Neuroprotective Effects of Bioactive Compounds and MAPK Pathway Modulation in “Ischemia”—Stressed PC12 Pheochromocytoma Cells



Abstract
:1. A Brief Introduction to Ischemia
1.1. Stroke (Cerebrovascular Insult-CVI)
1.2. Signaling Cascades Involved in CVI
1.2.1. MAPK Pathway Involvement in CVI
1.2.2. Antioxidants and Redox Potential Management in CVI
2. Experimental Models of Ischemia
2.1. Models of In Vivo Ischemia
2.2. Models of In Vitro Ischemia
PC12 Cells as a Representative In Vitro Ischemic Catecholaminergic Neuronal Model
3. Chemicals and Cell-Induced Neuroprotection in PC12 OGD/R Model
3.1. Drugs and Natural Products
3.2. Mesenchymal Stem Cells: A Prospective New Tool in Neuroprotection
3.3. Effect of Growth Factors on PC12 Cell Rescue after OGD/R Insult
4. Some Lessons from the PC12 OGD/R Model
Acknowledgments
Conflicts of Interest
Abbreviations
| AD3 | A PEG conjugate of Lipoic acid with Tempol |
| BDNF | brain-derived neurotrophic factor |
| b-FGF | Basic fibroblast growth factor |
| CCL2 | chemokine (C-C motif) ligand 2 |
| CVI | cerebrovascular insult |
| ELISA | enzyme-linked immunoassays |
| ERK | extracellular signal-regulated kinase |
| GDNF | glial cell derived neurotrophic factor |
| IHC | Immunohistochemistry |
| IL-6 | interleukin-6 |
| MAPK | mitogen activated protein kinase |
| MCAO | Middle Cerebral Artery Occlusion |
| MSC | mesenchymal stem cells |
| NGF | nerve growth factor |
| NT-3 | neurotrophin-3 |
| OGD | oxygen and glucose deprivation |
| OGD-R | oxygen glucose deprivation followed by reoxygenation |
| TGF-β1 | Transforming growth factor beta 1 |
| VEGF | vascular endothelial growth factor |
References
- Richard, M.J.; Saleh, T.M.; el Bahh, B.; Zidichouski, J.A. A novel method for inducing focal ischemia in vitro. J. Neurosci. Methods 2010, 190, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Guan, Y.M.; Huang, H.L.; Wang, Q.S. Human umbilical cord blood mesenchymal stem cell transplantation suppresses inflammatory responses and neuronal apoptosis during early stage of focal cerebral ischemia in rabbits. Acta Pharmacol. Sin. 2014, 35, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Sommer, C.J. Ischemic stroke: Experimental models and reality. Acta Neuropathol. 2017, 133, 245–261. [Google Scholar] [CrossRef] [PubMed]
- Holloway, P.M.; Gavins, F.N. Modeling Ischemic Stroke In Vitro: Status Quo and Future Perspectives. Stroke 2016, 47, 561–569. [Google Scholar] [CrossRef] [PubMed]
- Lipton, P. Ischemic cell death in brain neurons. Physiol. Rev. 1999, 79, 1431–1568. [Google Scholar] [CrossRef] [PubMed]
- Brouns, R.; de Deyn, P.P. The complexity of neurobiological processes in acute ischemic stroke. Clin. Neurol. Neurosurg. 2009, 111, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Beilharz, E.J.; Williams, C.E.; Dragunow, M.; Sirimanne, E.S.; Gluckman, P.D. Mechanisms of delayed cell death following hypoxic-ischemic injury in the immature rat: Evidence for apoptosis during selective neuronal loss. Mol. Brain Res. 1995, 29, 1–14. [Google Scholar] [CrossRef]
- Zheng, Z.; Zhao, H.; Steinberg, G.K.; Yenari, M.A. Cellular and molecular events underlying ischemia-induced neuronal apoptosis. Drug News Perspect. 2003, 16, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Bazan, N.G.; Marcheselli, V.L.; Cole-Edwards, K. Brain response to injury and neurodegeneration: Endogenous neuroprotective signaling. Ann. N. Y. Acad. Sci. 2005, 1053, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Adibhatla, R.M.; Hatcher, J.F. Phospholipase A2, reactive oxygen species, and lipid peroxidation in cerebral ischemia. Free Radic. Biol. Med. 2006, 40, 376–387. [Google Scholar] [CrossRef] [PubMed]
- Ginsberg, M.D. Neuroprotection for ischemic stroke: Past, present and future. Neuropharmacology 2008, 55, 363–389. [Google Scholar] [CrossRef] [PubMed]
- Bazan, N.G.; de Turco, E.B.R.; Allan, G. Mediators of injury in neurotrauma: Intracellular signal transduction and gene expression. J. Neurotrauma 1995, 12, 791–814. [Google Scholar] [CrossRef] [PubMed]
- Sanganalmath, S.K.; Gopal, P.; Parker, J.R.; Downs, R.K.; Parker, J.C., Jr.; Dawn, B. Global cerebral ischemia due to circulatory arrest: Insights into cellular pathophysiology and diagnostic modalities. Mol. Cell. Biochem. 2017, 426, 111–127. [Google Scholar] [CrossRef] [PubMed]
- Reinhart, W.H. Molecular biology and self-regulatory mechanisms of blood viscosity: A review. Biorheology 2001, 38, 203–212. [Google Scholar] [PubMed]
- Hallenbeck, J.M. Inflammatory reactions at the blood-endothelial interface in acute stroke. Adv. Neurol. 1996, 71, 281–297. [Google Scholar] [PubMed]
- Vila, N.; Castillo, J.; Davalos, A.; Chamorro, A. Proinflammatory cytokines and early neurological worsening in ischemic stroke. Stroke 2000, 31, 2325–2329. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Wang, W.; Zhang, Y.; Liu, S.; Liu, Y.; Zheng, D. Tumour necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced chemokine release in both TRAIL-resistant and TRAIL-sensitive cells via nuclear factor kappa B. FEBS J. 2009, 276, 581–593. [Google Scholar] [CrossRef] [PubMed]
