Possible Role of Inflammation and Galectin-3 in Brain Injury after Subarachnoid Hemorrhage



{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Inflammation Plays a Pivotal Role in EBI after SAH
3. Possible Molecular Mechanisms of Inflammation in EBI
3.1. Trigger Factors and Location of Inflammation in EBI
3.2. Involved Receptors in EBI
3.3. Major Inflammatory Signaling Pathways in EBI
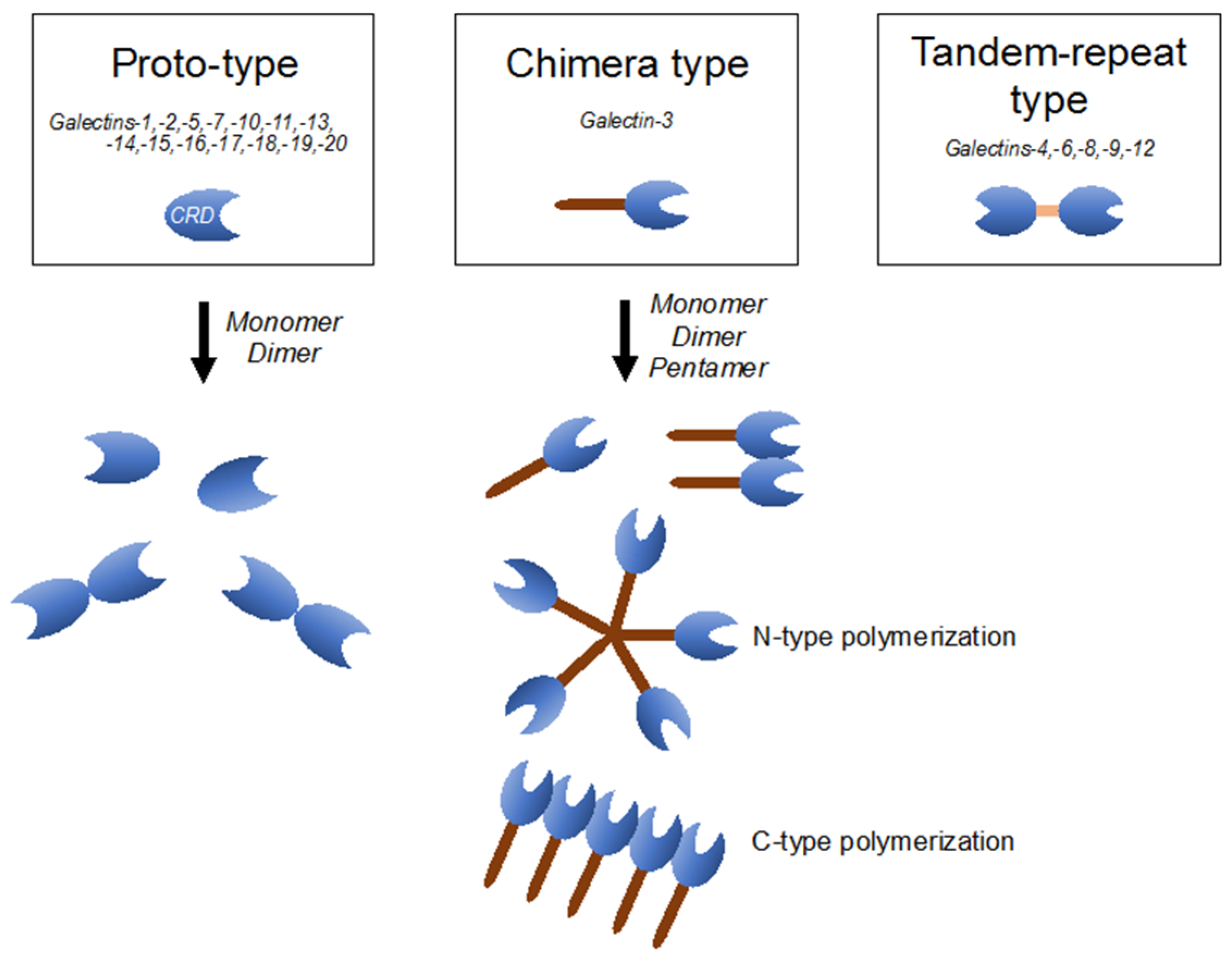
4. Galectin-3 Belongs to the Galectin Family, Exhibiting Unique Characteristics
5. Molecular Mechanisms and Clinical Implication of Galectin-3 in Systemic Diseases
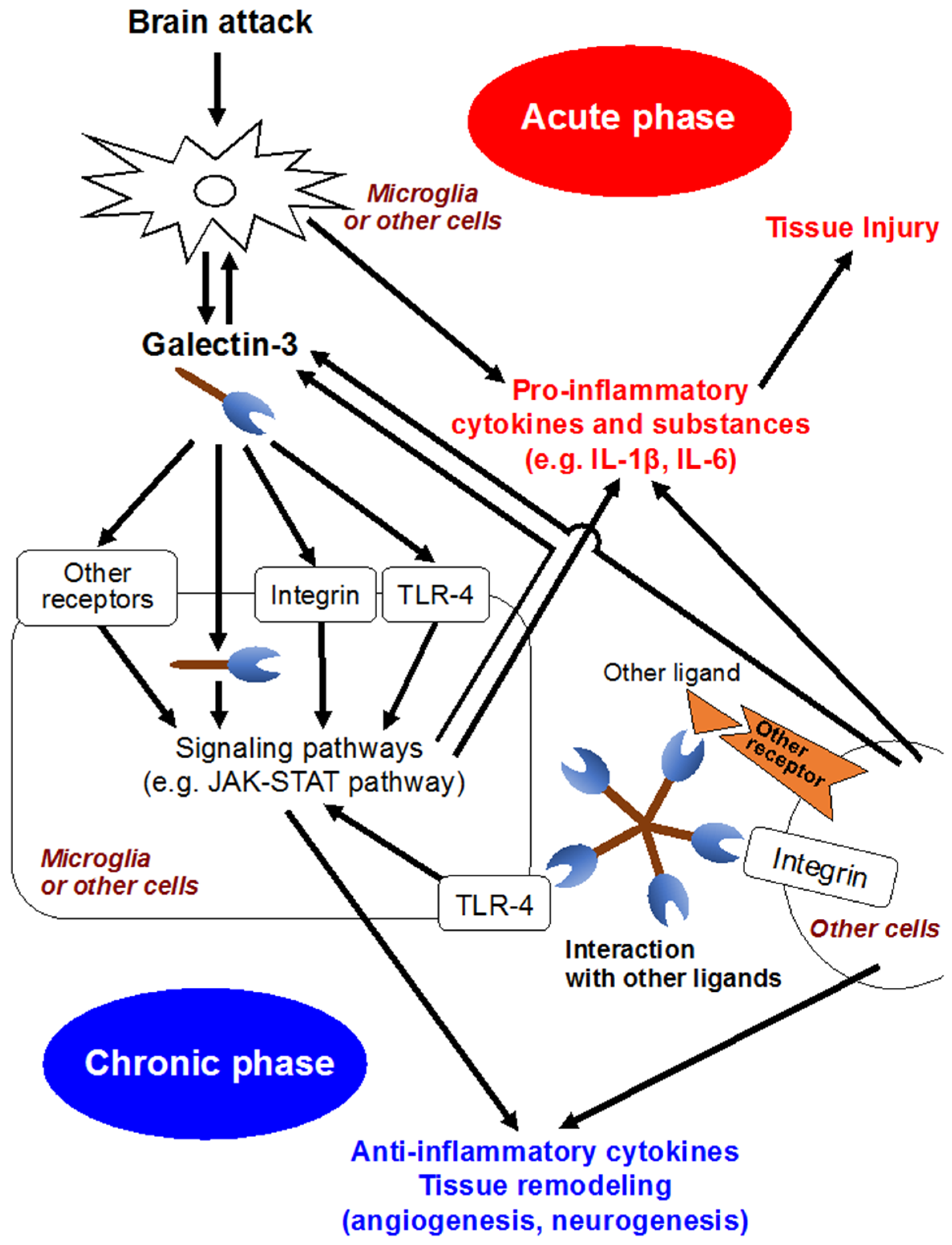
6. Galectin-3 Activates Microglia and Inflammatory Reactions in CNS
7. Clinical Implication of Galectin-3 in CNS
8. Galectin-3 Is a Novel Target for the Research of EBI
Author Contributions
Conflicts of Interest
References
- Zheng, V.Z.; Wong, G.K.C. Neuroinflammation responses after subarachnoid hemorrhage: A review. J. Clin. Neurosci. 2017, 42, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Venkatraman, A.; Hardas, S.; Patel, N.; Bajaj, N.S.; Arora, G.; Arora, P. Galectin-3: An emerging biomarker in stroke and cerebrovascular diseases. Eur. J. Neurol. 2018, 25, 238–246. [Google Scholar] [CrossRef] [PubMed]
- Nakatsuka, Y.; Kawakita, F.; Yasuda, R.; Umeda, Y.; Toma, N.; Sakaida, H.; Suzuki, H.; on behalf of the Prospective Registry for Searching Mediators of Neurovascular Events After Aneurysmal Subarachnoid Hemorrhage (pSEED) Group. Preventive effects of cilostazol against the development of shunt-dependent hydrocephalus after subarachnoid hemorrhage. J. Neurosurg. 2017, 127, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Nakatsuka, Y.; Shiba, M.; Nishikawa, H.; Terashima, M.; Kawakita, F.; Fujimoto, M.; Suzuki, H.; pSEED Group. Acute-phase plasma osteopontin as an independent predictor for poor outcome after aneurysmal subarachnoid hemorrhage. Mol. Neurobiol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Yancy, C.W.; Jessup, M.; Bozkurt, B.; Butler, J.; Casey, D.E., Jr.; Drazner, M.H.; Fonarow, G.C.; Geraci, S.A.; Horwich, T.; Januzzi, J.L. 2013 ACCF/AHA guideline for the management of heart failure: Executive summary: A report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation 2013, 128, 1810–1852. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H. What is early brain injury? Transl. Stroke Res. 2015, 6, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Shiba, M.; Nakatsuka, Y.; Nakano, F.; Nishikawa, H. Higher cerebrospinal fluid pH may contribute to the development of delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage. Transl. Stroke Res. 2017, 8, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Ishiguro, M.; Morielli, A.D.; Zvarova, K.; Tranmer, B.I.; Penar, P.L.; Wellman, G.C. Oxyhemoglobin-induced suppression of voltage-dependent k+ channels in cerebral arteries by enhanced tyrosine kinase activity. Circ. Res. 2006, 99, 1252–1260. [Google Scholar] [CrossRef] [PubMed]
