavla, M.; Paparella, G.; Bonanni, P.; Musumeci, O.; Brighina, E.; Hlavata, H.; Rossi, G.; Aghakhanyan, G.; et al. Clinical and Paraclinical Indicators of Motor System Impairment in Hereditary Spastic Paraplegia: A Pilot Study. PLoS ONE 2016, 11, e0153283. [Google Scholar] [CrossRef] [PubMed]
- Servelhere, K.R.; Rezende, T.J.R.; de Lima, F.D.; de Brito, M.R.; de Franca Nunes, R.F.; Casseb, R.F.; Pedroso, J.L.; Barsottini, O.G.P.; Cendes, F.; Franca, M.C., Jr. Brain Damage and Gene Expression Across Hereditary Spastic Paraplegia Subtypes. Mov. Disord. 2021. [Google Scholar] [CrossRef]
- Servelhere, K.R.; Casseb, R.F.; de Lima, F.D.; Rezende, T.J.R.; Ramalho, L.P.; Franca, M.C., Jr. Spinal Cord Gray and White Matter Damage in Different Hereditary Spastic Paraplegia Subtypes. AJNR Am. J. Neuroradiol. 2021. [Google Scholar] [CrossRef]
- Hedera, P.; Eldevik, O.P.; Maly, P.; Rainier, S.; Fink, J.K. Spinal cord magnetic resonance imaging in autosomal dominant hereditary spastic paraplegia. Neuroradiology 2005, 47, 730–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacinto-Scudeiro, L.A.; Dariva Machado, G.; Ayres, A.; Burguez, D.; Polese-Bonato, M.; Gonzalez-Salazar, C.; Siebert, M.; Cavalcante Franca, M., Jr.; Olchik, M.R.; Morales Saute, J.A. Are Cognitive Changes in Hereditary Spastic Paraplegias Restricted to Complicated Forms? Front. Neurol. 2019, 10, 508. [Google Scholar] [CrossRef]
- Abrahamsen, G.; Fan, Y.; Matigian, N.; Wali, G.; Bellette, B.; Sutharsan, R.; Raju, J.; Wood, S.A.; Veivers, D.; Sue, C.M.; et al. A patient-derived stem cell model of hereditary spastic paraplegia with SPAST mutations. Dis. Model Mech. 2013, 6, 489–502. [Google Scholar] [CrossRef] [Green Version]
- Stewart, R.; Kozlov, S.; Matigian, N.; Wali, G.; Gatei, M.; Sutharsan, R.; Bellette, B.; Wraith-Kijas, A.; Cochrane, J.; Coulthard, M.; et al. A patient-derived olfactory stem cell disease model for ataxia-telangiectasia. Hum. Mol. Genet. 2013, 22, 2495–2509. [Google Scholar] [CrossRef]
- Matigian, N.; Abrahamsen, G.; Sutharsan, R.; Cook, A.L.; Vitale, A.M.; Nouwens, A.; Bellette, B.; An, J.; Anderson, M.; Beckhouse, A.G.; et al. Disease-specific, neurosphere-derived cells as models for brain disorders. Dis. Model Mech. 2010, 3, 785–798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cook, A.L.; Vitale, A.M.; Ravishankar, S.; Matigian, N.; Sutherland, G.T.; Shan, J.; Sutharsan, R.; Perry, C.; Silburn, P.A.; Mellick, G.D.; et al. NRF2 activation restores disease related metabolic deficiencies in olfactory neurosphere-derived cells from patients with sporadic Parkinson’s disease. PLoS ONE 2011, 6, e21907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wali, G.; Sutharsan, R.; Fan, Y.; Stewart, R.; Tello Velasquez, J.; Sue, C.M.; Crane, D.I.; Mackay-Sim, A. Mechanism of impaired microtubule-dependent peroxisome trafficking and oxidative stress in SPAST-mutated cells from patients with Hereditary Spastic Paraplegia. Sci. Rep. 2016, 6, 27004. [Google Scholar] [CrossRef]
- Wali, G.; Liyanage, E.; Blair, N.F.; Sutharsan, R.; Park, J.S.; Mackay-Sim, A.; Sue, C.M. Oxidative Stress-Induced Axon Fragmentation Is a Consequence of Reduced Axonal Transport in Hereditary Spastic Paraplegia SPAST Patient Neurons. Front. Neurosci. 2020, 14, 401. [Google Scholar] [CrossRef]
