N-Acetyl Cysteine Supplement Minimize Tau Expression and Neuronal Loss in Animal Model of Alzheimer’s Disease


Abstract
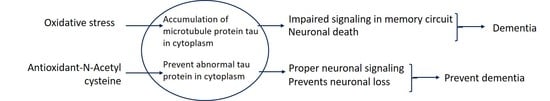
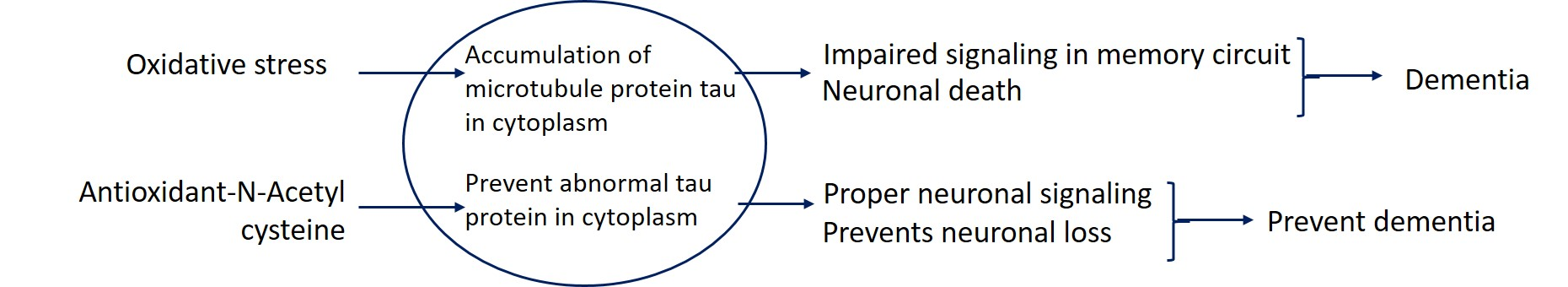
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Methods
2.1. Animals
2.2. Animal Groups
2.3. Chemicals
2.4. Surgery and Intracerebroventricular Administration of Colchicine
2.5. NAC Administration
2.6. Cognitive Assessment Using Passive Avoidance Task
2.7. Tissue Processing for Histopathological and Immuno Staining
2.8. Quantification of Neurons in Prefrontal Cortex and Hippocampus
2.9. Immunostaining and Estimation of Expression of Tau and Neurofibrillary Tangles Formations
2.10. Statistical Analysis
3. Results
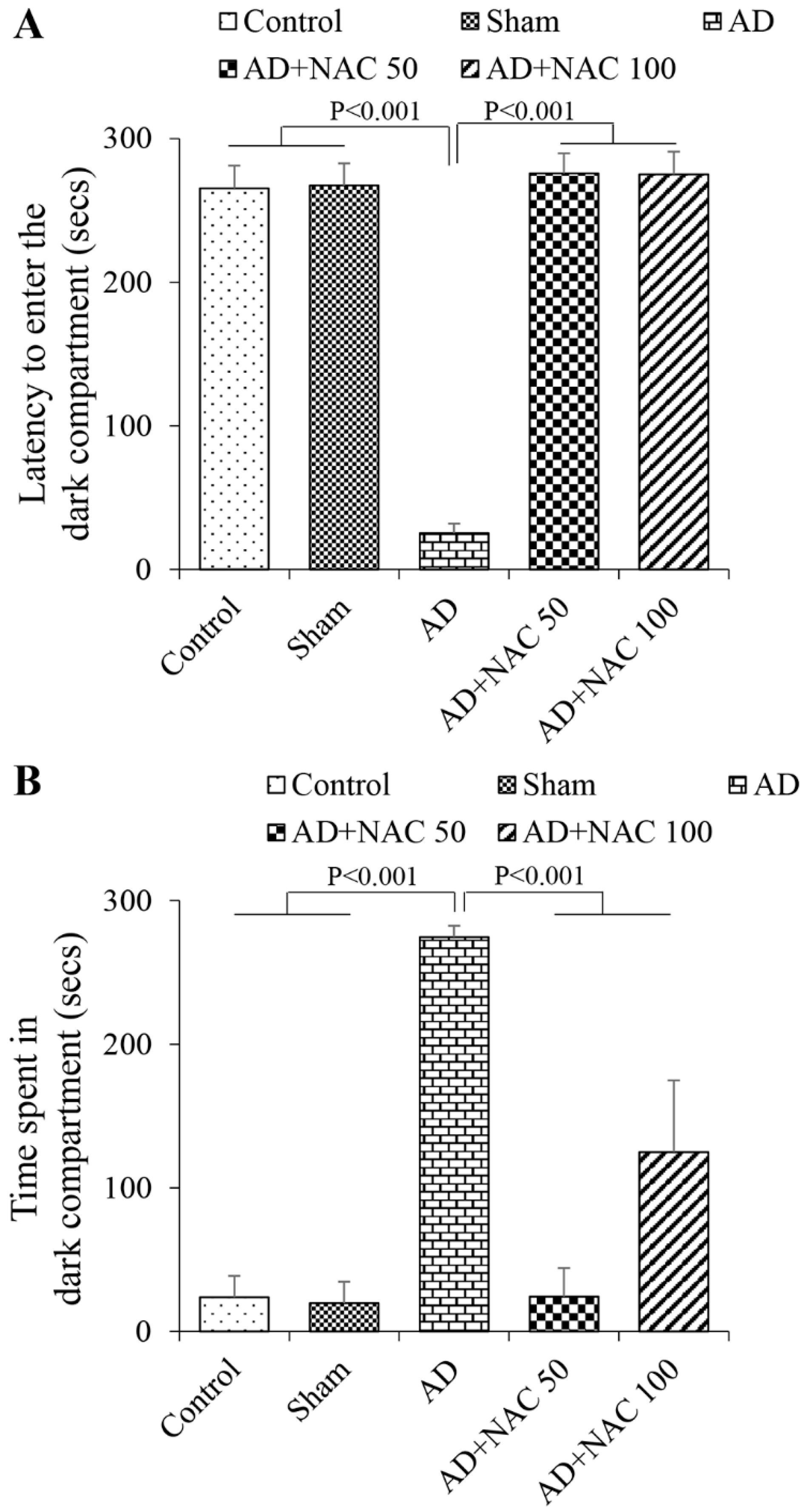
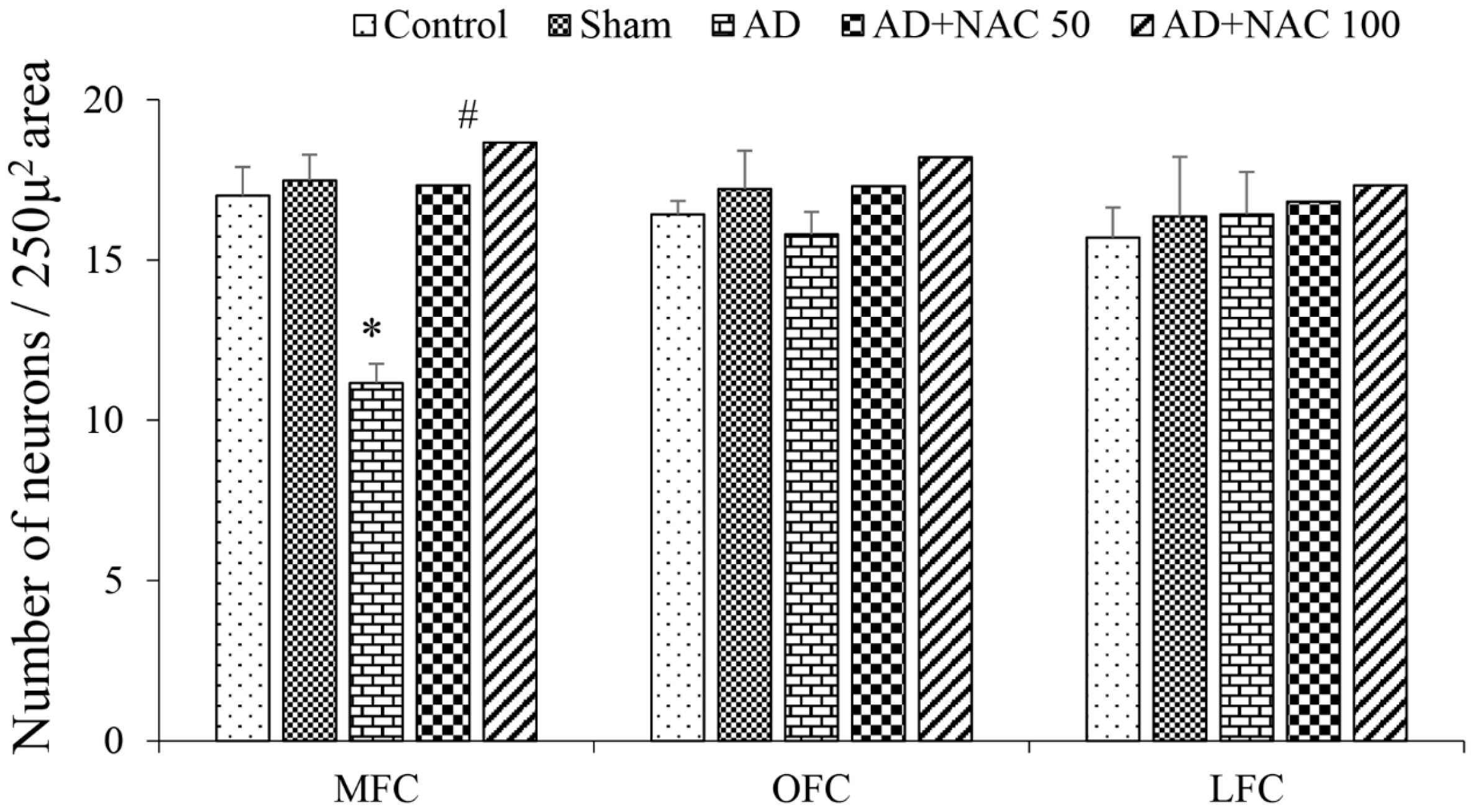
3.1. Passive Avoidance Test Performance
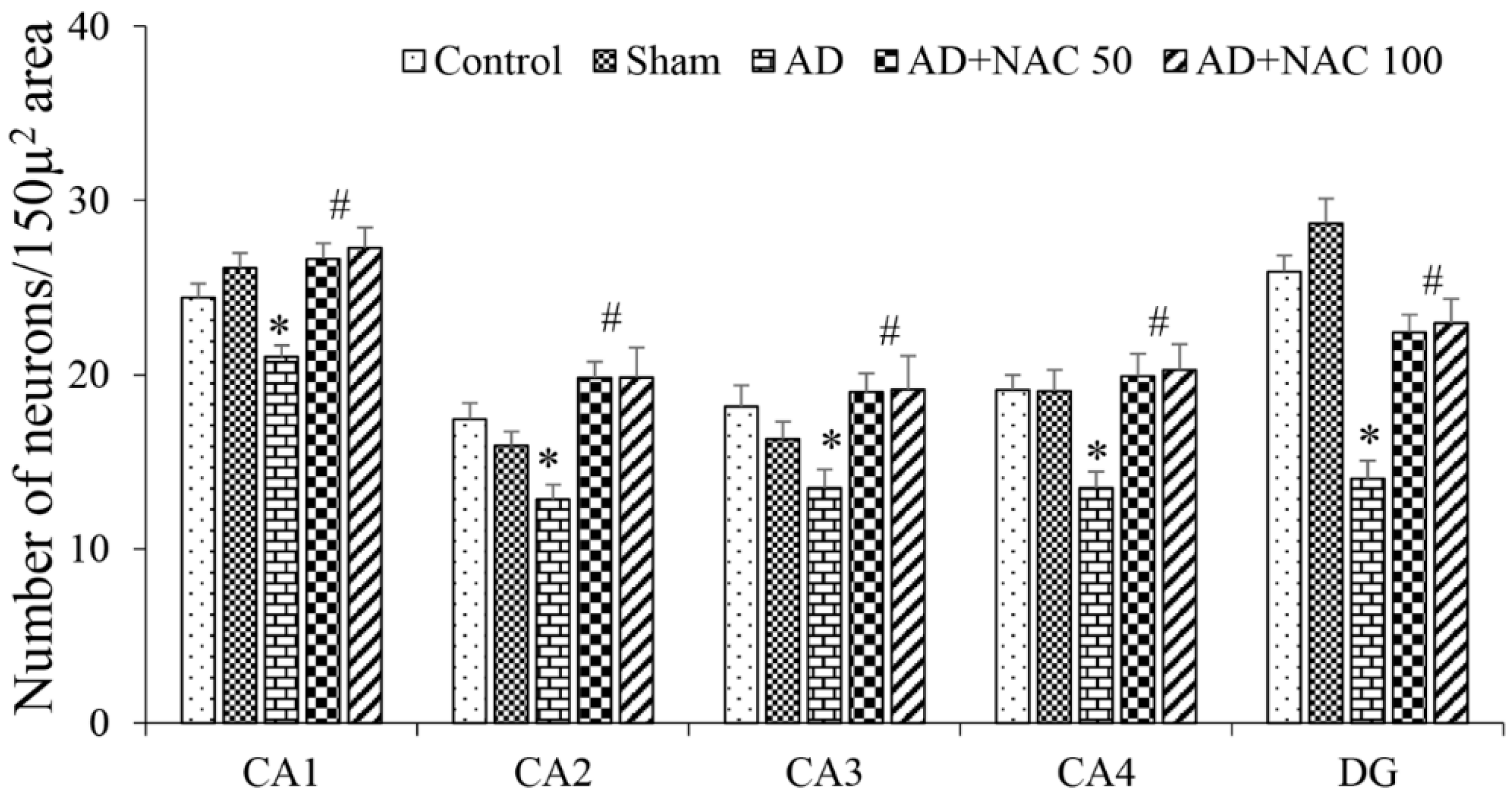
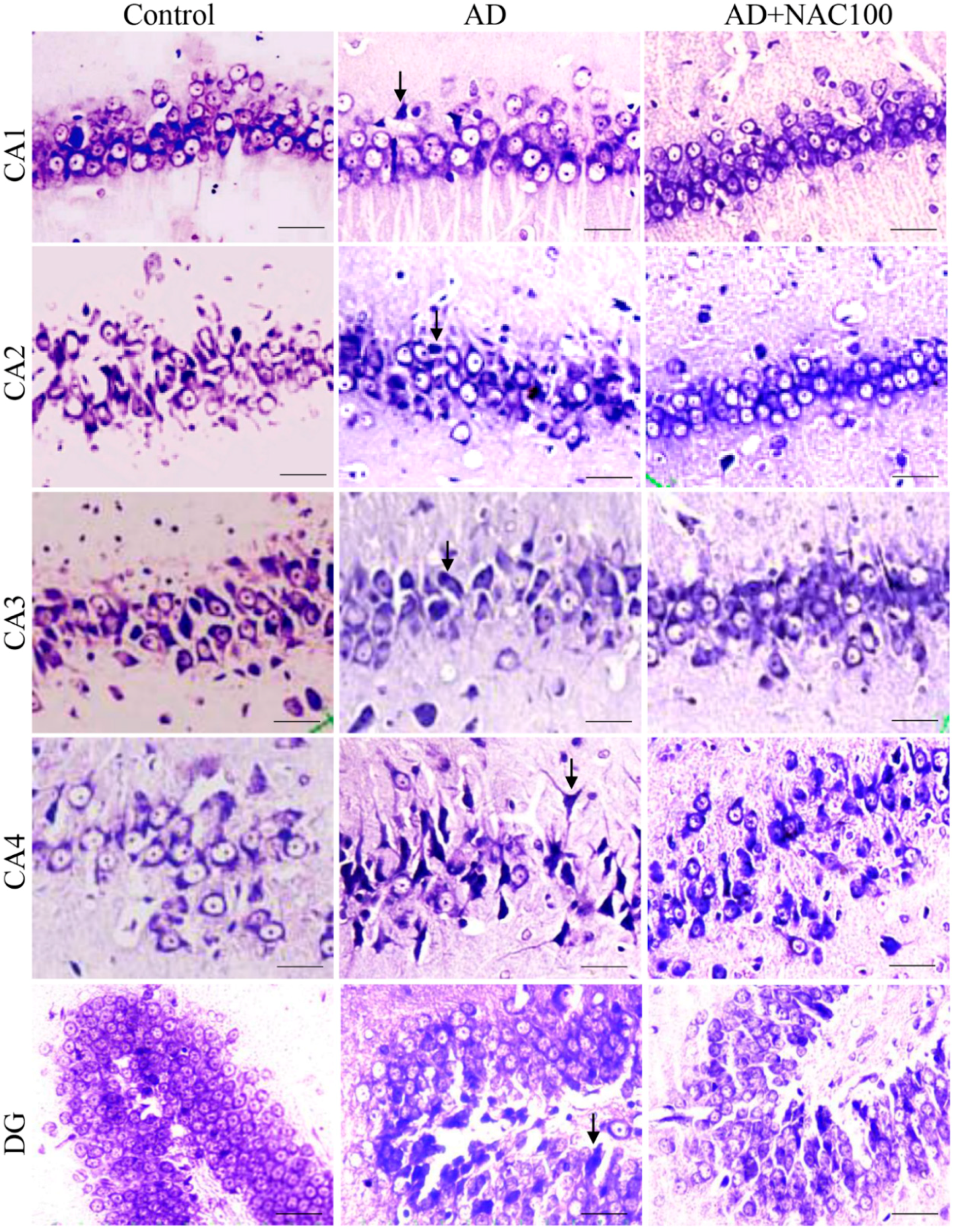
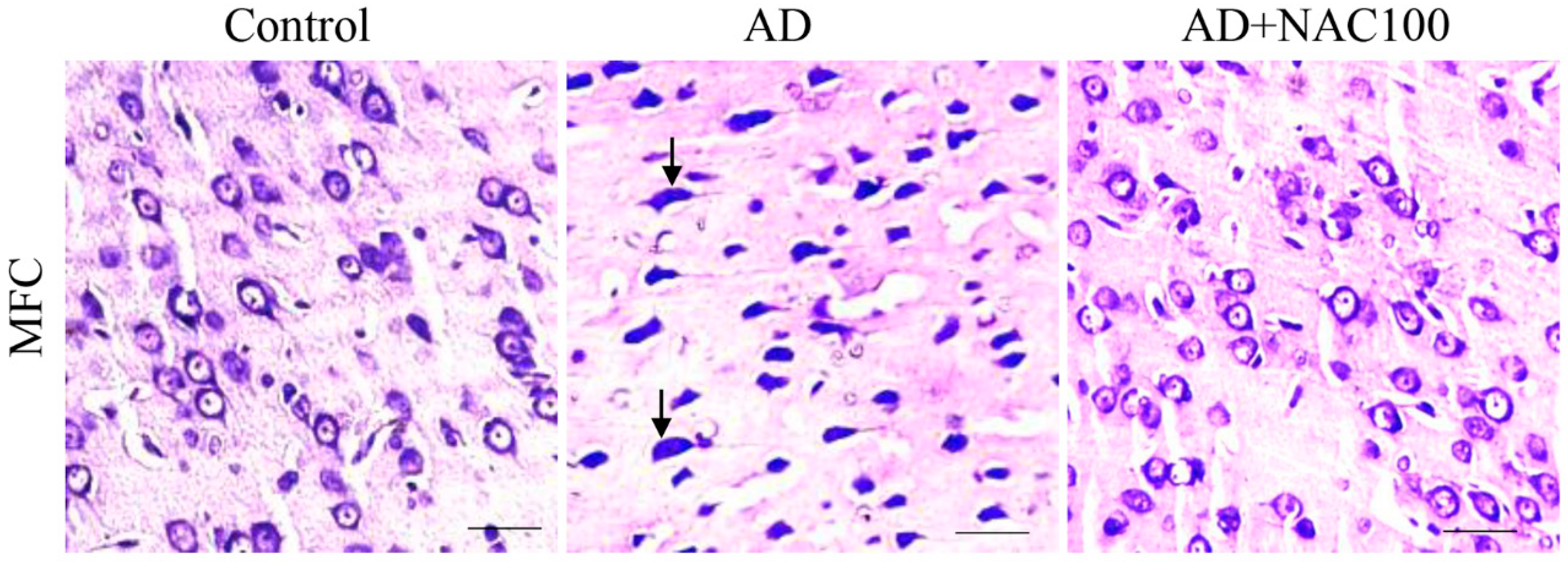
3.2. Neuronal Assay by Cresyl Violet Staining
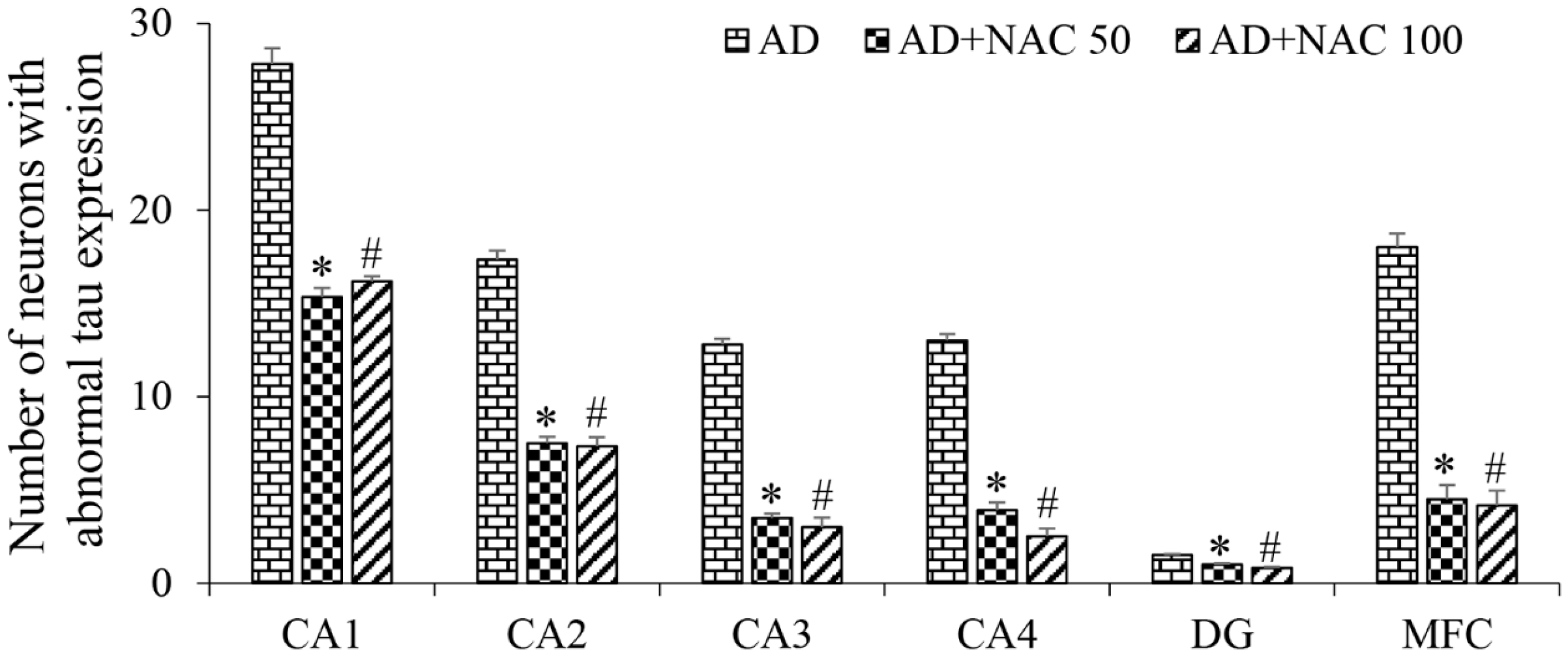
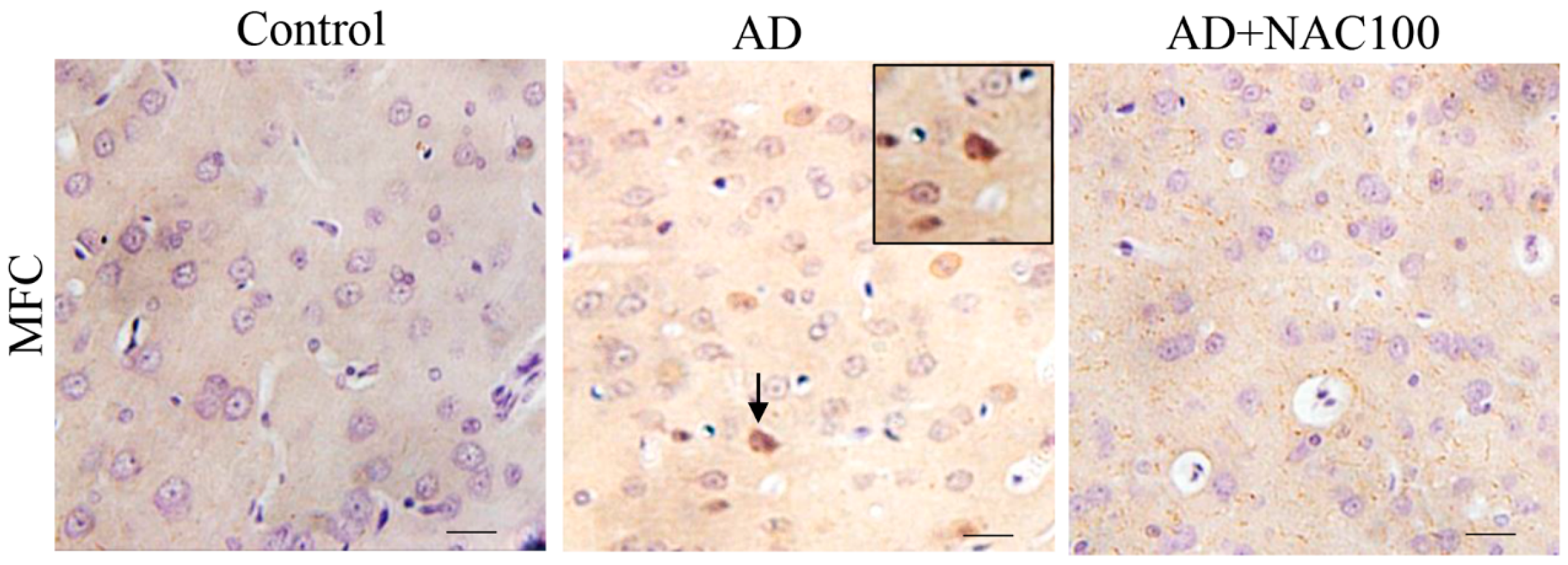
3.3. Expression of Tau and Neurofibrillary Tangles
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Huang, W.J.; Zhang, X.; Chen, W.W. Role of oxidative stress in Alzheimer’s disease. Biomed. Rep. 2016, 4, 519–522. [Google Scholar] [CrossRef] [PubMed]
- Torres, L.L.; Quaglio, N.B.; de Souza, G.T.; Garcia, R.T.; Dati, L.M.M.; Moreira, W.L.; de Melo Loureiro, A.P.; de souza-Talarico, J.N.; Smid, J.; Porto, C.S.; et al. Peripheral oxidative stress biomarkers in mild cognitive impairment and Alzheimer’s disease. J. Alzheimer’s Dis. 2011, 26, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Shigematsu, K.; McGeer, P.L. Accumulation of amyloid precursor protein in damaged neuronal processes and microglia following intracerebral administration of aluminum salts. Brain Res. 1992, 593, 117–123. [Google Scholar] [CrossRef]