- Woitzik, J.; Dreier, J.P.; Hecht, N.; Fiss, I.; Sandow, N.; Major, S.; Winkler, M.; Dahlem, Y.A.; Manville, J.; Diepers, M.; et al. Delayed cerebral ischemia and spreading depolarization in absence of angiographic vasospasm after subarachnoid hemorrhage. J. Cereb. Blood Flow Metab. 2012, 32, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Okada, T.; Suzuki, H. Toll-like receptor 4 as a possible therapeutic target for delayed brain injuries after aneurysmal subarachnoid hemorrhage. Neural Regen. Res. 2017, 12, 193–196. [Google Scholar] [PubMed]
- Kawakita, F.; Fujimoto, M.; Liu, L.; Nakano, F.; Nakatsuka, Y.; Suzuki, H. Effects of Toll-like receptor 4 antagonists against cerebral vasospasm after experimental subarachnoid hemorrhage in mice. Mol. Neurobiol. 2017, 54, 6624–6633. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Kawakita, F. Tenascin-C in aneurysmal subarachnoid hemorrhage: Deleterious or protective? Neural Regen. Res. 2016, 11, 230–231. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Luo, C.; Yu, S.; Gao, J.; Liu, C.; Wei, Z.; Zhang, Z.; Wei, L.; Yi, B. Erythropoietin ameliorates early brain injury after subarachnoid haemorrhage by modulating microglia polarization via the EPOR/JAK2-STAT3 pathway. Exp. Cell Res. 2017, 361, 342–352. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, M.; Kusaka, G.; Yamaguchi, N.; Sekizuka, E.; Nakadate, H.; Minamitani, H.; Shinoda, S.; Watanabe, E. Platelet and leukocyte adhesion in the microvasculature at the cerebral surface immediately after subarachnoid hemorrhage. Neurosurgery 2009, 64, 546–554. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.Y.; Sun, B.L.; Yang, M.F.; Li, D.W.; Fang, J.; Zhang, S. Carnosine attenuates early brain injury through its antioxidative and anti-apoptotic effects in a rat experimental subarachnoid hemorrhage model. Cell Mol. Neurobiol. 2015, 35, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Kawakita, F.; Fujimoto, M.; Nakano, F.; Imanaka-Yoshida, K.; Yoshida, T.; Suzuki, H. Role of periostin in early brain injury after subarachnoid hemorrhage in mice. Stroke 2017, 48, 1108–1111. [Google Scholar] [CrossRef] [PubMed]
- Shiba, M.; Fujimoto, M.; Imanaka-Yoshida, K.; Yoshida, T.; Taki, W.; Suzuki, H. Tenascin-C causes neuronal apoptosis after subarachnoid hemorrhage in rats. Transl. Stroke Res. 2014, 5, 238–247. [Google Scholar] [CrossRef] [PubMed]