- Kasher, P.R.; De Vos, K.J.; Wharton, S.B.; Manser, C.; Bennett, E.J.; Bingley, M.; Wood, J.D.; Milner, R.; McDermott, C.J.; Miller, C.C.; et al. Direct evidence for axonal transport defects in a novel mouse model of mutant spastin-induced hereditary spastic paraplegia (HSP) and human HSP patients. J. Neurochem. 2009, 110, 34–44. [Google Scholar] [CrossRef]
- Denton, K.R.; Lei, L.; Grenier, J.; Rodionov, V.; Blackstone, C.; Li, X.J. Loss of spastin function results in disease-specific axonal defects in human pluripotent stem cell-based models of hereditary spastic paraplegia. Stem. Cells 2014, 32, 414–423. [Google Scholar] [CrossRef] [Green Version]
- Rehbach, K.; Kesavan, J.; Hauser, S.; Ritzenhofen, S.; Jungverdorben, J.; Schule, R.; Schols, L.; Peitz, M.; Brustle, O. Multiparametric rapid screening of neuronal process pathology for drug target identification in HSP patient-specific neurons. Sci. Rep. 2019, 9, 9615. [Google Scholar] [CrossRef]
- Havlicek, S.; Kohl, Z.; Mishra, H.K.; Prots, I.; Eberhardt, E.; Denguir, N.; Wend, H.; Plotz, S.; Boyer, L.; Marchetto, M.C.; et al. Gene dosage-dependent rescue of HSP neurite defects in SPG4 patients’ neurons. Hum. Mol. Genet. 2014, 23, 2527–2541. [Google Scholar] [CrossRef]
- Casari, G.; Marconi, R. Spastic Paraplegia 7. In GeneReviews((R)); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Casari, G.; De Fusco, M.; Ciarmatori, S.; Zeviani, M.; Mora, M.; Fernandez, P.; De Michele, G.; Filla, A.; Cocozza, S.; Marconi, R.; et al. Spastic paraplegia and OXPHOS impairment caused by mutations in paraplegin, a nuclear-encoded mitochondrial metalloprotease. Cell 1998, 93, 973–983. [Google Scholar] [CrossRef] [Green Version]
- Ferreirinha, F.; Quattrini, A.; Pirozzi, M.; Valsecchi, V.; Dina, G.; Broccoli, V.; Auricchio, A.; Piemonte, F.; Tozzi, G.; Gaeta, L.; et al. Axonal degeneration in paraplegin-deficient mice is associated with abnormal mitochondria and impairment of axonal transport. J. Clin. Investig. 2004, 113, 231–242. [Google Scholar] [CrossRef]
- Wali, G.; Kumar, K.R.; Liyanage, E.; Davis, R.L.; Mackay-Sim, A.; Sue, C.M. Mitochondrial Function in Hereditary Spastic Paraplegia: Deficits in SPG7 but Not SPAST Patient-Derived Stem Cells. Front. Neurosci. 2020, 14, 820. [Google Scholar] [CrossRef]
- Arnoldi, A.; Tonelli, A.; Crippa, F.; Villani, G.; Pacelli, C.; Sironi, M.; Pozzoli, U.; D’Angelo, M.G.; Meola, G.; Martinuzzi, A.; et al. A clinical, genetic, and biochemical characterization of SPG7 mutations in a large cohort of patients with hereditary spastic paraplegia. Hum. Mutat. 2008, 29, 522–531. [Google Scholar] [CrossRef]