- Mehta, S.L.; Manhas, N.; Raghubir, R. Molecular targets in cerebral ischemia for developing novel therapeutics. Brain Res. Rev. 2007, 54, 34–66. [Google Scholar] [CrossRef] [PubMed]
- Pearson, G.; Robinson, F.; Gibson, T.B.; Xu, B.E.; Karandikar, M.; Berman, K.; Cobb, M.H. Mitogen-activated protein (MAP) kinase pathways: Regulation and physiological functions. Endocr. Rev. 2001, 22, 153–183. [Google Scholar] [CrossRef] [PubMed]
- Slevin, M.; Krupinski, J.; Slowik, A.; Rubio, F.; Szczudlik, A.; Gaffney, J. Activation of MAP kinase (ERK-1/ERK-2), tyrosine kinase and VEGF in the human brain following acute ischaemic stroke. Neuroreport 2000, 11, 2759–2764. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, S.; Schnellmann, R.G. A death-promoting role for extracellular signal-regulated kinase. J. Pharmacol. Exp. Ther. 2006, 319, 991–997. [Google Scholar] [CrossRef] [PubMed]
- Nozaki, K.; Nishimura, M.; Hashimoto, N. Mitogen-activated protein kinases and cerebral ischemia. Mol. Neurobiol. 2001, 23, 1–19. [Google Scholar] [CrossRef]
- Sugino, T.; Nozaki, K.; Hashimoto, N. Activation of mitogen-activated protein kinases in gerbil hippocampus with ischemic tolerance induced by 3-nitropropionic acid. Neurosci. Lett. 2000, 278, 101–104. [Google Scholar] [CrossRef]
- Shimada, K.; Nakamura, M.; Ishida, E.; Kishi, M.; Konishi, N. Roles of p38- and c-jun NH2-terminal kinase-mediated pathways in 2-methoxyestradiol-induced p53 induction and apoptosis. Carcinogenesis 2003, 24, 1067–1075. [Google Scholar] [CrossRef] [PubMed]
- Mielke, K.; Herdegen, T. JNK and p38 stresskinases–Degenerative effectors of signal-transduction-cascades in the nervous system. Prog. Neurobiol. 2000, 61, 45–60. [Google Scholar] [CrossRef]
- Irving, E.A.; Barone, F.C.; Reith, A.D.; Hadingham, S.J.; Parsons, A.A. Differential activation of MAPK/ERK and p38/SAPK in neurones and glia following focal cerebral ischaemia in the rat. Mol. Brain Res. 2000, 77, 65–75. [Google Scholar] [CrossRef]
- Deng, H.; Zuo, X.; Zhang, J.; Liu, X.; Liu, L.; Xu, Q.; Wu, Z.; Ji, A. Alphalipoic acid protects against cerebral ischemia/reperfusion-induced injury in rats. Mol. Med. Rep. 2015, 11, 3659–3665. [Google Scholar] [CrossRef] [PubMed]
- Shay, K.P.; Moreau, R.F.; Smith, E.J.; Smith, A.R.; Hagen, T.M. Alpha-lipoic acid as a dietary supplement: Molecular mechanisms and therapeutic potential. Biochim. Biophys. Acta 2009, 1790, 1149–1160. [Google Scholar] [CrossRef] [PubMed]
- Packer, L.; Tritschler, H.J.; Wessel, K. Neuroprotection by the metabolic antioxidant alpha-lipoic acid. Free Radic. Biol. Med. 1997, 22, 359–378. [Google Scholar] [CrossRef]
- Panigrahi, M.; Sadguna, Y.; Shivakumar, B.R.; Kolluri, S.V.; Roy, S.; Packer, L.; Ravindranath, V. α-Lipoic acid protects against reperfusion injury following cerebral ischemia in rats. Brain Res. 1996, 717, 184–188. [Google Scholar] [CrossRef]
- Ruszkiewicz, J.; Albrecht, J. Changes in the mitochondrial antioxidant systems in neurodegenerative diseases and acute brain disorders. Neurochem. Int. 2015, 88, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Gomes, E.C.; Silva, A.N.; de Oliveira, M.R. Oxidants, antioxidants, and the beneficial roles of exercise-induced production of reactive species. Oxidative Med. Cell. Longev. 2012, 2012, 756132. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, T.; Kawai, C.; Sano, M.; Minatogawa, Y. Peroxisomes exist in growth cones and move anterogradely and retrogradely in neurites of PC12D cells. Exp. Cell Res. 2001, 266, 260–269. [Google Scholar] [CrossRef] [PubMed]
- Gilgun-Sherki, Y.; Rosenbaum, Z.; Melamed, E.; Offen, D. Antioxidant therapy in acute central nervous system injury: Current state. Pharmacol. Rev. 2002, 54, 271–284. [Google Scholar] [CrossRef] [PubMed]
- Vatassery, G.T. Vitamin E. Neurochemistry and implications for neurodegeneration in Parkinson’s disease. Ann. N. Y. Acad. Sci. 1992, 669, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Agus, D.B.; Winfree, C.J.; Kiss, S.; Mack, W.J.; McTaggart, R.A.; Choudhri, T.F.; Kim, L.J.; Mocco, J.; Pinsky, D.J.; et al. Dehydroascorbic acid, a blood-brain barrier transportable form of vitamin C, mediates potent cerebroprotection in experimental stroke. Proc. Natl. Acad. Sci. USA 2001, 98, 11720–11724. [Google Scholar] [CrossRef] [PubMed]
- Gilgun-Sherki, Y.; Melamed, E.; Offen, D. Oxidative stress induced-neurodegenerative diseases: The need for antioxidants that penetrate the blood brain barrier. Neuropharmacology 2001, 40, 959–975. [Google Scholar] [CrossRef]
- Koenig, M.L.; Meyerhoff, J.L. In vitro neuroprotection against oxidative stress by pre-treatment with a combination of dihydrolipoic acid and phenyl-butyl nitrones. Neurotox. Res. 2003, 5, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Tuttolomondo, A.; di Sciacca, R.; di Raimondo, D.; Arnao, V.; Renda, C.; Pinto, A.; Licata, G. Neuron protection as a therapeutic target in acute ischemic stroke. Curr. Top. Med. Chem. 2009, 9, 1317–1334. [Google Scholar] [CrossRef] [PubMed]