- Hanafy, K.A. The role of microglia and the TLR4 pathway in neuronal apoptosis and vasospasm after subarachnoid hemorrhage. J. Neuroinflamm. 2013, 10, 868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schallner, N.; Pandit, R.; LeBlanc, R.; Thomas, A.J.; Ogilvy, C.S.; Zuckerbraun, B.S.; Gallo, D.; Otterbein, L.E.; Hanafy, K.A. Microglia regulate blood clearance in subarachnoid hemorrhage by heme oxygenase-1. J. Clin. Investig. 2015, 125, 2609–2625. [Google Scholar] [CrossRef] [PubMed]
- Schneider, U.C.; Davids, A.M.; Brandenburg, S.; Muller, A.; Elke, A.; Magrini, S.; Atangana, E.; Turkowski, K.; Finger, T.; Gutenberg, A.; et al. Microglia inflict delayed brain injury after subarachnoid hemorrhage. Acta Neuropathol. 2015, 130, 215–231. [Google Scholar] [CrossRef] [PubMed]
- Aloisi, F. Immune function of microglia. Glia 2001, 36, 165–179. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Li, P.; Guo, Y.; Wang, H.; Leak, R.K.; Chen, S.; Gao, Y.; Chen, J. Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke 2012, 43, 3063–3070. [Google Scholar] [CrossRef] [PubMed]
- Graeber, M.B. Changing face of microglia. Science 2010, 330, 783–788. [Google Scholar] [CrossRef] [PubMed]
- David, S.; Kroner, A. Repertoire of microglial and macrophage responses after spinal cord injury. Nat. Rev. Neurosci. 2011, 12, 388–399. [Google Scholar] [CrossRef] [PubMed]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Neurosci. 2008, 8, 958–969. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, M.M.; Hutchinson, M.; Watkins, L.R.; Yin, H. Toll-like receptor 4 in CNS pathologies. J. Neurochem. 2010, 114, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.; Wang, P.F.; Zhou, Y.; Wang, Y.C.; Yang, Q.W. Toll-like receptor 4 signaling in intracerebral hemorrhage-induced inflammation and injury. J. Neuroinflamm. 2013, 10, 27. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Zhang, D.; Wei, Y.; Ni, H.; Liang, W.; Zhang, H.; Hao, S.; Jin, W.; Li, K.; Hang, C.H. Inhibition of myeloid differentiation primary response protein 88 provides neuroprotection in early brain injury following experimental subarachnoid hemorrhage. Sci. Rep. 2017, 7, 15797. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Fujimoto, M.; Kawakita, F.; Nakano, F.; Imanaka-Yoshida, K.; Yoshida, T.; Suzuki, H. Anti-vascular endothelial growth factor treatment suppresses early brain injury after subarachnoid hemorrhage in mice. Mol. Neurobiol. 2016, 53, 4529–4538. [Google Scholar] [CrossRef] [PubMed]
