1. Introduction
Many important clinical conditions continue to elude effective treatments. Stroke is a notable example where over 100 clinical trials have failed to find a means to prevent cell death by neuroprotection [
1]. Similar strings of clinical trial failure have occurred with diseases such as myocardial infarction [
2] and acute nephrotic ischemia [
3]. We have argued that a key factor behind these failures is the lack of a general theory of biological cell injury.
We therefore introduced a nonlinear dynamical theory of acute cell injury [
4]. However, the original form of the theory possessed limitations as detailed in Ref. [
5]. This led us to reformulate the theory, which was introduced elsewhere [
5] but for which we here provide a deeper analysis. Technically, the original theory consisted of a system of
autonomous nonlinear ordinary differential equations. The reformulation of the theory consists of a system of
nonautonomous, nonlinear differential equations. The technical mathematical differences impact how the equations are solved and how the resulting solutions are interpreted in terms of acute cell injury. It is the purpose of this paper to illustrate our procedure for solving and interpreting the solutions of the nonautonomous theory of acute cell injury.
As the title indicates, constructing and understanding the theory, whatever mathematical form it takes, requires abstracting and idealizing the real world. Biology and the subdisciplines of biomedicine are generally descriptive. Advanced mathematics are not widely used in these sciences. On the other hand, the sciences that have allowed the most effective development of technology, notably physics, do not seek to literally describe its subject matter. Instead, physics idealizes and abstracts reality to mathematically formulate how things change. The second major aspect of the present work is to illustrate how idealization and abstraction can be used in biomedical research to model acute injury to biological cells.
In this regard, a key idea we wish to convey is that
mathematically abstracting a system allows all possible states of the system to be understood. Experiments usually involve high costs of time and resources and can only measure a finite number of parameter combinations. On the other hand, a theory allows us to see how the system behaves under all parameter conditions. Then, the goal of science is to assure congruency between specific solutions of the theory and specific experimental measurements. Experimental measurements made only to describe a phenomenon in the absence of mathematical theory are incomplete and are descriptive science [
6]. Rather, the measurements should seek to test a theory [
6].
Below we study one idealized cell type injured by one idealized injury mechanism. We shall see the enormous complexity in this one example. However, the complexity is not incomprehensible. The mathematics provide a systematic framework, a catalog of sorts, allowing all possible states of the system to be understood in an organized fashion. This has major implications for developing therapies for acute injuries such as stroke. We shall show that injuring one ideal cell type by one ideal injury mechanism, varying only the intensity of the injury, produces a continuum of states for which there is little appreciation in the paradigms that currently dominate biomedical research.
1.1 Cell Injury Idealized
There are two main categories of how cells become injured. There is either some identifiable injury applied to the cells or there is not. Examples of identifiable injuries would include mechanical trauma, chemical trauma (e.g., a poison), metabolic trauma (such as ischemia), and so on. The criteria being that there is a clearly identifiable exogenous agent or circumstance that injures the cell. Further, the intensity of the damage mechanism can be quantified: the minutes of ischemia, the concentration of a poison, the amount of force, and so on. We can abstract the quantitative aspect of injury intensity as a parameter I, and thereby abstract it from any specific injury mechanism. This stands in contrast to injuries such as cell transformation, or chronic neurodegenerative diseases in which the cause, let alone intensity, of the cell injury is not clearly identifiable, if even known. Thus, I represents the intensity of a clearly identifiable injury mechanism and we term this “acute cell injury”.
From these considerations, we can begin to construct an idealized picture of acute cell injury. A cell is injured by some acute injury mechanism, and in response it either lives or dies. This is an idealization because we do not specify what type of cell nor the specific injury. The cell and the injury are tokens that interact, and their interaction is quantified by the parameter I.
From the vast amount of empirical, descriptive studies, it has been shown that many biomolecular changes occur in cells after they have been acutely injured. These are generally expressed in terms of pathways: changes in phosphorylation or other signaling events, increases or decreases in the activity of specific proteins or pathways, changes in localization or amounts of ions, specific proteins, transcription factors, micro-RNAs, etc. It is these specific molecular events that constitute the complex network of changes in the injured intracellular milieu. It is a case of Humpty Dumpty: we injure cells (or tissues) then grind them up and identify the hundreds of changes in the biomolecules, but how to put them back together again to reconstruct how the cell dies? To date, such reconstruction efforts have generally been unsuccessful as attested by failed clinical trials in many fields of biomedicine.