- Atorino, L.; Silvestri, L.; Koppen, M.; Cassina, L.; Ballabio, A.; Marconi, R.; Langer, T.; Casari, G. Loss of m-AAA protease in mitochondria causes complex I deficiency and increased sensitivity to oxidative stress in hereditary spastic paraplegia. J. Cell Biol. 2003, 163, 777–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanderson, C.M.; Connell, J.W.; Edwards, T.L.; Bright, N.A.; Duley, S.; Thompson, A.; Luzio, J.P.; Reid, E. Spastin and atlastin, two proteins mutated in autosomal-dominant hereditary spastic paraplegia, are binding partners. Hum. Mol. Genet. 2006, 15, 307–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blackstone, C. Cellular pathways of hereditary spastic paraplegia. Annu. Rev. Neurosci. 2012, 35, 25–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, P.P.; Denton, K.R.; Pierson, T.M.; Li, X.J.; Blackstone, C. Pharmacologic rescue of axon growth defects in a human iPSC model of hereditary spastic paraplegia SPG3A. Hum. Mol. Genet. 2014, 23, 5638–5648. [Google Scholar] [CrossRef] [Green Version]
- Schulman, I.G. Liver X receptors link lipid metabolism and inflammation. FEBS Lett. 2017, 591, 2978–2991. [Google Scholar] [CrossRef] [Green Version]
- Lorbek, G.; Lewinska, M.; Rozman, D. Cytochrome P450s in the synthesis of cholesterol and bile acids--from mouse models to human diseases. FEBS J. 2012, 279, 1516–1533. [Google Scholar] [CrossRef]
- Li-Hawkins, J.; Lund, E.G.; Turley, S.D.; Russell, D.W. Disruption of the oxysterol 7alpha-hydroxylase gene in mice. J. Biol. Chem. 2000, 275, 16536–16542. [Google Scholar] [CrossRef] [Green Version]
- Schule, R.; Siddique, T.; Deng, H.X.; Yang, Y.; Donkervoort, S.; Hansson, M.; Madrid, R.E.; Siddique, N.; Schols, L.; Bjorkhem, I. Marked accumulation of 27-hydroxycholesterol in SPG5 patients with hereditary spastic paresis. J. Lipid Res. 2010, 51, 819–823. [Google Scholar] [CrossRef] [Green Version]
- Mishra, H.K.; Prots, I.; Havlicek, S.; Kohl, Z.; Perez-Branguli, F.; Boerstler, T.; Anneser, L.; Minakaki, G.; Wend, H.; Hampl, M.; et al. GSK3ss-dependent dysregulation of neurodevelopment in SPG11-patient induced pluripotent stem cell model. Ann. Neurol. 2016, 79, 826–840. [Google Scholar] [CrossRef] [Green Version]
- Perez-Branguli, F.; Mishra, H.K.; Prots, I.; Havlicek, S.; Kohl, Z.; Saul, D.; Rummel, C.; Dorca-Arevalo, J.; Regensburger, M.; Graef, D.; et al. Dysfunction of spatacsin leads to axonal pathology in SPG11-linked hereditary spastic paraplegia. Hum. Mol. Genet. 2014, 23, 4859–4874. [Google Scholar] [CrossRef]
- Pozner, T.; Schray, A.; Regensburger, M.; Lie, D.C.; Schlotzer-Schrehardt, U.; Winkler, J.; Turan, S.; Winner, B. Tideglusib Rescues Neurite Pathology of SPG11 iPSC Derived Cortical Neurons. Front. Neurosci. 2018, 12, 914. [Google Scholar] [CrossRef] [Green Version]
- Hansen, J.; Corydon, T.J.; Palmfeldt, J.; Durr, A.; Fontaine, B.; Nielsen, M.N.; Christensen, J.H.; Gregersen, N.; Bross, P. Decreased expression of the mitochondrial matrix proteases Lon and ClpP in cells from a patient with hereditary spastic paraplegia (SPG13). Neuroscience 2008, 153, 474–482. [Google Scholar] [CrossRef]
- Denton, K.; Mou, Y.; Xu, C.C.; Shah, D.; Chang, J.; Blackstone, C.; Li, X.J. Impaired mitochondrial dynamics underlie axonal defects in hereditary spastic paraplegias. Hum. Mol. Genet. 2018, 27, 2517–2530. [Google Scholar] [CrossRef] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Tsang, H.T.; Connell, J.W.; Brown, S.E.; Thompson, A.; Reid, E.; Sanderson, C.M. A systematic analysis of human CHMP protein interactions: Additional MIT domain-containing proteins bind to multiple components of the human ESCRT III complex. Genomics 2006, 88, 333–346. [Google Scholar] [CrossRef] [Green Version]
