
Contribution of the Degeneration of the Neuro-Axonal Unit to the Pathogenesis of Multiple Sclerosis


{kind=link}
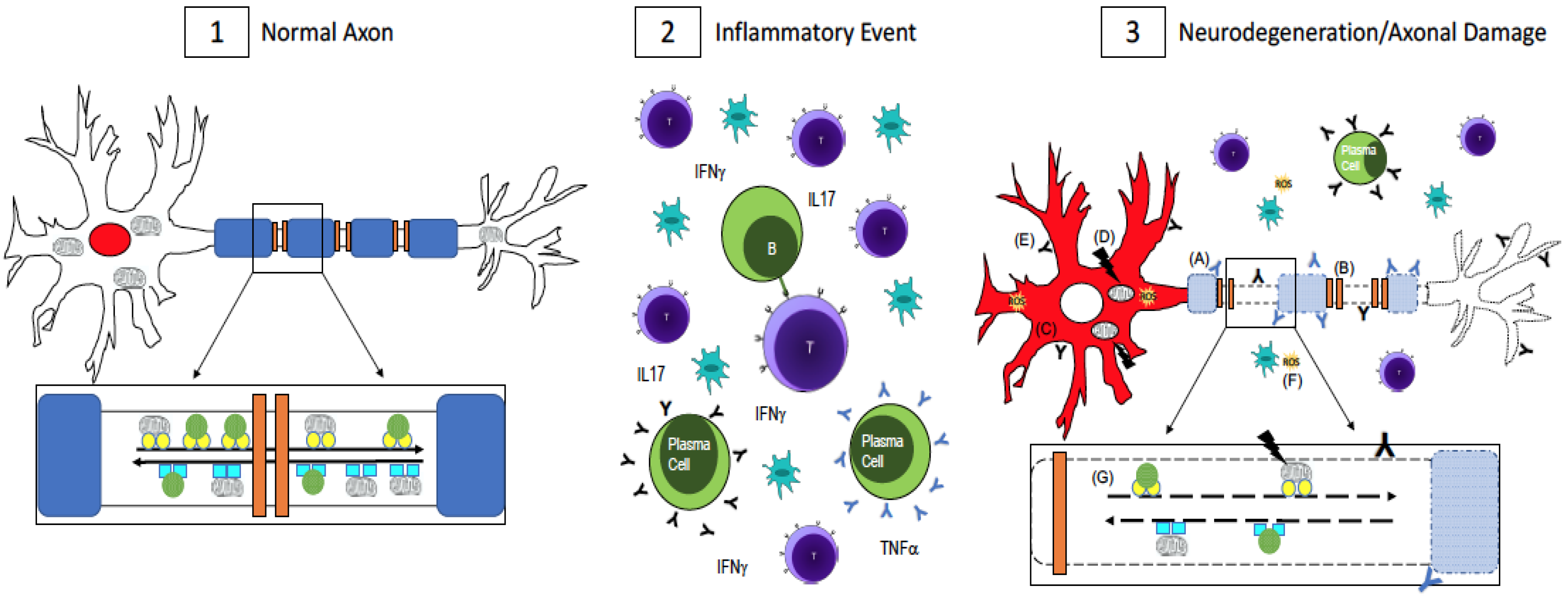
Abstract
:1. Multiple Sclerosis (MS)
2. hnRNP A1 and RNA Metabolism
3. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lassmann, H.; Brück, W.; Lucchinetti, C.F. The immunopathology of multiple sclerosis: An overview. Brain Pathol. 2007, 17, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Noseworthy, J.H.; Lucchinetti, C.; Rodriguez, M.; Weinshenker, B.G. Multiple sclerosis. N. Engl. J. Med. 2000, 343, 938–952. [Google Scholar] [CrossRef] [PubMed]
- Levin, M.C.; Lee, S.; Gardner, L.A.; Shin, Y.; Douglas, J.N.; Groover, C.J. Pathogenic mechanisms of neurodegeneration based on the phenotypic expression of progressive forms of immune-mediated neurologic disease. Degener. Neurol. Neuromuscul. Dis. 2012, 2, 175–187. [Google Scholar] [CrossRef]
- Milo, R.; Miller, A. Revised diagnostic criteria of multiple sclerosis. Autoimmun. Rev. 2014, 13, 518–524. [Google Scholar] [CrossRef] [PubMed]
- Confavreux, C.; Vukusic, S.; Moreau, T.; Adeleine, P. Relapses and progression of disability in multiple sclerosis. N. Engl. J. Med. 2000, 343, 1430–1438. [Google Scholar] [CrossRef] [PubMed]
- Love, S. Demyelinating diseases. J. Clin. Pathol. 2006, 59, 1151–1159. [Google Scholar] [CrossRef] [PubMed]
- Fisher, E.; Lee, J.C.; Nakamura, K.; Rudick, R.A. Gray matter atrophy in multiple sclerosis: A longitudinal study. Ann. Neurol. 2008, 64, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Fisniku, L.K.; Chard, D.T.; Jackson, J.S.; Anderson, V.M.; Altmann, D.R.; Miszkiel, K.A.; Thompson, A.J.; Miller, D.H. Gray matter atrophy is related to long-term disability in multiple sclerosis. Ann. Neurol. 2008, 64, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Kornek, B.; Storch, M.K.; Weissert, R.; Wallstroem, E.; Stefferl, A.; Olsson, T.; Linington, C.; Schmidbauer, M.; Lassmann, H. Multiple sclerosis and chronic autoimmune encephalomyelitis: A comparative quantitative study of axonal injury in active, inactive, and remyelinated lesions. Am. J. Pathol. 2000, 157, 267–276. [Google Scholar] [CrossRef]
- Kutzelnigg, A.; Lucchinetti, C.F.; Stadelmann, C.; Brück, W.; Rauschka, H.; Bergmann, M.; Schmidbauer, M.; Parisi, J.E.; Lassmann, H. Cortical demyelination and diffuse white matter injury in multiple sclerosis. Brain 2005, 128, 2705–2712. [Google Scholar] [CrossRef] [PubMed]
- DeLuca, G.C.; Williams, K.; Evangelou, N.; Ebers, G.C.; Esiri, M.M. The contribution of demyelination to axonal loss in multiple sclerosis. Brain 2006, 129, 1507–1516. [Google Scholar] [CrossRef] [PubMed]
- Nikić, I.; Merkler, D.; Sorbara, C.; Brinkoetter, M.; Kreutzfeldt, M.; Bareyre, F.M.; Brück, W.; Bishop, D.; Misgeld, T.; Kerschensteiner, M. A reversible form of axon damage in experimental autoimmune encephalomyelitis and multiple sclerosis. Nat. Med. 2011, 17, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Lassmann, H.; van Horssen, J. The molecular basis of neurodegeneration in multiple sclerosis. FEBS Lett. 2011, 585, 3715–3723. [Google Scholar] [CrossRef] [PubMed]