- Kumar, M.H.V.; Gupta, Y.K. Intracerebroventricular administration of colchicine produces cognitive impairment associated with oxidative stress in rats. Pharmacol. Biochem. Behav. 2002, 73, 565–571. [Google Scholar] [CrossRef]
- Zilka, N.; Novak, A. The tangled story of Alois Alzheimer. Bratisal. Lek. List. 2006, 107, 343–345. [Google Scholar]
- Meyers, C.A.; Kudelka, A.P.; Conrad, C.A.; Gelke, C.K.; Grove, W.; Pazdur, R. Neurotoxicity of CI-980, a novel mitotic inhibitor. Clin. Cancer Res. 1992, 3, 419–422. [Google Scholar]
- Ganguly, R.; Guha, D. Alteration of brain monoamines & EEG wave pattern in rat model of Alzheimer’s disease & protection by Moringa oleifera. Indian J. Med. Res. 2008, 128, 744–751. [Google Scholar] [PubMed]
- Nakayama, T.; Sawada, T. Involvement of microtubule integrity in memory impairment caused by colchicine. Pharmacol. Biochem. Behav. 2002, 71, 119–138. [Google Scholar] [CrossRef]
- Dahl, D.A.; Bignami, A.; Bich, N.T.; Chi, N.H. Immunohistochemical characterization of neurofibrillary tangles induced by mitotic spindle inhibitors. Acta Neuropathol. 1980, 51, 165–168. [Google Scholar] [CrossRef] [PubMed]
- Merrick, S.E.; Demoise, D.C.; Lee, V.M. Site-specific dephosphorylation of tau protein at Ser202/Thr205 in response to microtubule depolymerization in cultured human neurons involves protein phosphatase 2A. J. Biol. Chem. 1996, 271, 5589–5594. [Google Scholar] [CrossRef] [PubMed]
- Alavi Naini, S.M.; Soussi-Yanicostas, N. Tau Hyperphosphorylation and Oxidative Stress, a Critical Vicious Circle in Neurodegenerative Tauopathies? Oxid. Med. Cell. Longev. 2015, 151979. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.W.; Kim, S.J.; Kim, M.S. Oxidative stress with tau hyperphosphorylation in memory impaired 1,2-diacetylbenzene-treated mice. Toxicol. Lett. 2017, 279, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Kahn, I.; Andrews-Hanna, J.R.; Vincent, J.L.; Snyder, A.Z.; Buckner, R.L. Distinct cortical anatomy linked to subregions of the medial temporal lobe revealed by intrinsic functional connectivity. J. Neurophysiol. 