- Diotel, N.; Charlier, T.D.; Lefebvre d’Hellencourt, C.; Couret, D.; Trudeau, V.L.; Nicolau, J.C.; Meilhac, O.; Kah, O.; Pellegrini, E. Steroid Transport, Local Synthesis, and Signaling within the Brain: Roles in Neurogenesis, Neuroprotection, and Sexual Behaviors. Front Neurosci. 2018, 12, 84. [Google Scholar] [CrossRef] [Green Version]
- Gilep, A.A.; Sushko, T.A.; Usanov, S.A. At the crossroads of steroid hormone biosynthesis: The role, substrate specificity and evolutionary development of CYP17. Biochim. Biophys. Acta 2011, 1814, 200–209. [Google Scholar] [CrossRef]
- Westrate, L.M.; Lee, J.E.; Prinz, W.A.; Voeltz, G.K. Form follows function: The importance of endoplasmic reticulum shape. Annu. Rev. Biochem. 2015, 84, 791–811. [Google Scholar] [CrossRef]
- Phillips, M.J.; Voeltz, G.K. Structure and function of ER membrane contact sites with other organelles. Nat. Rev. Mol. Cell Biol. 2016, 17, 69–82. [Google Scholar] [CrossRef] [Green Version]
- Allison, R.; Lumb, J.H.; Fassier, C.; Connell, J.W.; Ten Martin, D.; Seaman, M.N.; Hazan, J.; Reid, E. An ESCRT-spastin interaction promotes fission of recycling tubules from the endosome. J. Cell Biol. 2013, 202, 527–543. [Google Scholar] [CrossRef] [Green Version]
- Montenegro, G.; Rebelo, A.P.; Connell, J.; Allison, R.; Babalini, C.; D’Aloia, M.; Montieri, P.; Schule, R.; Ishiura, H.; Price, J.; et al. Mutations in the ER-shaping protein reticulon 2 cause the axon-degenerative disorder hereditary spastic paraplegia type 12. J. Clin. Investig. 2012, 122, 538–544. [Google Scholar] [CrossRef] [PubMed]
- Gruenberg, J. Life in the lumen: The multivesicular endosome. Traffic 2020, 21, 76–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Tong, M.; Fu, Y.; Chen, F.; Zhang, S.; Chen, H.; Ma, X.; Li, D.; Liu, X.; Zhong, Q. Lipids and membrane-associated proteins in autophagy. Protein Cell 2020. [Google Scholar] [CrossRef]
- Lee, C.A.; Blackstone, C. ER morphology and endo-lysosomal crosstalk: Functions and disease implications. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 1865, 158544. [Google Scholar] [CrossRef] [PubMed]
- Allison, R.; Edgar, J.R.; Pearson, G.; Rizo, T.; Newton, T.; Gunther, S.; Berner, F.; Hague, J.; Connell, J.W.; Winkler, J.; et al. Defects in ER-endosome contacts impact lysosome function in hereditary spastic paraplegia. J. Cell Biol. 2017, 216, 1337–1355. [Google Scholar] [CrossRef] [PubMed]
- Rickman, O.J.; Baple, E.L.; Crosby, A.H. Lipid metabolic pathways converge in motor neuron degenerative diseases. Brain 2020, 143, 1073–1087. [Google Scholar] [CrossRef] [Green Version]
- Renvoise, B.; Malone, B.; Falgairolle, M.; Munasinghe, J.; Stadler, J.; Sibilla, C.; Park, S.H.; Blackstone, C. Reep1 null mice reveal a converging role for hereditary spastic paraplegia proteins in lipid droplet regulation. Hum. Mol. Genet. 2016, 25, 5111–5125. [Google Scholar] [CrossRef] [Green Version]