- Frischer, J.M.; Bramow, S.; Dal-Bianco, A.; Lucchinetti, C.F.; Rauschka, H.; Schmidbauer, M.; Laursen, H.; Sorensen, P.S.; Lassmann, H. The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain 2009, 132, 1175–1189. [Google Scholar] [CrossRef] [PubMed]
- Trapp, B.D.; Nave, K.A. Multiple sclerosis: An immune or neurodegenerative disorder? Annu. Rev. Neurosci. 2008, 31, 247–269. [Google Scholar] [CrossRef] [PubMed]
- Benarroch, E.E. Acquired axonal degeneration and regeneration. Neurology 2015, 84, 2076–2085. [Google Scholar] [CrossRef] [PubMed]
- Dziedzic, T.; Metz, I.; Dallenga, T.; König, F.B.; Müller, S.; Stadelmann, C.; Brück, W. Wallerian degeneration: A major component of early axonal pathology in multiple sclerosis. Brain Pathol. 2010, 20, 976–985. [Google Scholar] [CrossRef] [PubMed]
- Solowska, J.M.; Baas, P.W. Hereditary spastic paraplegia SPG4: What is known and not known about the disease. Brain 2015, 138, 2471–2484. [Google Scholar] [CrossRef] [PubMed]
- DeLuca, G.C.; Ebers, G.C.; Esiri, M.M. The extent of axonal loss in the long tracts in hereditary spastic paraplegia. Neuropathol. Appl. Neurobiol. 2004, 30, 576–584. [Google Scholar] [CrossRef] [PubMed]
- Denton, K.R.; Lei, L.; Grenier, J.; Rodionov, V.; Blackstone, C.; Li, X. Loss of spastin function results in disease-specific axonal defects in human pluripotent stem cell-based models of hereditary spastic paraplegia. Stem Cells 2014, 32, 414–423. [Google Scholar] [CrossRef] [PubMed]
- Bilsland, L.G.; Sahai, E.; Kelly, G.; Golding, M.; Greensmith, L.; Schiavo, G. Deficits in axonal transport precede ALS symptoms in vivo. Proc. Natl. Acad. Sci. USA 2010, 107, 20523–20528. [Google Scholar] [CrossRef] [PubMed]
- Shi, P.; Gal, J.; Kwinter, D.M.; Liu, X.; Zhu, H. Mitochondrial dysfunction in amyotrophic lateral sclerosis. Biochim. Biophys. Acta 2010, 1802, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Calkins, M.J.; Reddy, P.H. Amyloid beta impairs mitochondrial anterograde transport and degenerates synapses in Alzheimer’s disease neurons. Biochim. Biophys. Acta 2011, 1812, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H.V. Mitochondria in the aetiology and pathogenesis in Parkinson’s disease. Lancet Neurol. 2008, 7, 97–109. [Google Scholar] [CrossRef]
- Alami, N.H.; Smith, R.B.; Carrasco, M.A.; Williams, L.A.; Winborn, C.S.; Han, S.S.; Kiskinis, E.; Winborn, B.; Freibaum, B.D.; Kanagaraj, A.; et al. Axonal transport of TDP-43 mRNA granules is impaired by ALS-causing mutations. Neuron 2014, 81, 536–543. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Kim, N.C.; Wang, Y.D.; Scarboough, E.A.; Moore, J.; Diaz, Z.; MacLea, K.S.; Freibaum, B.; Li, S.; Molliex, A.; et al. Mutations in prion-like domains in hnRNP A2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 2013, 495, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Ling, S.C.; Polymenidou, M.; Cleveland, D.W. Converging mechanisms in ALS and FTD: Disrupted RNA and protein homeostasis. Neuron 2013, 79, 416–438. [Google Scholar] [CrossRef] [PubMed]
- Ranaswami, M.; Taylor, J.P.; Parker, R. Altered “ribostasis”: RNA-protein granule formation or persistence in the development of degenerative disorders. Cell 2013, 154, 727–735. [Google Scholar] [CrossRef] [PubMed]