- Candelario-Jalil, E. Injury and repair mechanisms in ischemic stroke: Considerations for the development of novel neurotherapeutics. Curr. Opin. Investig. Drugs 2009, 10, 644–654. [Google Scholar] [PubMed]
- Durukan, A.; Tatlisumak, T. Acute ischemic stroke: Overview of major experimental rodent models, pathophysiology, and therapy of focal cerebral ischemia. Pharmacol. Biochem. Behav. 2007, 87, 179–197. [Google Scholar] [CrossRef] [PubMed]
- Lalkovicova, M.; Danielisova, V. Neuroprotection and antioxidants. Neural Regen. Res. 2016, 11, 865–874. [Google Scholar] [CrossRef] [PubMed]
- Helmy, A.; Guilfoyle, M.R.; Carpenter, K.L.; Pickard, J.D.; Menon, D.K.; Hutchinson, P.J. Recombinant human interleukin-1 receptor antagonist in severe traumatic brain injury: A phase II randomized control trial. J. Cereb. Blood Flow Metab. 2014, 34, 845–851. [Google Scholar] [CrossRef] [PubMed]
- Xing, C.; Lo, E.H. Help-me signaling: Non-cell autonomous mechanisms of neuroprotection and neurorecovery. Prog. Neurobiol. 2017, 152, 181–199. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, A.N.; Siddiqui, N.; Khan, R.A.; Kalam, A.; Jabir, N.R.; Kamal, M.A.; Firoz, C.K.; Tabrez, S. Neuroprotective Role of Steroidal Sex Hormones: An Overview. CNS Neurosci. Ther. 2016, 22, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Bel, F.; Groenendaal, F. Drugs for neuroprotection after birth asphyxia: Pharmacologic adjuncts to hypothermia. Semin. Perinatol. 2016, 40, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Gupta, C.K.; Katiyar, G.P.; Gupta, S.P.; Agarwal, K.N.; Sen, P.C. Nitroblue tetrazolium dye test in septic meningitis. Indian J. Med. Res. 1975, 63, 266–272. [Google Scholar] [PubMed]
- Scheibe, F.; Klein, O.; Klose, J.; Priller, J. Mesenchymal stromal cells rescue cortical neurons from apoptotic cell death in an in vitro model of cerebral ischemia. Cell. Mol. Neurobiol. 2012, 32, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Regulski, M.J. Mesenchymal Stem Cells: “Guardians of Inflammation”. Wounds 2017, 29, 20–27. [Google Scholar] [PubMed]
- Sherman, L.S.; Shaker, M.; Mariotti, V.; Rameshwar, P. Mesenchymal stromal/stem cells in drug therapy: New perspective. Cytotherapy 2017, 19, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.L.; Zhang, H.T.; Cai, Y.Q.; Han, Y.J.; Yao, F.; Yuan, Z.H.; Wu, B.Y. Anti-inflammatory Effect of Mesenchymal Stromal Cell Transplantation and Quercetin Treatment in a Rat Model of Experimental Cerebral Ischemia. Cell. Mol. Neurobiol. 2016, 36, 1023–1034. [Google Scholar] [CrossRef] [PubMed]
- Palacios-Pelaez, R.; Lukiw, W.J.; Bazan, N.G. Omega-3 essential fatty acids modulate initiation and progression of neurodegenerative disease. Mol. Neurobiol. 2010, 41, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Walberer, M.; Rueger, M.A. The macrosphere model-an embolic stroke model for studying the pathophysiology of focal cerebral ischemia in a translational approach. Ann. Transl. Med. 2015, 3, 123. [Google Scholar] [CrossRef] [PubMed]
- Hata, R.; Mies, G.; Wiessner, C.; Fritze, K.; Hesselbarth, D.; Brinker, G.; Hossmann, K.A. A reproducible model of middle cerebral artery occlusion in mice: Hemodynamic, biochemical, and magnetic resonance imaging. J. Cereb. Blood Flow Metab. 1998, 18, 367–375. [Google Scholar] [CrossRef] [PubMed]
- Saleh, M.C.; Connell, B.J.; Saleh, T.M. Estrogen may contribute to ischemic tolerance through modulation of cellular stress-related proteins. Neurosci. Res. 2009, 63, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Huang, H.; He, Y.; Ruan, L.; Huang, J. Bumetanide protects focal cerebral ischemia-reperfusion injury in rat. Int. J. Clin. Exp. Pathol. 2014, 7, 1487–1494. [Google Scholar] [PubMed]
- Gerriets, T.; Li, F.; Silva, M.D.; Meng, X.; Brevard, M.; Sotak, C.H.; Fisher, M. The macrosphere model: Evaluation of a new stroke model for permanent middle cerebral artery occlusion in rats. J. Neurosci. Methods 2003, 122, 201–211. [Google Scholar] [CrossRef]
- Busch, E.; Kruger, K.; Hossmann, K.A. Improved model of thromboembolic stroke and rt-PA induced reperfusion in the rat. Brain Res. 1997, 778, 16–24. [Google Scholar] [CrossRef]
- Buchan, A.M.; Xue, D.; Slivka, A. A new model of temporary focal neocortical ischemia in the rat. Stroke 1992, 23, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Tamura, A.; Graham, D.I.; McCulloch, J.; Teasdale, G.M. Focal cerebral ischaemia in the rat: 1. Description of technique and early neuropathological consequences following middle cerebral artery occlusion. J. Cereb. Blood Flow Metab. 1981, 1, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Brint, S.; Jacewicz, M.; Kiessling, M.; Tanabe, J.; Pulsinelli, W. Focal brain ischemia in the rat: Methods for reproducible neocortical infarction using tandem occlusion of the distal middle cerebral and ipsilateral common carotid arteries. J. Cereb. Blood Flow Metab. 1988, 8, 474–485. [Google Scholar] [CrossRef] [PubMed]