- Papadopoulos, C.; Orso, G.; Mancuso, G.; Herholz, M.; Gumeni, S.; Tadepalle, N.; Jungst, C.; Tzschichholz, A.; Schauss, A.; Honing, S.; et al. Spastin binds to lipid droplets and affects lipid metabolism. PLoS Genet. 2015, 11, e1005149. [Google Scholar] [CrossRef] [Green Version]
- Arribat, Y.; Grepper, D.; Lagarrigue, S.; Qi, T.; Cohen, S.; Amati, F. Spastin mutations impair coordination between lipid droplet dispersion and reticulum. PLoS Genet. 2020, 16, e1008665. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.L.; Weigel, A.V.; Ioannou, M.S.; Pasolli, H.A.; Xu, C.S.; Peale, D.R.; Shtengel, G.; Freeman, M.; Hess, H.F.; Blackstone, C.; et al. Spastin tethers lipid droplets to peroxisomes and directs fatty acid trafficking through ESCRT-III. J. Cell Biol. 2019, 218, 2583–2599. [Google Scholar] [CrossRef]
- Fan, Y.; Wali, G.; Sutharsan, R.; Bellette, B.; Crane, D.I.; Sue, C.M.; Mackay-Sim, A. Low dose tubulin-binding drugs rescue peroxisome trafficking deficit in patient-derived stem cells in Hereditary Spastic Paraplegia. Biol. Open 2014, 3, 494–502. [Google Scholar] [CrossRef] [Green Version]
- Van der Kant, R.; Langness, V.F.; Herrera, C.M.; Williams, D.A.; Fong, L.K.; Leestemaker, Y.; Steenvoorden, E.; Rynearson, K.D.; Brouwers, J.F.; Helms, J.B.; et al. Cholesterol Metabolism Is a Druggable Axis that Independently Regulates Tau and Amyloid-beta in iPSC-Derived Alzheimer’s Disease Neurons. Cell Stem Cell 2019, 24, 363–375.e369. [Google Scholar] [CrossRef] [Green Version]
- Hong, C.; Tontonoz, P. Liver X receptors in lipid metabolism: Opportunities for drug discovery. Nat. Rev. Drug Discov. 2014, 13, 433–444. [Google Scholar] [CrossRef]
- Prufer, K.; Boudreaux, J. Nuclear localization of liver X receptor alpha and beta is differentially regulated. J. Cell. Biochem. 2007, 100, 69–85. [Google Scholar] [CrossRef]
- Barnat, M.; Benassy, M.N.; Vincensini, L.; Soares, S.; Fassier, C.; Propst, F.; Andrieux, A.; von Boxberg, Y.; Nothias, F. The GSK3-MAP1B pathway controls neurite branching and microtubule dynamics. Mol. Cell. Neurosci. 2016, 72, 9–21. [Google Scholar] [CrossRef]
- Boutry, M.; Pierga, A.; Matusiak, R.; Branchu, J.; Houllegatte, M.; Ibrahim, Y.; Balse, E.; El Hachimi, K.H.; Brice, A.; Stevanin, G.; et al. Loss of spatacsin impairs cholesterol trafficking and calcium homeostasis. Commun. Biol. 2019, 2, 380. [Google Scholar] [CrossRef] [Green Version]
- Bordt, E.A.; Clerc, P.; Roelofs, B.A.; Saladino, A.J.; Tretter, L.; Adam-Vizi, V.; Cherok, E.; Khalil, A.; Yadava, N.; Ge, S.X.; et al. The Putative Drp1 Inhibitor mdivi-1 Is a Reversible Mitochondrial Complex I Inhibitor that Modulates Reactive Oxygen Species. Dev. Cell. 2017, 40, 583–594.e586. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Zheng, Z.; Han, W.; Yuan, Y.; Li, Y.; Zhou, K.; Wang, Q.; Xie, L.; Xu, K.; Zhang, H.; et al. Metformin Promotes Axon Regeneration after Spinal Cord Injury through Inhibiting Oxidative Stress and Stabilizing Microtubule. Oxid. Med. Cell. Longev. 2020, 2020, 9741369. [Google Scholar] [CrossRef]
- Yoo, Y.E.; Ko, C.P. Treatment with trichostatin A initiated after disease onset delays disease progression and increases survival in a mouse model of amyotrophic lateral sclerosis. Exp. Neurol. 2011, 231, 147–159. [Google Scholar] [CrossRef]