- Ugras, S.E.; Shorter, J. RNA-binding proteins in amyotrophic lateral sclerosis and neurodegeneration. Neurol. Res. Int. 2012, 2012, 432780. [Google Scholar] [CrossRef] [PubMed]
- Lukong, K.E.; Chang, K.W.; Khandjian, E.W.; Richard, S. RNA-binding proteins in human genetic disease. Trends Genet. 2008, 24, 416–425. [Google Scholar] [CrossRef] [PubMed]
- Orr, H.T. FTD and ALS: Genetic ties that bind. Neuron 2011, 72, 189–190. [Google Scholar] [CrossRef] [PubMed]
- Black, J.A.; Newcombe, J.; Trapp, B.D.; Waxman, B.D. Sodium channel expression within chronic multiple sclerosis plaques. J. Neuropathol. Exp. Neurol. 2007, 66, 828–837. [Google Scholar] [CrossRef] [PubMed]
- Craner, M.J.; Newcombe, J.; Black, J.A.; Hartle, C.; Cuzner, M.L.; Waxman, S.G. Molecular changes in neurons in multiple sclerosis: Altered axonal expression of Nav1.2 and Nav1.6 sodium channels and Na+/Ca2+ exchanger. Proc. Natl. Acad. Sci. USA 2004, 101, 8168–8173. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, B.; Matyszak, M.K.; Esiri, M.M.; Perry, V.H. Axonal damage in acute multiple sclerosis lesions. Brain 1997, 120, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Friese, M.A.; Schattling, B.; Fugger, L. Mechanisms of neurodegeneration and axonal dysfuction in multiple sclerosis. Nat. Rev. Neurol. 2014, 10, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Sorbara, C.D.; Wagner, N.E.; Ladwig, A.; Nikić, I.; Merkler, D.; Kleele, T.; Marinković, P.; Naumann, R.; Godinho, L.; Bareyre, F.M.; et al. Pervasive axonal transport deficits in multiple sclerosis models. Neuron 2014, 84, 1183–1190. [Google Scholar] [CrossRef] [PubMed]
- De Vos, K.J.; Grierson, A.J.; Ackerley, S.; Miller, C.C.J. Role of axonal transport in neurodegenerative diseases. Annu. Rev. Neurosci. 2008, 31, 151–173. [Google Scholar] [CrossRef] [PubMed]
- Dutta, R.; McDonough, J.; Yin, X.; Peterson, J.; Chang, A.; Torres, T.; Gudz, T.; Macklin, W.B.; Lewis, D.A.; Fox, R.J.; et al. Mitochondrial dysfunction as a cause of axonal degeneration in multiple sclerosis patients. Ann. Neurol. 2006, 59, 478–489. [Google Scholar] [CrossRef] [PubMed]
- Campbell, G.R.; Ziabreva, I.; Reeve, A.K.; Krishnan, K.J.; Reynolds, R.; Howell, O.; Lassmann, H.; Turnbull, D.M.; Mahad, D.J. Mitochondrial DNA deletions and neurodegeneration in multiple sclerosis. Ann. Neurol. 2011, 69, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.T.; Sharma, R.; Lim, J.L.; Haider, L.; Frischer, J.M.; Drexhage, J.; Mahad, D.; Bradl, M.; van Horssen, J.; Lassmann, H. NADPH oxidase expression in active multiple sclerosis lesions in relation to oxidative tissue damage and mitochondrial injury. Brain 2012, 135, 886–899. [Google Scholar] [CrossRef] [PubMed]
- Haider, L.; Fischer, M.T.; Frischer, J.M.; Bauer, J.; Höftberger, R.; Botond, G.; Esterbauer, H.; Binder, C.J.; Witztum, J.L.; Lassmann, H. Oxidative damage in multiple sclerosis lesions. Brain 2011, 134, 1914–1924. [Google Scholar] [CrossRef] [PubMed]
- Satpute-Krishnan, P.; DeGiorgis, J.A.; Conley, M.P.; Jang, M.; Bearer, E.L. A peptide zipcode sufficient for anterograde transport within amyloid precursor protein. Proc. Natl. Acad. Sci. USA 2006, 103, 16532–16537. [Google Scholar] [CrossRef] [PubMed]