- Watson, B.D.; Dietrich, W.D.; Busto, R.; Wachtel, M.S.; Ginsberg, M.D. Induction of reproducible brain infarction by photochemically initiated thrombosis. Ann. Neurol. 1985, 17, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Ibayashi, S.; Sugimori, H.; Fujii, K.; Fujishima, M. Simplified model of krypton laser-induced thrombotic distal middle cerebral artery occlusion in spontaneously hypertensive rats. Stroke 1996, 27, 333–336. [Google Scholar] [CrossRef] [PubMed]
- Flierl, M.A.; Stahel, P.F.; Beauchamp, K.M.; Morgan, S.J.; Smith, W.R.; Shohami, E. Mouse closed head injury model induced by a weight-drop device. Nat. Protoc. 2009, 4, 1328–1337. [Google Scholar] [CrossRef] [PubMed]
- Northington, F.J. Brief update on animal models of hypoxic-ischemic encephalopathy and neonatal stroke. ILAR J. 2006, 47, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Magal, E.; Goldin, E.; Harel, S.; Yavin, E. Acute uteroplacental ischemic embryo: Lactic acid accumulation and prostaglandin production in the fetal rat brain. J. Neurochem. 1988, 51, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Graham, S.M.; McCullough, L.D.; Murphy, S.J. Animal models of ischemic stroke: Balancing experimental aims and animal care. Comp. Med. 2004, 54, 486–496. [Google Scholar] [PubMed]
- Nelson, P.; Ruffner, W.; Nirenberg, M. Neuronal tumor cells with excitable membranes grown in vitro. Proc. Natl. Acad. Sci. USA 1969, 64, 1004–1010. [Google Scholar] [CrossRef] [PubMed]
- Bottenstein, J.E. Proliferation of glioma cells in serum-free defined medium. Cancer Treat. Rep. 1981, 65 (Suppl. S2), 67–70. [Google Scholar] [PubMed]
- Richter-Landsberg, C.; Heinrich, M. OLN-93: A new permanent oligodendroglia cell line derived from primary rat brain glial cultures. J. Neurosci. Res. 1996, 45, 161–173. [Google Scholar] [CrossRef]
- Noraberg, J.; Poulsen, F.R.; Blaabjerg, M.; Kristensen, B.W.; Bonde, C.; Montero, M.; Meyer, M.; Gramsbergen, J.B.; Zimmer, J. Organotypic hippocampal slice cultures for studies of brain damage, neuroprotection and neurorepair. Curr. Drug Targets CNS Neurol. Disord. 2005, 4, 435–452. [Google Scholar] [CrossRef] [PubMed]
- Wohnsland, S.; Burgers, H.F.; Kuschinsky, W.; Maurer, M.H. Neurons and neuronal stem cells survive in glucose-free lactate and in high glucose cell culture medium during normoxia and anoxia. Neurochem. Res. 2010, 35, 1635–1642. [Google Scholar] [CrossRef] [PubMed]
- Nordstrom, T.; Jansson, L.C.; Louhivuori, L.M.; Akerman, K.E. Effects of acute hypoxia/acidosis on intracellular pH in differentiating neural progenitor cells. Brain Res. 2012, 1461, 10–23. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Yang, B.; Mo, X.; Zhou, F. HspB8 mediates neuroprotection against OGD/R in N2A cells through the phosphoinositide 3-kinase/Akt pathway. Brain Res. 2016, 1644, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Mahesh, R.; Jung, H.W.; Han, C.H.; Cho, C.W.; Park, Y.K. Joongpoongtang 05 (JP05) confers neuroprotection via anti-apoptotic activities in Neuro-2a cells during oxygen-glucose deprivation and reperfusion. Toxicol. In Vitro 2011, 25, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Seta, K.A.; Spicer, Z.; Yuan, Y.; Lu, G.; Millhorn, D.E. Responding to hypoxia: Lessons from a model cell line. Sci. STKE 2002, 2002, re11. [Google Scholar] [CrossRef] [PubMed]
- Tabakman, R.; Jiang, H.; Shahar, I.; Arien-Zakay, H.; Levine, R.A.; Lazarovici, P. Neuroprotection by NGF in the PC12 in vitro OGD model: Involvement of mitogen-activated protein kinases and gene expression. Ann. N. Y. Acad. Sci. 2005, 1053, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, M.P.; Choi, D.W. Combined oxygen and glucose deprivation in cortical cell culture: Calcium-dependent and calcium-independent mechanisms of neuronal injury. J. Neurosci. 1993, 13, 3510–3524. [Google Scholar] [PubMed]
- Zhang, L.M.; Zhao, X.C.; Sun, W.B.; Li, R.; Jiang, X.J. Sevoflurane post-conditioning protects primary rat cortical neurons against oxygen-glucose deprivation/resuscitation via down-regulation in mitochondrial apoptosis axis of Bid, Bim, Puma-Bax and Bak mediated by ERK1/2. J. Neurol. Sci. 2015, 357, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Andreev, D.E.; O’Connor, P.B.; Zhdanov, A.V.; Dmitriev, R.I.; Shatsky, I.N.; Papkovsky, D.B.; Baranov, P.V. Oxygen and glucose deprivation induces widespread alterations in mRNA translation within 20 minutes. Genome Biol. 2015, 16, 90. [Google Scholar] [CrossRef] [PubMed]
- Pichiule, P.; Chavez, J.C.; Schmidt, A.M.; Vannucci, S.J. Hypoxia-inducible factor-1 mediates neuronal expression of the receptor for advanced glycation end products following hypoxia/ischemia. J. Biol. Chem. 2007, 282, 36330–36340. [Google Scholar] [CrossRef] [PubMed]