2008, 100, 129–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laroche, S.; Davis, S.; Jay, T.M. Plasticity at hippocampal to prefrontal cortex synapses: Dual roles in working memory and consolidation. Hippocampus 2000, 10, 438–446. [Google Scholar] [CrossRef]
- Thompson, P.M.; Hayashi, K.M.; Dutton, R.A.; CHIANG, M.C.; Leow, A.D.; Sowell, E.R.; de Zubicaray, G.; Becker, J.T.; Lopez, O.L.; Aizenstein, H.J.; et al. Tracking Alzheimer’s disease. Ann. N. Y. Acad. Sci. 2007, 1097, 183–214. [Google Scholar] [CrossRef] [PubMed]
- Cente, M.; Filipcik, P.; Mandakova, S.; Zilka, N.; Krajciova, G.; Novak, M. Expression of a truncated human tau protein induces aqueous-phase free radicals in a rat model of tauopathy: Implications for targeted antioxidative therapy. J. Alzheimer’s Dis. 2009, 17, 913–920. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Kim, H.S.; Cho, E.K.; Kwon, B.Y.; Phark, S.; Hwang, K.W.; Sul, D. Curcumin protected PC12 cells against beta-amyloid-induced toxicity through the inhibition of oxidative damage and tau hyperphosphorylation. Food Chem. Toxicol. 2008, 46, 2881–2887. [Google Scholar] [CrossRef] [PubMed]
- Sen, O.; Caner, H.; Aydin, M.V.; Ozen, O.; Atalay, B.; Altinors, N.; Bavbek, M. The effect of mexiletine on the level of lipid peroxidation and apoptosis of endothelium following experimental subarachnoid hemorrhage. Neurol. Res. 2006, 28, 859–863. [Google Scholar] [CrossRef] [PubMed]
- Kerksick, C.; Willoughby, D. The Antioxidant role of glutathione and N-acetyl-cysteine supplements and exercise-induced oxidative stress. J. Int. Soc. Sports Nutr. 2005, 2, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Banaclocha, M.M. Therapeutic potential of N-acetyl-cysteine in age-related mitochondrial neurodegenerative diseases. Med. Hypothesis 2001, 56, 472–477. [Google Scholar] [CrossRef] [PubMed]