- Teng, Z.; Jiang, L.; Hu, Q.; He, Y.; Guo, Z.; Wu, Y.; Huang, Z.; Cao, F.; Cheng, C.; Sun, X.; et al. Peroxisome proliferator-activated receptor beta/delta alleviates early brain injury after subarachnoid hemorrhage in rats. Stroke 2016, 47, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Ji, C.; Chen, G. Signaling pathway in early brain injury after subarachnoid hemorrhage: News update. Acta Neurochir. Suppl. 2016, 121, 123–126. [Google Scholar] [PubMed]
- Wang, C.X.; Xie, G.B.; Zhou, C.H.; Zhang, X.S.; Li, T.; Xu, J.G.; Li, N.; Ding, K.; Hang, C.H.; Shi, J.X.; et al. Baincalein alleviates early brain injury after experimental subarachnoid hemorrhage in rats: Possible involvement of TLR4/NF-kappaB-mediated inflammatory pathway. Brain Res. 2015, 1594, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.H.; Wang, C.X.; Xie, G.B.; Wu, L.Y.; Wei, Y.X.; Wang, Q.; Zhang, H.S.; Hang, C.H.; Zhou, M.L.; Shi, J.X. Fisetin alleviates early brain injury following experimental subarachnoid hemorrhage in rats possibly by suppressing TLR4/NF-kappaB signaling pathway. Brain Res. 2015, 1629, 250–259. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Nan, G. The mitogen-activated protein kinase (MAPK) signaling pathway as a discovery target in stroke. J. Mol. Neurosci. 2016, 59, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Maddahi, A.; Ansar, S.; Chen, Q.; Edvinsson, L. Blockade of the MEK/ERK pathway with a raf inhibitor prevents activation of pro-inflammatory mediators in cerebral arteries and reduction in cerebral blood flow after subarachnoid hemorrhage in a rat model. J. Cereb. Blood Flow Metab. 2011, 31, 144–154. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhao, X.D.; Shi, J.X.; Yin, H.X. Inhibition of the p38 mitogen-activated protein kinase (MAPK) pathway attenuates cerebral vasospasm following experimental subarachnoid hemorrhage in rabbits. Ann. Clin. Lab. Sci. 2011, 41, 244–250. [Google Scholar] [PubMed]
- Huang, L.; Wan, J.; Chen, Y.; Wang, Z.; Hui, L.; Li, Y.; Xu, D.; Zhou, W. Inhibitory effects of p38 inhibitor against mitochondrial dysfunction in the early brain injury after subarachnoid hemorrhage in mice. Brain Res. 2013, 1517, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.F.; Song, J.N.; Ma, X.D.; Zhao, Y.L.; Liu, Z.W.; Li, Y.; Sun, P.; Li, D.D.; Pang, H.G.; Huang, T.Q. Etanercept alleviates early brain injury following experimental subarachnoid hemorrhage and the possible role of tumor necrosis factor-α and c-Jun N-terminal kinase pathway. Neurochem. Res. 2015, 40, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.H.; Yen, T.L.; Hsu, C.Y.; Thomas, P.A.; Sheu, J.R.; Jayakumar, T. Multi-targeting andrographolide, a novel NF-kappaB inhibitor, as a potential therapeutic agent for stroke. Int. J. Mol. Sci. 2017, 18, 1638. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.S.; Kwon, Y.W.; Yang, H.M.; Kim, S.H.; Kim, T.Y.; Hur, J.; Park, K.W.; Cho, H.J.; Kang, H.J.; Park, Y.B.; et al. New mechanism of rosiglitazone to reduce neointimal hyperplasia: Activation of glycogen synthase kinase-3beta followed by inhibition of MMP-9. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 472–479. [Google Scholar] [CrossRef] [PubMed]