- Jaiswal, M.K. Riluzole and edaravone: A tale of two amyotrophic lateral sclerosis drugs. Med. Res. Rev. 2019, 39, 733–748. [Google Scholar] [CrossRef]
- Fanara, P.; Banerjee, J.; Hueck, R.V.; Harper, M.R.; Awada, M.; Turner, H.; Husted, K.H.; Brandt, R.; Hellerstein, M.K. Stabilization of hyperdynamic microtubules is neuroprotective in amyotrophic lateral sclerosis. J. Biol. Chem. 2007, 282, 23465–23472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perlman, S.L. Update on the Treatment of Ataxia: Medication and Emerging Therapies. Neurotherapeutics 2020. [Google Scholar] [CrossRef] [PubMed]
- Bernard-Marissal, N.; Medard, J.J.; Azzedine, H.; Chrast, R. Dysfunction in endoplasmic reticulum-mitochondria crosstalk underlies SIGMAR1 loss of function mediated motor neuron degeneration. Brain 2015, 138, 875–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregianin, E.; Pallafacchina, G.; Zanin, S.; Crippa, V.; Rusmini, P.; Poletti, A.; Fang, M.; Li, Z.; Diano, L.; Petrucci, A.; et al. Loss-of-function mutations in the SIGMAR1 gene cause distal hereditary motor neuropathy by impairing ER-mitochondria tethering and Ca2+ signalling. Hum. Mol. Genet. 2016, 25, 3741–3753. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, T.; Su, T.P. Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca(2+) signaling and cell survival. Cell 2007, 131, 596–610. [Google Scholar] [CrossRef] [Green Version]
- Panagaki, T.; Michael, M.; Holscher, C. Liraglutide restores chronic ER stress, autophagy impairments and apoptotic signalling in SH-SY5Y cells. Sci. Rep. 2017, 7, 16158. [Google Scholar] [CrossRef]
- Yamamoto, W.R.; Bone, R.N.; Sohn, P.; Syed, F.; Reissaus, C.A.; Mosley, A.L.; Wijeratne, A.B.; True, J.D.; Tong, X.; Kono, T.; et al. Endoplasmic reticulum stress alters ryanodine receptor function in the murine pancreatic beta cell. J. Biol. Chem. 2019, 294, 168–181. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical SPG Classifications and HSP Genes | |||||
|---|---|---|---|---|---|
| HSP | Inheritance | Gene Name | HSP | Inheritance | Gene Name |
| Classification | Classification | ||||
| SPG01 | XL | L1CAM * | SPG41 | AD | Gene Locus |
| SPG02 | XL | PLP1 * | SPG42 | AD | SLC33A1 ** |
| SPG03A | AD | ATL1 *** | SPG43 | AR | C10orf12 * |
| SPG04 | AD | SPAST *** | SPG44 | AR | GJC2 * |
| SPG05A | AR | CYP7B1*** | SPG45 | AR | NT5C2 * |
| SPG06 | AD | NIPA1 ** | SPG46 | AR | GBA2 * |
| SPG07 | AR | SPG7 * | SPG47 | AR | AP4B1 * |
| SPG08 | AD | WASHC5 ** | SPG48 | AR | AP5Z1 ** |
| SPG09 | AR | ALDH18A1 | SPG49 | AR | TECPR2 * |
| SPG10 | AD | KIF5A * | SPG50 | AR | AP4M1 * |
| SPG11 | AR | SPG11 * | SPG51 | AR | AP4E1 * |
| SPG12 | AD | RTN2*** | SPG52 | AR | AP4S1 * |
| SPG13 | AD | HSPD1 ** | SPG53 | AR | VPS37A * |
| SPG14 | AR | Gene Locus | SPG54 | AR | DDH2 ** |
| SPG15 | AR | ZFYVE26 * | SPG55 | AR | C19ORF65R |
| SPG16 | XL | Gene Locus | SPG56 | AR | CYP2U1 * |
| SPG17 | AD | BSCL2 * | SPG57 | AR | TFG * |
| SPG18 | AR | ERLIN2 * | SPG58 | AR | KIF1C * |
| SPG19 | AD | Gene Locus | SPG59 | AR | USP8 ** |
| SPG20 | AR | SPART * | SPG60 | AR | WDR48 * |
| SPG21 | AR | SPG21 * | SPG61 | EX | ARL6IP1 * |