- Schluesener, H.J.; Sobel, R.A.; Linington, C.; Weiner, H.L. A monoclonal antibody against a myelin oligodendrocyte glycoprotein induces relapses and demyelination in central nervous system autoimmune disease. J. Immunol. 1987, 139, 4016–4021. [Google Scholar] [PubMed]
- Vanguri, P.; Koski, C.L.; Silverman, B.; Shin, M.L. Complement activation by isolated myelin: Activation of the classical pathway in the absence of myelin-specific antibodies. Proc. Natl. Acad. Sci. USA 1982, 79, 3290–3294. [Google Scholar] [CrossRef] [PubMed]
- Vanguri, P.; Shin, M.L. Activation of complement by myelin: Identification of C1-binding proteins of human myelin from central nervous tissue. J. Neurochem. 1986, 46, 1535–1541. [Google Scholar] [CrossRef] [PubMed]
- Huizinga, R.; Heijmans, N.; Schubert, P.; Gschmeissner, S.; ’t Hart, B.A.; Herrmann, H.; Amor, S. Immunization with neurofilament light protein induces spastic paresis and axonal degeneration in Biozzi ABH mice. J. Neuropathol. Exp. Neurol. 2007, 66, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Huizinga, R.; Gerritsen, W.; Heijmans, N.; Amor, S. Axonal loss and gray matter pathology as a direct result of autoimmunity to neurofilaments. Neurobiol. Dis. 2008, 32, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Mathey, E.K.; Derfuss, T.; Storch, M.K.; Williams, K.R.; Hales, K.; Woolley, D.R.; Al-Hayani, A.; Davies, S.N.; Rasband, M.N.; Olsson, T.; et al. Neurofascin as a novel target for autoantibody-mediated axonal injury. J. Exp. Med. 2007, 204, 2363–2372. [Google Scholar] [CrossRef] [PubMed]
- Levin, M.C.; Lee, S.; Gardner, L.A.; Shin, Y.; Douglas, J.N.; Cooper, C. Autoantibodies to non-myelin antigens as contributors to the pathogenesis of multiple sclerosis. J. Clin. Cell Immunol. 2013, 30, 4. [Google Scholar] [CrossRef] [PubMed]
- Johri, A.; Beal, M.F. Mitochondrial dysfunction in neurodegenerative diseases. J. Pharmacol. Exp. Ther. 2012, 342, 619–630. [Google Scholar] [CrossRef] [PubMed]
- Peterson, J.W.; Bö, L.; Mörk, S.; Chang, A.; Trapp, B.D. Transected neurites, apoptotic neurons, and reduced inflammation in cortical multiple sclerosis lesions. Ann. Neurol. 2001, 50, 389–400. [Google Scholar] [CrossRef] [PubMed]
- Vercellino, M.; Plano, F.; Votta, B.; Mutani, R.; Giordana, M.T.; Cavalla, P. Grey matter pathology in multiple sclerosis. J. Neuropathol. Exp. Neurol. 2005, 64, 1101–1107. [Google Scholar] [CrossRef] [PubMed]
- Booss, J.; Esiri, M.M.; Tourtellotte, W.W.; Mason, D.Y. Immunohistological analaysis of T lymphocyte subsets in the central nervous system in chronic progressive multiple sclerosis. J. Neurol. Sci. 1983, 62, 219–232. [Google Scholar] [CrossRef]
- Lassmann, H.; Bradl, M. Multiple sclerosis: Experimental models and reality. Acta Neuropathol. 2017, 133, 223–244. [Google Scholar] [CrossRef] [PubMed]
- Morandi, B.; Bramanti, P.; Bonaccorsi, I.; Montalto, E.; Oliveri, D.; Pezzino, G.; Navarra, M.; Ferlazzo, G. Role of natural killer cells in the pathogenesis and progression of multiple sclerosis. Pharmacol. Res. 2008, 57, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Barnett, M.H.; Parratt, J.D.; Cho, E.S.; Prineas, J.W. Immunoglobulins and complement in postmortem multiple sclerosis tissue. Ann. Neurol. 2009, 65, 32–46. [Google Scholar] [CrossRef] [PubMed]