- Madhyastha, S.; Somayaji, S.N.; Rao, M.S.; Nalini, K.; Bairy, K.L. Hippocampal brain amines in methotrexate-induced learning and memory deficit. Can. J. Physiol. Pharmacol. 2002, 80, 1076–1084. [Google Scholar] [CrossRef] [PubMed]
- Farr, S.A.; Poon, H.F.; Dogrukaol-Ak, D.; Drake, J.; Banks, W.A.; Eyerman, E.; Butterfield, D.A.; Morley, J.E. The antioxidants α-lipoic acid and N-acetylcysteine reverse memory impairment and brain oxidative stree in aged SAMP8 mice. J. Neurochem. 2003, 84, 1173–1183. [Google Scholar] [CrossRef] [PubMed]
- Bures, J.; Burešová, O.; Huston, J.P. Techniques and Basic Experiments for the Study of Brain and Behavior; Elsevier Science Publishers: Amsterdam, NY, USA, 1983; p. 155. [Google Scholar]
- Pelligrino, L.J.; Pelligrino, A.S.; Cushman, A.J. Stereotaxic Atlas of the Rat Brain, 2nd ed.; Plenum Press: New York, NY, USA, 1981; pp. 41–43. [Google Scholar]
- Lu, F.; Li, X.; Suo, A.Q.; Zhang, J.W. Inhibition of tau hyperphosphorylation and beta amyloid production in rat brain by oral administration of atorvastatin. Chin. Med. J. 2010, 123, 1864–1870. [Google Scholar] [PubMed]
- Kumar, A.; Dogra, S.; Prakash, A. Neuroprotective Effects of Centella asiatica against Intracerebroventricular Colchicine-Induced Cognitive Impairment and Oxidative Stress. Int. J. Alzheimer’s Dis. 2009, 972178. [Google Scholar] [CrossRef]
- Cantuti-Castelvetri, I.; Shukitt-Hale, B.; Joseph, J.A. Neurobehavioral aspects of antioxidants in aging. Int. J. Dev. Neurosci. 2000, 18, 367–381. [Google Scholar] [CrossRef]
- Kumar, A.; Naidu, P.S.; Seghal, N.; Padi, S.S.V. Effect of curcumin on intracerebroventricular colchicine-induced cognitive impairment and oxidative stress in rats. J. Med. Food 2007, 10, 486–494. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, A.R.; Soliman, G.Y.; Ismail, C.A.; Mannaa, H.F. Neuroprotective role of vitamin D3 in colchicine-induced Alzheimer’s disease in rats. Alexandria J. Med. 2015, 51, 127–136. [Google Scholar] [CrossRef]