Instead of a literal reconstruction of the events, we idealize as follows. We know a priori that some of these biomolecular changes harm the cell but that others serve to protect the cell. Let all the cell-damaging changes be represented by the variable D, the total damage in the cell. Likewise, represent all the pro-survival changes in the cell by the variable S, the total stress response. Theoretically, there is a third category of changes in the acutely injured cell: those changes that have no effect on damaging the cell or helping it survive, which we denote by an empty set [ ]. Thus, all the complex molecular changes in the cell fall into these three general categories: D, S, or [ ]. There are no other possibilities. Since [ ] has no effect on outcome, we can ignore it here.
The concepts of D and S are key abstractions and idealizations of acute cell injury. We assert they provide an incontrovertible generalization that covers all possibilities. In mainstream biomedical research, the goal is to discover which molecules or pathways damage, and which enhance cell survival. Further, do some molecules that, at one point in the post-injury time course foster survival, transform into damaging influences? We submit that such questions lead down blind alleys. As abstractions, D and S already implicitly subsume these possibilities. From a theoretical perspective, we do not need to know the specific molecules of which D and S consist at any moment in time any more than we need to know the exact velocity of each air molecule when we measure air temperature.
Thus, we can fill in our ideal picture further: There is a cell. It is acutely injured with intensity I. In response, inside the cell, D and S assume nonzero values that change with time. The changes in D and S directly determine the survival or death outcome.
1.2 What Causes Cell Death?
The main use to which the concepts of D and S are applied is to define the cause of cell death. The core assumption of the mathematical idealization of acute cell injury is this: if S > D, the cell lives but if D > S, the cell dies.
A metaphor may help illustrate the concept. Imagine kicking a ball up a hill. The ball starts at a position at the bottom of the hill. This is the uninjured cell. A force is applied to the ball and it rolls up the hill. The force is the application of injury I to the cell. If the force is weak, the ball will roll to some point on the facing hillside, then roll back down to where it started. This is the survival case where S > D. However, if the force of I is greater than some specific value, the ball will go over the top of the hill and roll down the other side. This is the state D > S, and the other side of the hill is the state of death.
To be more precise, we are suggesting that acute cell injury is a tipping point phenomenon. This is already recognized in other terms by the concept of “cell death threshold”. The “cell death threshold” is the amount of injury that causes the cell to die. The “cell death threshold” concept does not describe a threshold but instead indicates a tipping point between survival and death. Thresholds and tipping points are different mathematical entities, as illustrated below. The tipping point between survival and death is quantitatively determined by the intensity of injury I.
Another useful way to give meaning to D and S is to recognize that, before injury, the cell is in a homeostatic steady-state. Application of injury intensity, I, “knocks” the cell out of homeostasis. The variable S represents changes inside the cell seeking to bring the cell back into homeostasis. D represents changes that disrupt homeostasis. If the disruption of homeostasis is greater than the cell’s ability to re-achieve homeostasis (e.g., D > S), the cell dies.
1.3 Survival and Death Outcomes after Acute Injury
These ideas intimately link sublethal and lethal injuries which, however, are generally studied separately in mainstream biomedical studies, and generally treated as different phenomena. On the sublethal side is survival after injury, often accompanied by a preconditioning effect [
7,
8,
9,
10,
11]. Preconditioning is specifically defined here to mean that a cell subjected to a sublethal injury can, after a specified time, survive a lethal injury. On the lethal side is cell death which can take on different qualitative forms.
With respect to cell death after brain ischemia, necrosis and delayed neuronal death (DND) [
12,
13] are observed. They appear different by many criteria and therefore are thought to be due to different causes. Thus, one finds terms such as apoptosis, necrosis, and necroptosis, and other variants, described by lists of qualitative features such as cell appearance during death or which molecular pathways are activated [
14,
15,
16,
17,
18,
19,
20,
21,
22,
23].
With respect to survival and preconditioning, empirical evidence shows that the time between the sublethal and lethal injuries required to achieve optimal survival is finely tuned [
24,
25,
26]. Thus, there are degrees of preconditioning, and the preconditioning effect is not permanent but fades with time after the sublethal injury. Further, different tissues display different forms of preconditioning (e.g., rapid preconditioning in heart, and delayed preconditioning in brain [
27]).
Rapid or delayed preconditioning, necrosis and DND are qualitative distinctions. By defining acute cell injury in terms of the dynamics of D and S we build a framework where these seemingly different phenomena are points on a continuum of responses of cells to acute injury. The continuum is injury intensity, I. The dynamics of D and S vary quantitatively as a function of I as we will show below. This allows us to understand clearly that the specific qualitative forms of preconditioning or cell death are but cross sections of a continuum of dynamical behaviors.