| SPG22 | XL | SLC16A2 * | SPG62 | AR | ERLIN1 * |
| SPG23 | AR | DSTYK * | SPG63 | AR | AMPD2 |
| SPG24 | AR | Gene Locus | SPG64 | AR | ENTPD1 * |
| SPG25 | AR | Gene Locus | SPG65 | AR | NT5C2 * |
| SPG26 | AR | B4GALNT1* * | SPG66 | AR | ARSI * |
| SPG27 | AR | Gene Locus | SPG67 | AR | PGAP1 * |
| SPG28 | AR | DDHD1 * | SPG68 | AR | FLRT1 * |
| SPG29 | AD | Gene Locus | SPG69 | AR | RAB3GAP2 * |
| SPG30 | AR | KIF1A * | SPG70 | AR | MARS1 |
| SPG31 | AD | REEP1 *** | SPG71 | AR | ZFR ** |
| SPG32 | AD | Gene Locus | SPG72 | AR | REEP2 * |
| SPG33 | AD | ZFYVE27 *** | SPG73 | AD | CPT1C * |
| SPG34 | XL | SPG34 | SPG74 | AR | IBA57 * |
| SPG35 | AR | FA2H * | SPG75 | AR | MAG |
| SPG36 | AD | SPG36 | SPG76 | AR | CAPN1 |
| SPG37 | AD | SPG37 | SPG77 | AR | FARS2 |
| SPG38 | AD | SPG38 | SPG78 | AR | ATP13A |
| SPG39 | AR | PNPLA6 * | SPG79 | AR | UCHL1 |
| SPG40 | AD | Gene Locus | SPG80 | AD | UBAP1 ** |
| 6 Most Common Pure SPGs | 15 Pure SPGs | 42 Complicated SPGs | 57 Pure and Uncomplicated SPGs |
|---|---|---|---|
| Biological process | |||
| endoplasmic reticulum tubular network organization | endoplasmic reticulum tubular network organization | retrograde neuronal dense core vesicle transport | endoplasmic reticulum tubular network |
| endomembrane system organization | endomembrane system organization | intracellular transport | intracellular transport |
| cytoskeleton-dependent cytokinesis | axo-dendritic transport | cellular localization | |
| cellular localization | lipid metabolic process | amide transport | |
| establishment of localization | intracellular protein transport | axo-dendritic transport | |
| localization | negative regulation of cholesterol biosynthetic process | endomembrane system organization | |
| protein localization | anterograde neuronal dense core vesicle transport | establishment of protein localization | |
| endosomal transport | DREBP signaling pathway | localization | |
| intracellular transport | cytosolic transport | establishment of localization | |
| amide transport | establishment of vesicle location | retrograde neuronal dense core vesicle transport | |
| Cellular Component | |||
| endoplasmic reticulum tubular network | endoplasmic reticulum tubular network | organelle subcompartment | organelle subcompartment |
| endoplasmic reticulum membrane | endosome | AP4-adaptor complex | endoplasmic reticulum membrane |
| endoplasmic reticulum subcompartment | endomembrane system | organelle membrane | endoplasmic reticulum subcompartment |
| axon | endoplasmic reticulum membrane | endoplasmic reticulum membrane | organelle membrane |
| integral component of endoplasmic reticulum membrane | endoplasmic reticulum subcompartment | endoplasmic reticulum subcompartment | endomembrane system |
| endoplasmic reticulum | endomembrane system | endoplasmic reticulum | |
| organelle membrane | endoplasmic reticulum | AP-4 adaptor complex | |
| early endosome | endosome lumen | endosome | |
| cytoplasm | trans-Golgi network membrane | AP-type membrane coat adaptor complex | |
| neuron projection | membrane | endoplasmic reticulum tubular network |