- Raya, S.A.; Trembovler, V.; Shohami, E.; Lazarovici, P. A tissue culture ischemic device to study eicosanoid release by pheochromocytoma PC12 cultures. J. Neurosci. Methods 1993, 50, 197–203. [Google Scholar] [CrossRef]
- Tabakman, R.; Lazarovici, P.; Kohen, R. Neuroprotective effects of carnosine and homocarnosine on pheochromocytoma PC12 cells exposed to ischemia. J. Neurosci. Res. 2002, 68, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Hollig, A.; Schug, A.; Fahlenkamp, A.V.; Rossaint, R.; Coburn, M.; Organo-Protective, N.A. Argon: Systematic review on neuro- and organoprotective properties of an “inert” gas. Int. J. Mol. Sci. 2014, 15, 18175–18196. [Google Scholar] [CrossRef] [PubMed]
- Tasca, C.I.; Dal-Cim, T.; Cimarosti, H. In vitro oxygen-glucose deprivation to study ischemic cell death. Methods Mol. Biol. 2015, 1254, 197–210. [Google Scholar] [PubMed]
- Cummins, T.R.; Agulian, S.K.; Haddad, G.G. Oxygen tension clamp around single neurons in vitro: A computerized method for studies on O2 deprivation. J. Neurosci. Methods 1993, 46, 183–189. [Google Scholar] [CrossRef]
- Goldberg, W.J.; Kadingo, R.M.; Barrett, J.N. Effects of ischemia-like conditions on cultured neurons: Protection by low Na+, low Ca2+ solutions. J. Neurosci. 1986, 6, 3144–3151. [Google Scholar] [PubMed]
- Lahiani, A.; Hidmi, A.; Katzhendler, J.; Yavin, E.; Lazarovici, P. Novel Synthetic PEGylated Conjugate of alpha-Lipoic Acid and Tempol Reduces Cell Death in a Neuronal PC12 Clonal Line Subjected to Ischemia. ACS Chem. Neurosci. 2016, 7, 1452–1462. [Google Scholar] [CrossRef] [PubMed]
- Lahiani, A.; Zahavi, E.; Netzer, N.; Ofir, R.; Pinzur, L.; Raveh, S.; Arien-Zakay, H.; Yavin, E.; Lazarovici, P. Human placental eXpanded (PLX) mesenchymal-like adherent stromal cells confer neuroprotection to nerve growth factor (NGF)-differentiated PC12 cells exposed to ischemia by secretion of IL-6 and VEGF. Biochim. Biophys. Acta 2015, 1853, 422–430. [Google Scholar] [CrossRef] [PubMed]
- Lecht, S.; Rotfeld, E.; Arien-Zakay, H.; Tabakman, R.; Matzner, H.; Yaka, R.; Lelkes, P.I.; Lazarovici, P. Neuroprotective effects of nimodipine and nifedipine in the NGF-differentiated PC12 cells exposed to oxygen-glucose deprivation or trophic withdrawal. Int. J. Dev. Neurosci. 2012, 30, 465–469. [Google Scholar] [CrossRef] [PubMed]
- Smirnov, V.V.; Mishenkova, E.L.; Baraboi, V.A.; Petrenko, G.T.; Volynets, N.N. Antioxidant effect of the antineoplastic antibiotic obtained from a plant of the family Asteraceae. Ukr. Biokhimicheskii Zhurnal 1989, 61, 117–119. [Google Scholar]
- Arien-Zakay, H.; Lecht, S.; Bercu, M.M.; Tabakman, R.; Kohen, R.; Galski, H.; Nagler, A.; Lazarovici, P. Neuroprotection by cord blood neural progenitors involves antioxidants, neurotrophic and angiogenic factors. Exp. Neurol. 2009, 216, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Greene, L.A.; Tischler, A.S. Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc. Natl. Acad. Sci. USA 1976, 73, 2424–2428. [Google Scholar] [CrossRef] [PubMed]
- Rudkin, B.B.; Lazarovici, P.; Levi, B.Z.; Abe, Y.; Fujita, K.; Guroff, G. Cell cycle-specific action of nerve growth factor in PC12 cells: Differentiation without proliferation. EMBO J. 1989, 8, 3319–3325. [Google Scholar] [PubMed]
- Greene, L.A.; Rein, G. Release, storage and uptake of catecholamines by a clonal cell line of nerve growth factor (NGF) responsive pheo-chromocytoma cells. Brain Res. 1977, 129, 247–263. [Google Scholar] [CrossRef]
- Martin, T.F.; Grishanin, R.N. PC12 cells as a model for studies of regulated secretion in neuronal and endocrine cells. Methods Cell Biol. 2003, 71, 267–286. [Google Scholar] [PubMed]
- Anderson, D.J. Molecular control of cell fate in the neural crest: The sympathoadrenal lineage. Ann. Rev. Neurosci. 1993, 16, 129–158. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K.; Lazarovici, P.; Guroff, G. Regulation of the differentiation of PC12 pheochromocytoma cells. Environ. Health Perspect. 1989, 80, 127–142. [Google Scholar] [CrossRef] [PubMed]
- Vaudry, D.; Stork, P.J.; Lazarovici, P.; Eiden, L.E. Signaling pathways for PC12 cell differentiation: Making the right connections. Science 2002, 296, 1648–1649. [Google Scholar] [CrossRef] [PubMed]
- Kritis, A.A.; Stamoula, E.G.; Paniskaki, K.A.; Vavilis, T.D. Researching glutamate-induced cytotoxicity in different cell lines: A comparative/collective analysis/study. Front. Cell. Neurosci. 2015, 9, 91. [Google Scholar] [CrossRef] [PubMed]
- Shimoke, K.; Chiba, H. Nerve growth factor prevents 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced cell death via the Akt pathway by suppressing caspase-3-like activity using PC12 cells: Relevance to therapeutical application for Parkinson’s disease. J. Neurosci. Res. 2001, 63, 402–409. [Google Scholar] [CrossRef]