Thus, through idealization and abstraction, we define the cause of cell death in a general fashion, independent of any specific cell type or specific injury mechanism, by focusing on and abstracting features common to all cells. By converting these ideas into a mathematical theory, we intimately link survival and death outcomes in a single continuum of dynamical behaviors.
1.4. A Mathematical Theory of Acute Cell Injury
We have developed the refined theory elsewhere [
5] and therefore give only a brief summary here. We begin with our idealized picture of acute cell injury: there is a cell which is acutely injured with intensity
I.
D and
S accumulate in the cell and change with time. If
D >
S, the cell irreversibly exits homeostasis and dies.
By definition, D and S are mutually antagonistic. Stress responses seek to eliminate damage, but damage reactions can destroy the mediators of stress responses. This has a specific mathematical meaning: D and S are inversely related. The inverse relationships between D and S can be quantified using the Hill function, which is the function that gives S-shaped curves. The Hill function defines a threshold, Θ, where a 50% effect occurs (e.g. like LD50). Thus, there is some amount of S, ΘS, which causes a 50% decline in D. Similarly, there is some amount of D, ΘD, which causes a 50% reduction in S. ΘD and ΘS are true mathematical thresholds, meaning they are the values of D and S at the 50% reduction point.
The key assumption of our theory is that
ΘD and
ΘS change as a function of injury intensity,
I.
Equation (1) posits specific functional forms: ΘD increases and ΘS decreases exponentially with I. These constitute assumptions that require empirical verification. However, for the sake of theory building, they provide a simple, plausible relationship. The parameters cD, cS, λD, and λS are constants of proportionality, required to correctly express the proportionalities between the thresholds and I. Their meanings are discussed in the next section.
To model the changes in
D and
S with time, the following equation is used
As indicated, this is a well-known system of differential equations for modeling the common sense understanding that the net rate equals the difference between the rate of formation and the rate of decay [
28]. The rates of formation are given by Hill functions expressing the inverse relationship between
D and
S, scaled by a velocity parameter
v. In Equation (2), the rates of decay are assumed to be linear, with a decay parameter
k.
The original theory substituted Equation (1) into Equation (2) [
4]. We present here a refined theory using two additional assumptions. In addition to the assumption embodied by Equation (1) we now assume that (a) the velocity parameter decreases exponentially with time (Equation (3)), and (b) the decay parameter is a function of the instantaneous difference of
D and
S (Equation (4)). The rationalization of these assumptions was discussed previously [
5].