- Costa, A.P.; Tramontina, A.C.; Biasibetti, R.; Batassini, C.; Lopes, M.W.; Wartchow, K.M.; Bernardi, C.; Tortorelli, L.S.; Leal, R.B.; Gonçalves, C.-A. Neuroglial alterations in rats submitted to the okadaic acid-induced model of dementia. Behav. Brain Res. 2012, 226, 420–427. [Google Scholar] [CrossRef] [PubMed]
- Borza, L.R. A review on the cause-effect relationship between oxidative stress and toxic proteins in the pathogenesis of neurodegenerative diseases. Med. Surg. J. 2014, 118, 19–27. [Google Scholar]
- Iqbal, K.; Liu, F.; Gong, C.X.; Grundke-Iqbal, I. Tau in Alzheimer disease and related tauopathies. Curr. Alzheimer Res. 2010, 7, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Lace, G.; Savva, G.M.; Forster, G.; De Silva, R.; Brayne, C.; Matthews, F.E.; Barclay, J.J.; Dakin, L.; Ince, P.G.; Wharton, S.B. Hippocampal tau pathology is related to neuroanatomical connections: An ageing population-based study. Brain 2009, 132, 1324–1334. [Google Scholar] [CrossRef] [PubMed]
- Jucker, M.; Walker, L.C. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 2013, 501, 45–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llorens-Martin, M.; Blazquez-Llorca, L.; Benavides-Piccione, R.; Rabano, A.; Hernandez, F.; Avila, J.; DeFelipe, J. Selective alterations of neurons and circuits related to early memory loss in Alzheimer’s disease. Front. Neuroanat. 2014, 8, 38. [Google Scholar] [CrossRef] [PubMed]
- Blazquez-Llorca, L.; Garcia-Marin, V.; Merino-Serrais, P.; Ávila, J.; DeFelipe, J. Abnormal tau phosphorylation in the thorny excrescences of CA3 hippocampal neurons in patients with Alzheimer’s disease. J. Alzheimer’s Dis. 2011, 26, 683–698. [Google Scholar] [CrossRef] [PubMed]
- Mufson, E.J.; Ginsberg, S.D.; Ikonomovic, M.D.; DeKosky, S.T. Human cholinergic basal forebrain: Chemoanatomy and neurologic dysfunction. J. Chem. Neuroanat. 2003, 26, 233–242. [Google Scholar] [CrossRef]