- Kaltschmidt, B.; Widera, D.; Kaltschmidt, C. Signaling via NF-kappaB in the nervous system. Biochim. Biophys. Acta 2005, 1745, 287–299. [Google Scholar] [CrossRef] [PubMed]
- You, W.C.; Wang, C.X.; Pan, Y.X.; Zhang, X.; Zhou, X.M.; Zhang, X.S.; Shi, J.X.; Zhou, M.L. Activation of nuclear factor-κB in the brain after experimental subarachnoid hemorrhage and its potential role in delayed brain injury. PLoS ONE 2013, 8, e60290. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Liang, G.; Ma, T.; Li, J.; Wang, P.; Liu, L.; Yu, B.; Liu, Y.; Xue, Y. Blood-brain barrier permeability change and regulation mechanism after subarachnoid hemorrhage. Metab. Brain Dis. 2015, 30, 597–603. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.T.; Rabinovich, G.A. Galectins: Regulators of acute and chronic inflammation. Ann. N. Y. Acad. Sci. 2010, 1183, 158–182. [Google Scholar] [CrossRef] [PubMed]
- Arikawa, T.; Simamura, E.; Shimada, H.; Nakamura, T.; Hatta, T.; Shoji, H. Significance of sugar chain recognition by galectins and its involvement in disease-associated glycosylation. Congenit. Anom. (Kyoto) 2014, 54, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.B.; Yoon, H.J.; Chang, C.Y.; Koh, H.S.; Jeon, S.H.; Park, E.J. Galectin-3 exerts cytokine-like regulatory actions through the JAK-STAT pathway. J. Immunol. 2010, 185, 7037–7046. [Google Scholar] [CrossRef] [PubMed]
- Lalancette-Hebert, M.; Swarup, V.; Beaulieu, J.M.; Bohacek, I.; Abdelhamid, E.; Weng, Y.C.; Sato, S.; Kriz, J. Galectin-3 is required for resident microglia activation and proliferation in response to ischemic injury. J. Neurosci. 2012, 32, 10383–10395. [Google Scholar] [CrossRef] [PubMed]
- Burguillos, M.A.; Svensson, M.; Schulte, T.; Boza-Serrano, A.; Garcia-Quintanilla, A.; Kavanagh, E.; Santiago, M.; Viceconte, N.; Oliva-Martin, M.J.; Osman, A.M.; et al. Microglia-secreted galectin-3 acts as a Toll-like receptor 4 ligand and contributes to microglial activation. Cell Rep. 2015, 10, 1626–1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, T. The pleiotropic effects of galectin-3 in neuroinflammation: A review. Acta Histochem. 2013, 115, 407–411. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.Y.; Rabinovich, G.A.; Liu, F.T. Galectins: Structure, function and therapeutic potential. Expert Rev. Mol. Med. 2008, 10, e17. [Google Scholar] [CrossRef] [PubMed]