Substituting Equations (1), (3) and (4) into Equation (2) gives our current theory of the nonlinear dynamics of acute cell injury:
2. Methods
2.1. Preliminary Considerations
When the term “prediction” is used in a scientific context, it does not refer to qualitative statements. The term “prediction” specifically means that one of the mathematical solutions of a mathematical theory fits a dataset intended to measure the theory. A necessary precondition to data fitting is to interpret the theory in terms of the relevant physical system. Our goal now is to display a typical solution of Equation (5) and how it can be interpreted with respect to experimental situations.
To summarize what is detailed below: Equation (5) takes input numbers (the “input vector”) and outputs time courses of
D and
S. A time course can begin at
D = 0 and
S = 0, corresponding to an uninjured system, or it can begin at any value of
D or
S. Where the time courses start are the
initial conditions, (
D0,
S0), which are part of the input vector. For a given pair of
D and
S time courses output by Equation (5), either
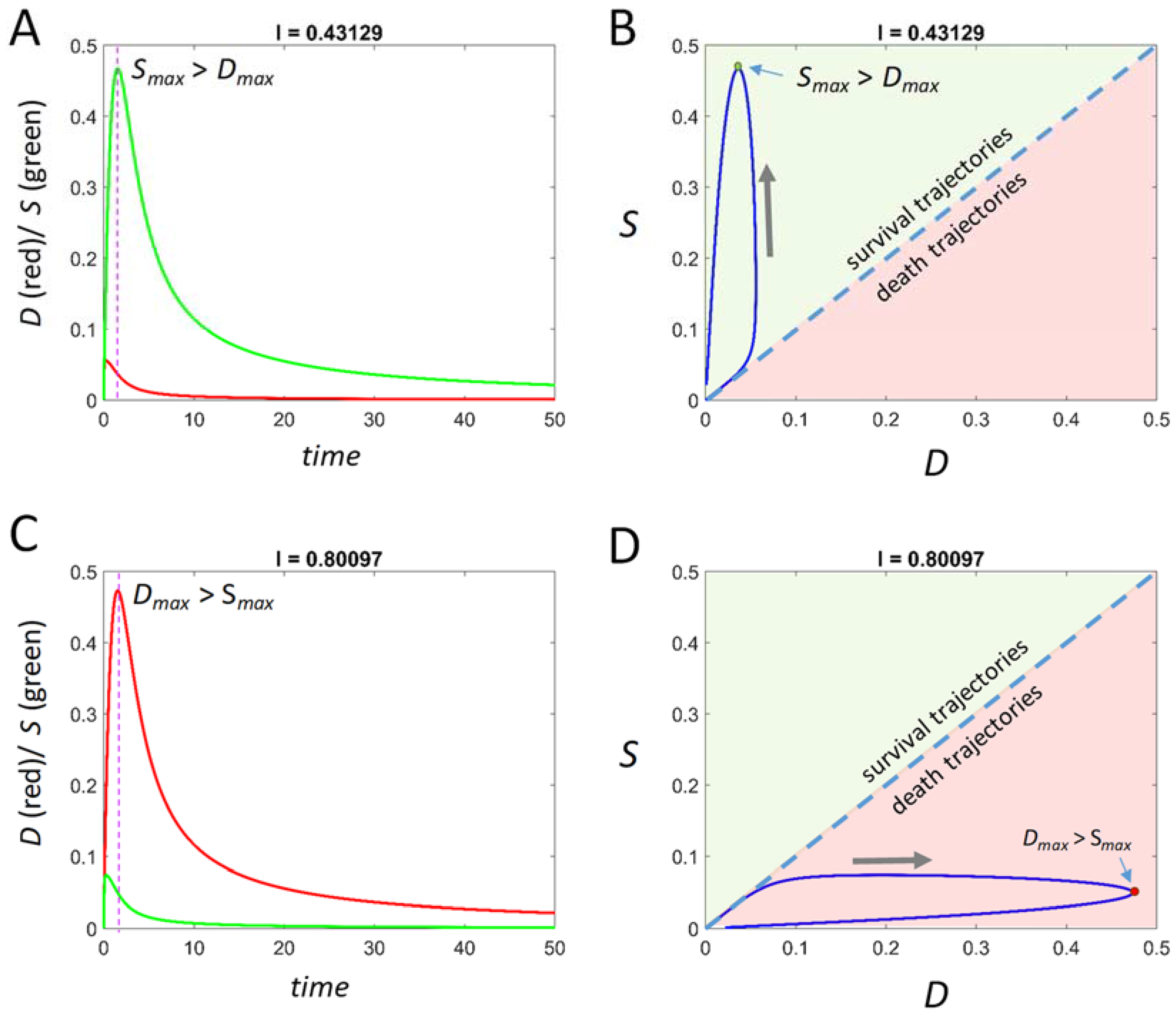
D or
S achieves a maximum value (
Dmax and
Smax, respectively). Above we spoke of
S >
D or
D >
S. In the time courses output by Equation (5), these inequalities take the form
Smax >
Dmax or
Dmax >
Smax, corresponding to survival and death outcomes, respectively. The
control parameter is
I, injury intensity, and all other parameters are held constants to examine how the system behaves as a function of injury intensity. The series of time course along the continuum of
I we have termed an
injury course [
4]. We show below how to express the solutions of Equation (5) as injury courses calculated across a range of initial conditions.
2.2 The Input Vector
Equation (5) has 9 parameters and two initial conditions (
D0,
S0), giving the following input vector:
A brief description of the parameters now follows. v0 is the initial velocity of D and S formation at time zero. As Equation (3) indicates, the initial velocity decreases exponentially with time, meaning that the rate at which D and S form decreases with time after the injury. c1 is a decay constant indicating how quickly v0 decreases with time; the larger c1, the faster v0 decreases. c2 sets the rate that D and S decay; the larger c2, the faster are the D and S decay rates. In general, if v0 and c2 are set to 1, the solutions to Equation (5) stay in the unit plane (i.e., D and S range only between 0 and 1).
Four parameters,
cD,
λD,
cS, and
λS, characterize the qualitative aspects of the system. As described in detail elsewhere [
29],
cD and
λD represent the injury mechanism, and
cS and
λS represent the cell type. The parameter
n, typically called the Hill coefficient, can, in the context of our theory, be taken to represent how “tightly” the nodes of the molecular networks defined by
D and
S are linked. As stated above,
I, injury intensity, is the control parameter. All these parameters can vary over large ranges and produce sensible output from Equation (5).