- Wevers, A.; Witter, B.; Moser, N.; Burghaus, L.; Banerjee, C.; Steinlein, O.K.; Schütz, U.; de Vos, R.A.I.; Steur, E.N.H.J.; Lindstrom, J.; et al. Classical Alzheimer features and cholinergic dysfunction: Towards a unifying hypothesis? Acta Neurol. Scand. 2000, 176, 42–48. [Google Scholar] [CrossRef]
- Kim, H.J.; Moon, W.J.; Han, S.H. Differential cholinergic pathway involvement in Alzheimer’s disease and subcortical ischemic vascular dementia. J. Alzheimer’s Dis. 2013, 35, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.L.; Kesner, R.P. Cholinergic modulation of the hippocampus during encoding and retrieval of tone/shock-induced fear conditioning. Learn. Mem. 2004, 11, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Funahashi, S. Thalamic mediodorsal nucleus and its participation in spatial working memory processes: Comparison with the prefrontal cortex. Front. Syst. Neurosci. 2013, 7, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomita, H.; Ohbayashi, M.; Nakahara, K.; Hasegawa, I.; Miyashita, Y. Top-down signal from prefrontal cortex in executive control of memory retrieval. Nature 1999, 401, 699–703. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.A.; Hayrapetyan, V.Y.; Maimon, B.; Mak, K.; Je, H.S.; Yin, H.H. Prefrontal cortical mechanisms underlying delayed alternation in mice. J. Neurophysiol. 2012, 108, 1211–1222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Euston, D.R.; Gruber, A.J.; Mcnaughton, B.L. The role of medial prefrontal cortex in memory and decision making. Neuron 2012, 76, 1057–1070. [Google Scholar] [CrossRef] [PubMed]
- Farovik, A.; Dupont, L.M.; Arce, M.; Eichenbaum, H. Medial prefrontal cortex supports recollection, but not familiarity, in the rat. J. Neurosci. 2008, 28, 13428–13434. [Google Scholar] [CrossRef] [PubMed]