- Sato, S. Galectins as molecules of danger signal, which could evoke an immune response to infection. Trends Glycosci. Glycotechnol. 2002, 14, 285–301. [Google Scholar] [CrossRef]
- Song, L.; Tang, J.W.; Owusu, L.; Sun, M.Z.; Wu, J.; Zhang, J. Galectin-3 in cancer. Clin. Chim. Acta 2014, 431, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Funasaka, T.; Raz, A.; Nangia-Makker, P. Galectin-3 in angiogenesis and metastasis. Glycobiology 2014, 24, 886–891. [Google Scholar] [CrossRef] [PubMed]
- De Boer, R.A.; van Veldhuisen, D.J.; Gansevoort, R.T.; Muller Kobold, A.C.; van Gilst, W.H.; Hillege, H.L.; Bakker, S.J.; van der Harst, P. The fibrosis marker galectin-3 and outcome in the general population. J. Intern. Med. 2012, 272, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Calvier, L.; Martinez-Martinez, E.; Miana, M.; Cachofeiro, V.; Rousseau, E.; Sadaba, J.R.; Zannad, F.; Rossignol, P.; Lopez-Andres, N. The impact of galectin-3 inhibition on aldosterone-induced cardiac and renal injuries. JACC Heart Fail. 2015, 3, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Kolatsi-Joannou, M.; Price, K.L.; Winyard, P.J.; Long, D.A. Modified citrus pectin reduces galectin-3 expression and disease severity in experimental acute kidney injury. PLoS ONE 2011, 6, e18683. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Simpson, J.L.; Zhang, J.; Gibson, P.G. Galectin-3: Its role in asthma and potential as an anti-inflammatory target. Respir. Res. 2013, 14, 136. [Google Scholar] [CrossRef] [PubMed]
- Zuberi, R.I.; Hsu, D.K.; Kalayci, O.; Chen, H.Y.; Sheldon, H.K.; Yu, L.; Apgar, J.R.; Kawakami, T.; Lilly, C.M.; Liu, F.T. Critical role for galectin-3 in airway inflammation and bronchial hyperresponsiveness in a murine model of asthma. Am. J. Pathol. 2004, 165, 2045–2053. [Google Scholar] [CrossRef]
- Arad, U.; Madar-Balakirski, N.; Angel-Korman, A.; Amir, S.; Tzadok, S.; Segal, O.; Menachem, A.; Gold, A.; Elkayam, O.; Caspi, D. Galectin-3 is a sensor-regulator of toll-like receptor pathways in synovial fibroblasts. Cytokine 2015, 73, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Papaspyridonos, M.; McNeill, E.; de Bono, J.P.; Smith, A.; Burnand, K.G.; Channon, K.M.; Greaves, D.R. Galectin-3 is an amplifier of inflammation in atherosclerotic plaque progression through macrophage activation and monocyte chemoattraction. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Xin, M.; Dong, X.W.; Guo, X.L. Role of the interaction between galectin-3 and cell adhesion molecules in cancer metastasis. Biomed. Pharmacother. 2015, 69, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Markowska, A.I.; Liu, F.T.; Panjwani, N. Galectin-3 is an important mediator of VEGF- and bFGF-mediated angiogenic response. J. Exp. Med. 2010, 207, 1981–1993. [Google Scholar] [CrossRef] [PubMed]