2.3 Initial Conditions
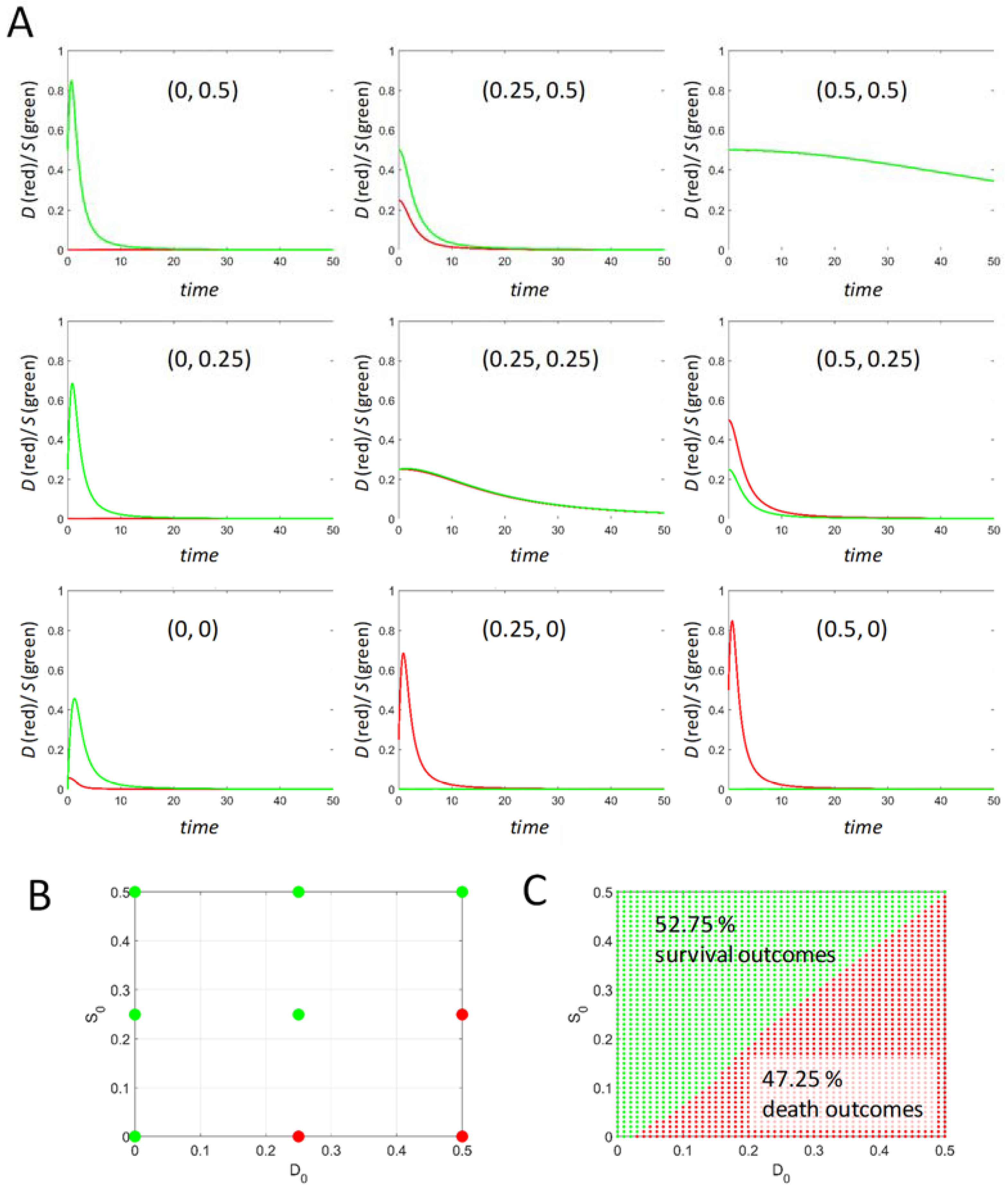
Initial conditions (D0, S0) were described above but merit further discussion because they (1) link to experimental designs commonly encountered in biomedical studies, and (2) provide an example where a mathematical theory can study situations that are limited by time and resources in the laboratory.
In the laboratory, there is generally an uninjured control condition that is compared against the injured cells. A typical example would be a cell culture given a poison (e.g., thapsigargin that inhibits the endoplasmic reticulum SERCA pump). In this instance, thapsigargin is the injury mechanism, and its concentration is the injury intensity, I, which can be sublethal or lethal. For such a study there will always be a control cell culture given only the vehicle in which the thapsigargin is dissolved. Prior to administering thapsigargin, the experimental cells are identical to the control cells. This is an example of beginning an injury from initial conditions (D0,S0) = (0,0). Prior to drug administration, there is no cell damage and no active stress responses in the cell culture.