| 6 Most Common Pure SPGs | 15 Pure SPGs | 42 Complicated SPGs | 57 Pure and Uncomplicated SPGs |
|---|---|---|---|
| Biological Process | |||
| multivesicular body assembly | endosomal transport | organic substance catabolic process | intracellular transport |
| viral budding | amide transport | intracellular transport | establishment of protein localization |
| endomembrane system organization | establishment of protein localization | catabolic process | amide transport |
| steroid biosynthesis process | protein transport | cellular macromolecule catabolic process | protein transport |
| septum digestion after cytokinesis | intracellular transport | cellular catabolic process | cellular localization |
| viral budding via host ESCRT complex | protein localization | protein targeting | establishment of localization |
| viral life cycle | virion assembly | establishment of protein localization | protein localization |
| ESCRT III complex disassembly | cellular localization | intracellular protein transport | macromolecular localization |
| androgen biosynthetic process | organic substance transport | establishment of protein localization to endoplasmic reticulum | endomembrane system organization |
| vacuolar transport | endosome organization | protein transport | localization |
| Cellular Component | |||
| organelle membrane | endoplasmic reticulum tubular network | organelle subcompartment | organelle subcompartment |
| ESCRT III | endosome | AP4-adaptor complex | endoplasmic reticulum membrane |
| endomembrane system | endomembrane system | organelle membrane | endoplasmic reticulum subcompartment |
| endoplasmic reticulum membrane | endoplasmic reticulum membrane | endoplasmic reticulum membrane | organelle membrane |
| endoplasmic reticulum subcompartment | endoplasmic reticulum subcompartment | endoplasmic reticulum subcompartment | endomembrane system |
| endosome membrane | endoplasmic reticulum | endomembrane system | endoplasmic reticulum |
| late endosome membrane | organelle membrane | endoplasmic reticulum | AP-4 adaptor complex |
| endoplasmic reticulum tubular network | early endosome | endosome lumen | endosome |
| endosome | cytoplasm | trans-Golgi network membrane | AP-type membrane coat adaptor complex |
| endoplasmic reticulum | neuron projection | membrane | endoplasmic reticulum tubular network |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mackay-Sim, A. Hereditary Spastic Paraplegia: From Genes, Cells and Networks to Novel Pathways for Drug Discovery. Brain Sci. 2021, 11, 403. https://doi.org/10.3390/brainsci11030403
Mackay-Sim A. Hereditary Spastic Paraplegia: From Genes, Cells and Networks to Novel Pathways for Drug Discovery. Brain Sciences. 2021; 11(3):403. https://doi.org/10.3390/brainsci11030403
Chicago/Turabian StyleMackay-Sim, Alan. 2021. "Hereditary Spastic Paraplegia: From Genes, Cells and Networks to Novel Pathways for Drug Discovery" Brain Sciences 11, no. 3: 403. https://doi.org/10.3390/brainsci11030403
APA StyleMackay-Sim, A. (2021). Hereditary Spastic Paraplegia: From Genes, Cells and Networks to Novel Pathways for Drug Discovery. Brain Sciences, 11(3), 403. https://doi.org/10.3390/brainsci11030403