- Meinl, E.; Derfuss, T.; Krumbholz, M.; Pröbstel, A.K.; Hohlfeld, R. Humoral autoimmunity in multiple sclerosis. J. Neurol. Sci. 2010, 306, 180–182. [Google Scholar] [CrossRef] [PubMed]
- Mead, R.J.; Neal, J.W.; Griffiths, M.R.; Linington, C.; Botto, M.; Lassmann, H.; Morgan, B.M. Deficiency of the complement regulator CD59a enhances disease severity, demyelination and axonal injury in murine acute experimental allergic encephalomyelitis. Lab. Investig. 2004, 84, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Levin, M.C.; Lee, S.; Kalume, F.; Morcos, Y.; Dohan, F.C.; Hasty, K.A.; Callaway, J.C.; Zunt, J.; Desiderio, D.M.; Stuart, J.M. Autoimmunity due to molecular mimicry as a cause of neurological disease. Nat. Med. 2002, 8, 509–513. [Google Scholar] [CrossRef] [PubMed]
- Jean-Philippe, J.; Paz, S.; Caputi, M. hnRNP A1: The Swiss army knife of gene expression. Int. J. Mol. Sci. 2013, 14, 18999–19024. [Google Scholar] [CrossRef] [PubMed]
- Jernigan, M.; Morcos, Y.; Lee, S.M.; Dohan, F.C., Jr.; Raine, C.; Levin, M.C. IgG in brain correlates with clinicopathological damage in HTLV-1 associated neurologic disease. Neurology 2003, 60, 1320–1327. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.M.; Dunnavant, F.D.; Jang, H.; Zunt, J.; Levin, M.C. Autoantibodies that recognize functional domains of hnRNPA1 implicate molecular mimicry in the pathogenesis of neurologic disease. Neurosci. Lett. 2006, 401, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Jean-Philippe, J.; Paz, S.; Lu, M.L.; Caputi, M. A truncated hnRNP A1 isoform, lacking the RGG-box RNA binding domain, can efficiently regulate HIV-1 splicing and replication. Biochim. Biophys. Acta 2014, 1839, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Xu, L.; Shin, Y.; Gardner, L.; Hartzes, A.; Dohan, F.C.; Raine, C.; Homayouni, R.; Levin, M.C. A potential link between autoimmunity and neurodegeneration in immune-mediated neurological disease. J. Neuroimmunol. 2011, 235, 56–69. [Google Scholar] [CrossRef] [PubMed]
- Yukitake, M.; Sueoka, E.; Sueoka-Aragane, N.; Sato, A.; Ohashi, H.; Yakushiji, Y.; Saito, M.; Osame, M.; Izumo, S.; Kuroda, Y. Significantly increased antibody response to heterogeneous nuclear ribonucleoproteins in cerebrospinal fluid of multiple sclerosis patients not in patients with human T-lymphotropic virus I-associated myelopathy/tropical spastic paraparesis. J. Neurovirol. 2008, 14, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Douglas, J.N.; Gardner, L.A.; Salapa, H.E.; Levin, M.C. Antibodies to the RNA binding protein heterogeneous nuclear ribonucleoprotein A1 colocalize to stress granules resulting in altered RNA and protein levels in a model of neurodegeneration in multiple sclerosis. J. Clin. Cell. Immunol. 2016, 7, 402. [Google Scholar] [PubMed]
- Douglas, J.N.; Gardner, L.A.; Levin, M.C. Antibodies to an intracellular antigen penetrate neuronal cells and cause deleterious effects. J. Clin. Cell. Immunol. 2013, 4, 134. [Google Scholar] [CrossRef]