However, what if the cells were first transfected to express, say, HSP70 protein? HSP70 is an important pro-survival stress response protein. One would hypothesize that the transfected cells should survive a higher concentration of thapsigargin than the un-transfected cells. Increasing HSP70 protein before administering thapsigargin means that S0 is no longer zero but is now a positive value greater than zero. Transfecting HSP70 therefore represents a change in the S initial conditions of the cells. In the typical case, the experimental group would be transfected plus thapsigargin, and the control would be transfected plus vehicle. However, the transfected control is different from the untreated, un-transfected control. It is generally assumed that it is good enough to take the transfected plus vehicle as the control, and the effect of transfecting with HSP70 is not taken into account in the study design. However, in the scope of our model, the transfection of the control cells is a change in initial conditions. As we show below, changing initial conditions can radically alter the dynamics.
In general, experimental manipulations such as the example given above are not recognized as changes in initial conditions, and hence, these manipulations are not systematically accounted for in the empirical biomedical literature. The study of varied initial conditions provides a systematic handle on such circumstances. Equation (5) can be studied over ranges of initial conditions that would otherwise require prohibitive amounts of time and resources to empirically study in the laboratory. Thus, at any value of I, we also study the behavior of Equation (5) over a range of initial conditions.
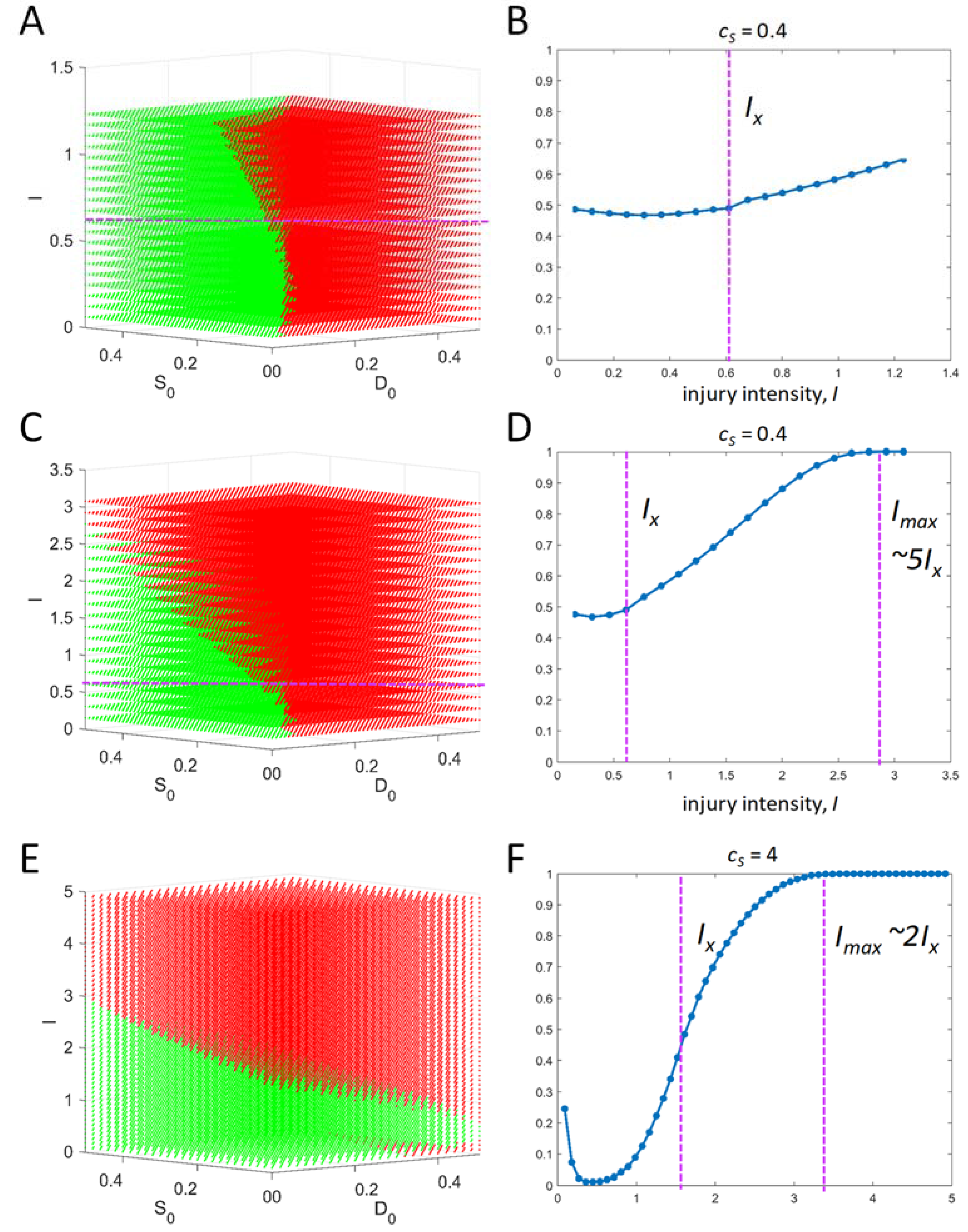
It is theoretically relevant to ask: what dictates the range of initial conditions? Across an injury course, there will be one time course that gives the maximum possible value for D and another that gives the maximum possible value of S. The maximum value of S across all time courses can be interpreted as the maximum possible total stress responses for the specific cell type. Therefore, S cannot exceed this value. Thus, any initial condition of S must be less than or equal to this maximum value. For example, if the maximum S across all time courses = 1, then the initial condition for S must range as 0 < S0 < 1. The same logic holds for D0. There are other possibilities for setting the initial condition ranges, but this one will serve in the present analysis.