- Kahn, J.B.; Ward, R.D.; Kahn, L.W.; Rudy, N.M.; Kandel, E.R.; Balsam, P.D.; Simpson, E.H. Medial prefrontal lesions in mice impair sustained attention but spare maintenance of information in working memory. Learn. Mem. 2012, 19, 513–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, T.A.; Lee, H.P.; Rolston, R.K.; Zhu, X.; Marlatt, M.W.; Castellani, R.J.; Nunomura, A.; Casadesus, G.; Smith, M.A.; Lee, H.; et al. Oxidative stress and its implications for future treatments and management of Alzheimer disease. Int. J. Biomed. Sci. 2010, 6, 225–227. [Google Scholar] [PubMed]
- Underwood, B.R.; Imarisio, S.; Fleming, A.; Rose, C.; Krishna, G.; Heard, P.; Quick, M.; Korolchuk, V.I.; Renna, M.; Sarkar, S.; et al. Antioxidants can inhibit basal autophagy and enhance neurodegeneration in models of polyglutamine disease. Hum. Mol. Genet. 2010, 19, 3413–3429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aoyama, K.; Suh, S.W.; Hamby, A.M.; Liu, J.; Chan, W.Y.; Chen, Y.; Swanson, R.A. Neuronal glutathione deficiency and age-dependent neurodegeneration in the EAAC1 deficient mouse. Nat. Neurosci. 2006, 9, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Tchantchou, F.; Graves, M.; Rogers, E.; Ortiz, D.; Shea, T.B. N-acteylcysteine alleviates oxidative damage to central nervous system of ApoE-deficient mice following folate and vitamin E-deficiency. J. Alzheimer’s Dis. 2005, 7, 135–138. [Google Scholar] [CrossRef]
- Tucker, S.; Ahl, M.; Bush, A.; Westaway, D.; Huang, X.; Rogers, J.T. Pilot study of the reducing effect on amyloidosis in vivo by three FDA pre-approved drugs via the Alzheimer’s APP 5′ untranslated region. Curr. Alzheimer Res. 2005, 2, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; West, M.B.; Cheng, W.; Ewert, D.L.; Li, W.; Saunders, D.; Towner, R.A.; Floyd, R.A.; Kopke, R.D. Ameliorative Effects of Antioxidants on the Hippocampal Accumulation of Pathologic Tau in a Rat Model of Blast-Induced Traumatic Brain Injury. Oxid. Med. Cell. Longev. 2016, 2016, 4159357. [Google Scholar] [CrossRef] [PubMed]