- Douglas, J.N.; Gardner, L.A.; Salapa, H.E.; Lalor, S.J.; Lee, S.; Segal, B.M.; Sawchenko, P.E.; Levin, M.C. Antibodies to the RNA-binding protein hnRNP A1 contribute to neurodegeneration in a model of central nervous system autoimmune inflammatory disease. J. Neuroinflamm. 2016, 13, 178. [Google Scholar] [CrossRef] [PubMed]
- Arima, Y.; Harada, M.; Kamimura, D.; Park, J.H.; Kawano, F.; Yull, F.E.; Kawamoto, T.; Iwakura, Y.; Betz, U.A.; Márquez, G.; et al. Regional neural activation defines a gateway for autoreactive T cells to cross the blood-brain barrier. Cell 2012, 148, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Kamimura, D.; Yamada, M.; Harada, M.; Sabharwal, L.; Meng, J.; Bando, H.; Ogura, H.; Atsumi, T.; Arima, Y.; Murakami, M. The gateway theory: Bridging neural and immune interactions in the CNS. Front. Neurosci. 2013, 29, 204. [Google Scholar] [CrossRef] [PubMed]
- Solowska, J.M.; Morfini, G.; Falnikar, A.; Himes, B.T.; Brady, S.T.; Huang, D.; Baas, P.W. Quantitative and functional analyses of spastin in the nervous system: Implications for hereditary spastic paraplegia. J. Neurosci. 2008, 28, 2147–2157. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, P.A.; Crosby, A.H.; Turner, C.; Bradley, L.J.; Ginsberg, L.; Wood, N.W.; Schapira, A.H.; Warner, T.T. A clinical, genetic, and biochemical study of SPG7 mutations in hereditary spastic paraplegia. Brain 2004, 127, 973–980. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Levin, M.C. Novel somatic single nucleotide variants within the RNA binding protein hnRNP A1 in multiple sclerosis patient. F1000Research 2014, 3, 132. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.L.; Shin, Y.; Levin, M.C.; University of Tennessee Health Science Center, Memphis, TN. Unpublished work. 2017.
- Kremenchutzky, M.; Rice, G.P.A.; Baskerville, J.; Wingerchuk, D.M.; Ebers, G.C. The natural history of multiple sclerosis: A geographically based study 9: Observations on the progressive phase of the disease. Brain 2006, 129, 584–594. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.H.; Leary, S.M. Primary-progressive multiple sclerosis. Lancet Neurol. 2007, 6, 903–912. [Google Scholar] [CrossRef]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salapa, H.E.; Lee, S.; Shin, Y.; Levin, M.C. Contribution of the Degeneration of the Neuro-Axonal Unit to the Pathogenesis of Multiple Sclerosis. Brain Sci. 2017, 7, 69. https://doi.org/10.3390/brainsci7060069
Salapa HE, Lee S, Shin Y, Levin MC. Contribution of the Degeneration of the Neuro-Axonal Unit to the Pathogenesis of Multiple Sclerosis. Brain Sciences. 2017; 7(6):69. https://doi.org/10.3390/brainsci7060069
Chicago/Turabian StyleSalapa, Hannah E., Sangmin Lee, Yoojin Shin, and Michael C. Levin. 2017. "Contribution of the Degeneration of the Neuro-Axonal Unit to the Pathogenesis of Multiple Sclerosis" Brain Sciences 7, no. 6: 69. https://doi.org/10.3390/brainsci7060069
APA StyleSalapa, H. E., Lee, S., Shin, Y., & Levin, M. C. (2017). Contribution of the Degeneration of the Neuro-Axonal Unit to the Pathogenesis of Multiple Sclerosis. Brain Sciences, 7(6), 69. https://doi.org/10.3390/brainsci7060069