2.4. Summary of Input Vector
For the present exercise, we hold [
v0,
c1,
c2,
cD,
λD,
cS,
λS,
n] constant. We then vary
I over the range 0 <
I <
Imax, where
Imax is the injury intensity beyond which the cells are incapable of mustering any stress response (e.g.,
S ~ 0) for the entire post injury time course. The behavior of the system over the range 0 <
I <
Imax constitutes the
injury course. The
I-range is determined relative to
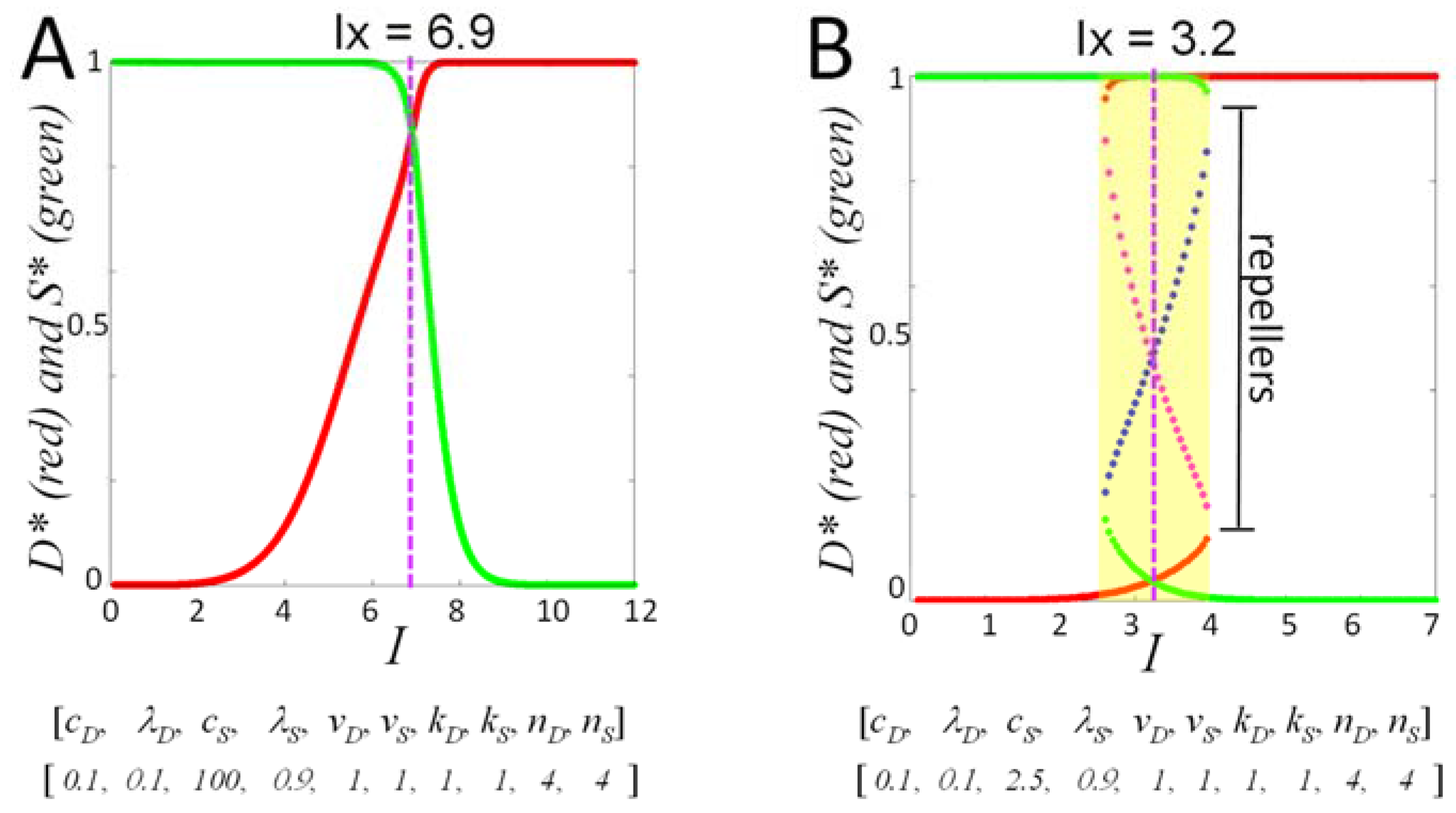
IX, the tipping point value of injury intensity [
4]:
Finally, at each value of I, within the I-range, we study Equation (5) over ranges of initial conditions. To repeat, if v0 and c2 each equal 1, then the values of D and S over time never exceed 1, and so our initial conditions can be confined to the range 0–1. From initial conditions (0,0), all time courses from 0 < I< IX will have Smax > Dmax (survival outcome), and all time courses from IX < I < Imax will have Dmax > Smax (death outcome). This statement, however, does not hold in general at other initial conditions. We show below how to represent outcomes across ranges of initial conditions at each value of I.
To summarize, the values of parameters and initial conditions used in our example are:
[v0, c1, c2, cD, λD, cS, λS, n] = [1, 1, 1, 0.1, 0.25, 0.4, 2, 4].
I-range centered at IX = 0.6161 (as calculated from the parameters given in 1).
Maximum possible range of initial conditions: 0 < D0 < 1 and 0 < S0 < 1.
5. Measuring the Theory
The main purposes of this paper have been: (1) to show how to solve the nonautonomous theory and interpret the solutions in terms of acute cell injury, and (2) illustrate how to think of injured cells in abstract, ideal terms. We have not focused on how we would measure or test the theory. In the following we make a few comments along these lines.
The link between the theory and real injured cells or tissues are the concepts of D and S. When a brain or heart is injured by ischemia, or when cultured cells are injured by thapsigargin (or any other injury mechanism), one must envision that D and S are real phenomena occurring inside the cells and that the amount of each follows time courses as calculated by the theory. Ideally, one would then measure how D and S change with time and determine if the theory accurately predicts the D and S time courses.