- Harvey, A.; Montezano, A.C.; Lopes, R.A.; Rios, F.; Touyz, R.M. Vascular fibrosis in aging and hypertension: Molecular mechanisms and clinical implications. Can. J. Cardiol. 2016, 32, 659–668. [Google Scholar] [CrossRef] [PubMed]
- Henderson, N.C.; Mackinnon, A.C.; Farnworth, S.L.; Poirier, F.; Russo, F.P.; Iredale, J.P.; Haslett, C.; Simpson, K.J.; Sethi, T. Galectin-3 regulates myofibroblast activation and hepatic fibrosis. Proc. Natl. Acad. Sci. USA 2006, 103, 5060–5065. [Google Scholar] [CrossRef] [PubMed]
- Vergaro, G.; Prud’homme, M.; Fazal, L.; Merval, R.; Passino, C.; Emdin, M.; Samuel, J.L.; Cohen Solal, A.; Delcayre, C. Inhibition of galectin-3 pathway prevents isoproterenol-induced left ventricular dysfunction and fibrosis in mice. Hypertension 2016, 67, 606–612. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.H.; Chou, C.H.; Wu, X.M.; Chang, Y.Y.; Hung, C.S.; Chen, Y.H.; Tzeng, Y.L.; Wu, V.C.; Ho, Y.L.; Hsieh, F.J.; et al. Aldosterone induced galectin-3 secretion in vitro and in vivo: From cells to humans. PLoS ONE 2014, 9, e95254. [Google Scholar] [CrossRef] [PubMed]
- Filipe, M.D.; Meijers, W.C.; Rogier van der Velde, A.; de Boer, R.A. Galectin-3 and heart failure: Prognosis, prediction & clinical utility. Clin. Chim. Acta 2015, 443, 48–56. [Google Scholar] [PubMed]
- Pasquini, L.A.; Millet, V.; Hoyos, H.C.; Giannoni, J.P.; Croci, D.O.; Marder, M.; Liu, F.T.; Rabinovich, G.A.; Pasquini, J.M. Galectin-3 drives oligodendrocyte differentiation to control myelin integrity and function. Cell Death Differ. 2011, 18, 1746–1756. [Google Scholar] [CrossRef] [PubMed]
- Comte, I.; Kim, Y.; Young, C.C.; van der Harg, J.M.; Hockberger, P.; Bolam, P.J.; Poirier, F.; Szele, F.G. Galectin-3 maintains cell motility from the subventricular zone to the olfactory bulb. J. Cell Sci. 2011, 124, 2438–2447. [Google Scholar] [CrossRef] [PubMed]
- Yip, P.K.; Carrillo-Jimenez, A.; King, P.; Vilalta, A.; Nomura, K.; Chau, C.C.; Egerton, A.M.; Liu, Z.H.; Shetty, A.J.; Tremoleda, J.L.; et al. Galectin-3 released in response to traumatic brain injury acts as an alarmin orchestrating brain immune response and promoting neurodegeneration. Sci. Rep. 2017, 7, 41689. [Google Scholar] [CrossRef] [PubMed]