- Troy, C.M.; Rabacchi, S.A.; Xu, Z.; Maroney, A.C.; Connors, T.J.; Shelanski, M.L.; Greene, L.A. beta-Amyloid-induced neuronal apoptosis requires c-Jun N-terminal kinase activation. J. Neurochem. 2001, 77, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Menei, P.; Pean, J.M.; Nerriere-Daguin, V.; Jollivet, C.; Brachet, P.; Benoit, J.P. Intracerebral implantation of NGF-releasing biodegradable microspheres protects striatum against excitotoxic damage. Exp. Neurol. 2000, 161, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Pan, Z.; Sampath, D.; Jackson, G.; Werrbach-Perez, K.; Perez-Polo, R. Nerve growth factor and oxidative stress in the nervous system. Adv. Exp. Med. Biol. 1997, 429, 173–193. [Google Scholar] [PubMed]
- Rukenstein, A.; Rydel, R.E.; Greene, L.A. Multiple agents rescue PC12 cells from serum-free cell death by translation- and transcription-independent mechanisms. J. Neurosci. 1991, 11, 2552–2563. [Google Scholar] [PubMed]
- Batistatou, A.; Greene, L.A. Aurintricarboxylic acid rescues PC12 cells and sympathetic neurons from cell death caused by nerve growth factor deprivation: Correlation with suppression of endonuclease activity. J. Cell Biol. 1991, 115, 461–471. [Google Scholar] [CrossRef] [PubMed]
- Fischer, S.J.; Podratz, J.L.; Windebank, A.J. Nerve growth factor rescue of cisplatin neurotoxicity is mediated through the high affinity receptor: Studies in PC12 cells and p75 null mouse dorsal root ganglia. Neurosci. Lett. 2001, 308, 1–4. [Google Scholar] [CrossRef]
- Tabakman, R.; Jiang, H.; Levine, R.A.; Kohen, R.; Lazarovici, P. Apoptotic characteristics of cell death and the neuroprotective effect of homocarnosine on pheochromocytoma PC12 cells exposed to ischemia. J. Neurosci. Res. 2004, 75, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Hanefeld, M.; Naumann, H.J.; Stotzner, H.; Haller, H. Studies on the influence of body weight and form of therapy on the development of fatty liver in maturity-onset diabetes. Z. Gesamte Inn. Med. Grenzgeb. 1975, 30, 59–60. [Google Scholar]
- Abu-Raya, S.; Blaugrund, E.; Trembovler, V.; Shilderman-Bloch, E.; Shohami, E.; Lazarovici, P. Rasagiline, a monoamine oxidase-B inhibitor, protects NGF-differentiated PC12 cells against oxygen-glucose deprivation. J. Neurosci. Res. 1999, 58, 456–463. [Google Scholar] [CrossRef]
- Tabakman, R.; Jiang, H.; Schaefer, E.; Levine, R.A.; Lazarovici, P. Nerve growth factor pretreatment attenuates oxygen and glucose deprivation-induced c-Jun amino-terminal kinase 1 and stress-activated kinases p38α and p38β activation and confers neuroprotection in the pheochromocytoma PC12 Model. J. Mol. Neurosci. 2004, 22, 237–250. [Google Scholar] [CrossRef]
- Xiong, S.; Xu, Y.; Ma, M.; Wang, H.; Wei, F.; Gu, Q.; Xu, X. Neuroprotective effects of a novel peptide, FK18, under oxygen-glucose deprivation in SH-SY5Y cells and retinal ischemia in rats via the Akt pathway. Neurochem. Int. 2017, 108, 78–90. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Zheng, S.; Leng, J.; Wang, S.; Zhao, T.; Liu, J. Inhibition of mitochondrial calcium uniporter protects neurocytes from ischemia/reperfusion injury via the inhibition of excessive mitophagy. Neurosci. Lett. 2016, 628, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.B.; Ju, X.N.; Wang, X.Y.; Wang, M.H.; Kong, F.; Sun, C.; Bi, J.Z. Pretreatment with baicalin attenuates hypoxia and glucose deprivation-induced injury in SH-SY5Y cells. Chin. J. Integr. Med. 2016, 22, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.R.; Hu, L.S.; Li, G.Y. SH-SY5Y human neuroblastoma cell line: In vitro cell model of dopaminergic neurons in Parkinson’s disease. Chin. Med. J. 2010, 123, 1086–1092. [Google Scholar] [PubMed]
- Tremblay, R.G.; Sikorska, M.; Sandhu, J.K.; Lanthier, P.; Ribecco-Lutkiewicz, M.; Bani-Yaghoub, M. Differentiation of mouse Neuro 2A cells into dopamine neurons. J. Neurosci. Methods 2010, 186, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Shipley, M.M.; Mangold, C.A.; Szpara, M.L. Differentiation of the SH-SY5Y Human Neuroblastoma Cell Line. J. Vis. Exp. 2016, 108, 53193. [Google Scholar] [CrossRef] [PubMed]
- Jensen, L.M. Phenotypic differentiation of aphidicolin-selected human neuroblastoma cultures after long-term exposure to nerve growth factor. Dev. Biol. 1987, 120, 56–64. [Google Scholar] [CrossRef]
- Algarni, A.S.; Hargreaves, A.J.; Dickenson, J.M. Activation of transglutaminase 2 by nerve growth factor in differentiating neuroblastoma cells: A role in cell survival and neurite outgrowth. Eur. J. Pharmacol. 2018, 820, 113–129. [Google Scholar] [CrossRef] [PubMed]
- Ross, R.A.; Spengler, B.A.; Biedler, J.L. Coordinate morphological and biochemical interconversion of human neuroblastoma cells. J. Natl. Cancer Inst. 1983, 71, 741–747. [Google Scholar] [PubMed]
- Kovalevich, J.; Langford, D. Considerations for the use of SH-SY5Y neuroblastoma cells in neurobiology. Methods Mol. Biol. 2013, 1078, 9–21. [Google Scholar] [PubMed]