The empirical question thus comes down to measuring D and S. Recalling their definitions, D is the total damage and S is the total-induced stress responses. Thus, to measure them would be to measure every single form of post-injury damage at a point in time and sum them together to obtain D at that time point. Similarly, every induced stress response would be measured and summed to obtain S. In practice, given today’s technology and our incomplete understanding of cell physiology, this is impossible.
However, taking this approach is analogous to measuring temperature by attempting to measure the velocity of every single particle in the medium and average them, which is also impossible. Instead, we measure temperature via e.g., the expansion volume of a liquid, typically mercury. This provides a surrogate measure that correlates perfectly with the average velocity of the particles making up the medium whose temperature we seek to know.
A similar approach is required to estimate D and S. There must be specific changes inside the injured cells that estimate or track the real values of D and S. We have hypothesized that the gene changes inside the cell track S and that some general form of cell damage, such as protein aggregates, track the total damage D. We have made these measurements and are in the process of preparing our results for publication. We have not included our empirical work here because it is outside the scope of the present work, and the present work is a necessary prelude for reporting our empirical results.
The important point to emphasize here is that there is a chasm between the mainstream descriptive approach to cell injury that assumes it can discover some qualitative feature that causes cell death, and our theoretical approach that dictates a priori what needs to be measured to characterize cell injury. In our theory, D and S are a priori concepts, and the theory indicates that empirical work needs to be directed first to discover how to quantitatively estimate D and S, and then determine their time courses. Ultimately, it is the failure of the descriptive approach that has motivated us, and we believe, in the long run, both approaches will be necessary much in the same manner that theory and experiment co-exist in physics.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