- Farr, S.A.; Ripley, J.L.; Sultana, R.; Zhang, Z.; Niehoff, M.L.; Platt, T.L.; Murphy, M.P.; Morley, J.E.; Kumar, V.; Butterfield, D.A. Antisense oligonucleotide against GSK-3B in brain of SAMP8 mice improves learning and memory and decreases oxidative stress: Involvement of transcription factor Nrf2 and implication for Alzheimer disease. Free Radic. Biol. Med. 2016, 7, 387–395. [Google Scholar]
- Cohen, T.J.; Friedmann, D.; Hwang, A.W.; Marmorstein, R.; Lee, V.M. The microtubuleassociated tau protein has intrinsic acetyltransferase activity. Nat. Struct. Mol. Biol. 2013, 20, 756–762. [Google Scholar] [CrossRef] [PubMed]
- Wegmann, S.; Medalsy, I.D.; Mandelkow, E.; Müller, D.J. The fuzzy coat of pathological human Tau fibrils is a two-layered polyelectric brush. Proc. Natl. Acad. Sci. USA 2012, 110, E313–E321. [Google Scholar] [CrossRef] [PubMed]
- Aldini, G.; Altomare, A.; Baron, G.; Vistoli, G.; Carini, M.; Borsani, L.; Sergio, F. N-Acetylcysteine as an antioxidant and disulphide breaking agent: The reasons why. Free Radic. Res. 2018, 52, 751–762. [Google Scholar] [CrossRef] [PubMed]








© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Joy, T.; Rao, M.S.; Madhyastha, S. N-Acetyl Cysteine Supplement Minimize Tau Expression and Neuronal Loss in Animal Model of Alzheimer’s Disease. Brain Sci. 2018, 8, 185. https://doi.org/10.3390/brainsci8100185
Joy T, Rao MS, Madhyastha S. N-Acetyl Cysteine Supplement Minimize Tau Expression and Neuronal Loss in Animal Model of Alzheimer’s Disease. Brain Sciences. 2018; 8(10):185. https://doi.org/10.3390/brainsci8100185
Chicago/Turabian StyleJoy, Teresa, Muddanna S. Rao, and Sampath Madhyastha. 2018. "N-Acetyl Cysteine Supplement Minimize Tau Expression and Neuronal Loss in Animal Model of Alzheimer’s Disease" Brain Sciences 8, no. 10: 185. https://doi.org/10.3390/brainsci8100185
APA StyleJoy, T., Rao, M. S., & Madhyastha, S. (2018). N-Acetyl Cysteine Supplement Minimize Tau Expression and Neuronal Loss in Animal Model of Alzheimer’s Disease. Brain Sciences, 8(10), 185. https://doi.org/10.3390/brainsci8100185