- Heusinkveld, H.J.; Westerink, R.H.S. Comparison of different in vitro cell models for the assessment of pesticide-induced dopaminergic neurotoxicity. Toxicol. In Vitro 2017, 45 Pt 1, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Popova, D.; Karlsson, J.; Jacobsson, S.O.P. Comparison of neurons derived from mouse P19, rat PC12 and human SH-SY5Y cells in the assessment of chemical- and toxin-induced neurotoxicity. BMC Pharmacol. Toxicol. 2017, 18, 42. [Google Scholar] [CrossRef] [PubMed]
- Rahbar-Roshandel, N.; Razavi, L.; Tavakoli-Far, B.; Mahmoudian, M. Mebudipine and dibudipine protect PC12 cells against oxygen-glucose deprivation and glutamate-induced cell death. Pathophysiology 2008, 15, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Bai, Y.; Zhang, C.; Zhang, X.; Zhang, Y.X.; Chen, J.; Xiong, L.; Shi, M.; Zhao, G. Cinepazide maleate protects PC12 cells against oxygen-glucose deprivation-induced injury. Neurol. Sci. 2014, 35, 875–881. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.G.; Lu, T.S.; Yuan, H.Y. Neuroprotective effects of ultra-low-molecular-weight heparin in vitro and vivo models of ischemic injury. Fundam. Clin. Pharmacol. 2011, 25, 300–303. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.J.; Wang, D.M.; Hu, J.F.; Sun, M.N.; Li, G.; Li, Z.P.; Wu, D.H.; Liu, G.; Chen, N.H. IMM-H004, a novel courmarin derivative, protects against oxygen-and glucose-deprivation/restoration-induced apoptosis in PC12 cells. Eur. J. Pharmacol. 2014, 723, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.M.; Yu, J.Y.; Ou, H.C.; Jeng, K.C. Effect of naloxone on the induction of immediately early genes following oxygen- and glucose-deprivation in PC12 cells. Neurosci. Lett. 2008, 438, 252–256. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, K.; Kawahara, K.; Biswas, K.K.; Ito, T.; Tancharoen, S.; Morimoto, Y.; Matsuda, F.; Oyama, Y.; Takenouchi, K.; Miura, N.; et al. Minocycline attenuates both OGD-induced HMGB1 release and HMGB1-induced cell death in ischemic neuronal injury in PC12 cells. Biochem. Biophys. Res. Commun. 2009, 385, 132–136. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.Y.; Zhou, X.Y.; Hou, J.C.; Zhu, H.; Wang, Z.; Liu, J.X.; Zheng, Y.Q. Ginsenoside Rd promotes neurogenesis in rat brain after transient focal cerebral ischemia via activation of PI3K/Akt pathway. Acta Pharmacol. Sin. 2015, 36, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Tao, X.; Sun, X.; Yin, L.; Han, X.; Xu, L.; Qi, Y.; Xu, Y.; Li, H.; Lin, Y.; Liu, K.; et al. Dioscin ameliorates cerebral ischemia/reperfusion injury through the downregulation of TLR4 signaling via HMGB-1 inhibition. Free Radic. Biol. Med. 2015, 84, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Fu, Y.; Tang, X.C. Huperzine A protects rat pheochromocytoma cells against oxygen-glucose deprivation. Neuroreport 2001, 12, 2073–2077. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Fu, Y.; Tang, X.C. Huperzine A and donepezil protect rat pheochromocytoma cells against oxygen-glucose deprivation. Neurosci. Lett. 2001, 306, 53–56. [Google Scholar] [CrossRef]
- Liu, X.; Zhu, X.; Chen, M.; Ge, Q.; Shen, Y.; Pan, S. Resveratrol protects PC12 cells against OGD/R-induced apoptosis via the mitochondrial-mediated signaling pathway. Acta Biochim. Biophys. Sin. 2016, 48, 342–353. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, M.; Kumar, V.; Kashyap, M.P.; Khanna, V.K.; Randhawa, G.S.; Pant, A.B. Ischemic insult induced apoptotic changes in PC12 cells: Protection by trans resveratrol. Eur. J. Pharmacol. 2011, 666, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, M.; Kumar, V.; Singh, A.K.; Kashyap, M.P.; Khanna, V.K.; Siddiqui, M.A.; Pant, A.B. trans-Resveratrol protects ischemic PC12 Cells by inhibiting the hypoxia associated transcription factors and increasing the levels of antioxidant defense enzymes. ACS Chem. Neurosci. 2013, 4, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Chang, R.; Zhou, R.; Qi, X.; Wang, J.; Wu, F.; Yang, W.; Zhang, W.; Sun, T.; Li, Y.; Yu, J. Protective effects of aloin on oxygen and glucose deprivation-induced injury in PC12 cells. Brain Res. Bull. 2016, 121, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.L.; Hsia, T.C.; Yin, M.C. s-Methyl cysteine enhanced survival of nerve growth factor differentiated PC12 cells under hypoxic conditions. Food Funct. 2014, 5, 1125–1133. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Zhang, H.; Zhang, Z.; Wang, T.; Niu, J.; Cui, D.; Xu, S. Neuroprotective effects of pre-treatment with l-carnitine and acetyl-l-carnitine on ischemic injury in vivo and in vitro. Int. J. Mol. Sci. 2012, 13, 2078–2090. [Google Scholar] [CrossRef] [PubMed]
- Sawe, N.; Steinberg, G.; Zhao, H. Dual roles of the MAPK/ERK1/2 cell signaling pathway after stroke. J. Neurosci. Res. 2008, 86, 1659–1669. [Google Scholar] [CrossRef] [PubMed]
- Nijboer, C.H.; van der Kooij, M.A.; van Bel, F.; Ohl, F.; Heijnen, C.J.; Kavelaars, A. Inhibition of the JNK/AP-1 pathway reduces neuronal death and improves behavioral outcome after neonatal hypoxic-ischemic brain injury. Brain Behav. Immun. 2010, 24, 812–821. [Google Scholar] [CrossRef] [PubMed]