- Wesley, U.V.; Vemuganti, R.; Ayvaci, E.R.; Dempsey, R.J. Galectin-3 enhances angiogenic and migratory potential of microglial cells via modulation of integrin linked kinase signaling. Brain Res. 2013, 1496, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.P.; Lang, B.T.; Vemuganti, R.; Dempsey, R.J. Galectin-3 mediates post-ischemic tissue remodeling. Brain Res. 2009, 1288, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.J.; Yu, G.F.; Jie, Y.Q.; Fan, X.F.; Huang, Q.; Dai, W.M. Role of galectin-3 in plasma as a predictive biomarker of outcome after acute intracerebral hemorrhage. J. Neurol. Sci. 2016, 368, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.F.; Yu, W.H.; Dong, X.Q.; Du, Q.; Yang, D.B.; Wu, G.Q.; Zhang, Z.Y.; Wang, H.; Jiang, L. The change of plasma galectin-3 concentrations after traumatic brain injury. Clin. Chim. Acta 2016, 456, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Jagodzinski, A.; Havulinna, A.S.; Appelbaum, S.; Zeller, T.; Jousilahti, P.; Skytte-Johanssen, S.; Hughes, M.F.; Blankenberg, S.; Salomaa, V. Predictive value of galectin-3 for incident cardiovascular disease and heart failure in the population-based FINRISK 1997 cohort. Int. J. Cardiol. 2015, 192, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Liu, Y.; Zhao, J.; Liu, H.; He, S. Prognostic value of plasma galectin-3 levels after aneurysmal subarachnoid hemorrhage. Brain Behav. 2016, 6, e00543. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, H.; Nakatsuka, Y.; Shiba, M.; Kawakita, F.; Fujimoto, M.; Suzuki, H.; pSEED Group. Increased plasma galectin-3 preceding the development of delayed cerebral infarction and eventual poor outcome in non-severe aneurysmal subarachnoid hemorrhage. Transl. Stroke Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, H.; Suzuki, H. Implications of periostin in the development of subarachnoid hemorrhage-induced brain injuries. Neural Regen. Res. 2017, 12, 1982–1984. [Google Scholar] [PubMed]
- Suzuki, H.; Nakano, F. To improve translational research in subarachnoid hemorrhage. Transl. Stroke Res. 2018, 9, 1–3. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nishikawa, H.; Suzuki, H. Possible Role of Inflammation and Galectin-3 in Brain Injury after Subarachnoid Hemorrhage. Brain Sci. 2018, 8, 30. https://doi.org/10.3390/brainsci8020030
Nishikawa H, Suzuki H. Possible Role of Inflammation and Galectin-3 in Brain Injury after Subarachnoid Hemorrhage. Brain Sciences. 2018; 8(2):30. https://doi.org/10.3390/brainsci8020030
Chicago/Turabian StyleNishikawa, Hirofumi, and Hidenori Suzuki. 2018. "Possible Role of Inflammation and Galectin-3 in Brain Injury after Subarachnoid Hemorrhage" Brain Sciences 8, no. 2: 30. https://doi.org/10.3390/brainsci8020030
APA StyleNishikawa, H., & Suzuki, H. (2018). Possible Role of Inflammation and Galectin-3 in Brain Injury after Subarachnoid Hemorrhage. Brain Sciences, 8(2), 30. https://doi.org/10.3390/brainsci8020030