- Barone, F.C.; Irving, E.A.; Ray, A.M.; Lee, J.C.; Kassis, S.; Kumar, S.; Badger, A.M.; Legos, J.J.; Erhardt, J.A.; Ohlstein, E.H.; et al. Inhibition of p38 mitogen-activated protein kinase provides neuroprotection in cerebral focal ischemia. Med. Res. Rev. 2001, 21, 129–145. [Google Scholar] [CrossRef]
- De Almeida, L.M.; Leite, M.C.; Thomazi, A.P.; Battu, C.; Nardin, P.; Tortorelli, L.S.; Zanotto, C.; Posser, T.; Wofchuk, S.T.; Leal, R.B.; et al. Resveratrol protects against oxidative injury induced by H2O2 in acute hippocampal slice preparations from Wistar rats. Arch. Biochem. Biophys. 2008, 480, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Xue, L.X.; Xu, Z.H.; Wang, J.Q.; Cui, Y.; Liu, H.Y.; Liang, W.Z.; Ji, Q.Y.; He, J.T.; Shao, Y.K.; Mang, J.; et al. Activin A/Smads signaling pathway negatively regulates Oxygen Glucose Deprivation-induced autophagy via suppression of JNK and p38 MAPK pathways in neuronal PC12 cells. Biochem. Biophys. Res. Commun. 2016, 480, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Lin, F.; Zhu, P.; Wu, C.; Zhao, S.; Han, Q.; Li, X. Extract of Spatholobus suberctus Dunn ameliorates ischemia-induced injury by targeting miR-494. PLoS ONE 2017, 12, e0184348. [Google Scholar] [CrossRef] [PubMed]
- Connell, B.J.; Saleh, M.; Khan, B.V.; Saleh, T.M. Lipoic acid protects against reperfusion injury in the early stages of cerebral ischemia. Brain Res. 2011, 1375, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Mitsui, Y.; Schmelzer, J.D.; Zollman, P.J.; Mitsui, M.; Tritschler, H.J.; Low, P.A. Alpha-lipoic acid provides neuroprotection from ischemia-reperfusion injury of peripheral nerve. J. Neurol. Sci. 1999, 163, 11–16. [Google Scholar] [CrossRef]
- Zhang, L.; Xing, G.Q.; Barker, J.L.; Chang, Y.; Maric, D.; Ma, W.; Li, B.S.; Rubinow, D.R. Alpha-lipoic acid protects rat cortical neurons against cell death induced by amyloid and hydrogen peroxide through the Akt signalling pathway. Neurosci. Lett. 2001, 312, 125–128. [Google Scholar] [CrossRef]
- Jalali-Nadoushan, M.; Roghani, M. Alpha-lipoic acid protects against 6-hydroxydopamine-induced neurotoxicity in a rat model of hemi-parkinsonism. Brain Res. 2013, 1505, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, D.; Reljanovic, M.; Mehnert, H.; Gries, F.A. Alpha-lipoic acid in the treatment of diabetic polyneuropathy in Germany: Current evidence from clinical trials. Exp. Clin. Endocrinol. Diabetes 1999, 107, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Ankel, E.G.; Lai, C.S.; Hopwood, L.E.; Zivkovic, Z. Cytotoxicity of commonly used nitroxide radical spin probes. Life Sci. 1987, 40, 495–498. [Google Scholar] [CrossRef]
- Berliner, J.L.; Fujii, H. Magnetic resonance imaging of biological specimens by electron paramagnetic resonance of nitroxide spin labels. Science 1985, 227, 517–519. [Google Scholar] [CrossRef] [PubMed]
- Krishna, M.C.; Russo, A.; Mitchell, J.B.; Goldstein, S.; Dafni, H.; Samuni, A. Do nitroxide antioxidants act as scavengers of O2 or as SOD mimics? J. Biol. Chem. 1996, 271, 26026–26031. [Google Scholar] [CrossRef] [PubMed]
- Marciniak, A.; Walczyna, B.; Rajtar, G.; Marciniak, S.; Wojtak, A.; Lasiecka, K. Tempol, a Membrane-Permeable Radical Scavenger, Exhibits Anti-Inflammatory and Cardioprotective Effects in the Cerulein-Induced Pancreatitis Rat Model. Oxidative Med. Cell. Longev. 2016, 2016, 4139851. [Google Scholar] [CrossRef] [PubMed]
- Lipman, T.; Tabakman, R.; Lazarovici, P. Neuroprotective effects of the stable nitroxide compound Tempol on 1-methyl-4-phenylpyridinium ion-induced neurotoxicity in the Nerve Growth Factor-differentiated model of pheochromocytoma PC12 cells. Eur. J. Pharmacol. 2006, 549, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Numa, R.; Baron, M.; Kohen, R.; Yaka, R. Tempol attenuates cocaine-induced death of PC12 cells through decreased oxidative damage. Eur. J. Pharmacol. 2011, 650, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Deng-Bryant, Y.; Singh, I.N.; Carrico, K.M.; Hall, E.D. Neuroprotective effects of tempol, a catalytic scavenger of peroxynitrite-derived free radicals, in a mouse traumatic brain injury model. J. Cereb. Blood Flow Metab. 2008, 28, 1114–1126. [Google Scholar] [CrossRef] [PubMed]
- Rak, R.; Chao, D.L.; Pluta, R.M.; Mitchell, J.B.; Oldfield, E.H.; Watson, J.C. Neuroprotection by the stable nitroxide Tempol during reperfusion in a rat model of transient focal ischemia. J. Neurosurg. 2000, 92, 646–651. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, C.S. Effects of tempol and redox-cycling nitroxides in models of oxidative stress. Pharmacol. Ther. 2010, 126, 119–145. [Google Scholar] [CrossRef] [PubMed]
- Son, Y.; Kim, S.; Chung, H.T.; Pae, H.O. Reactive oxygen species in the activation of MAP kinases. Methods Enzymol. 2013, 528, 27–48. [Google Scholar] [PubMed]
- Pinnen, F.; Cacciatore, I.; Cornacchia, C.; Sozio, P.; Iannitelli, A.; Costa, M.; Pecci, L.; Nasuti, C.; Cantalamessa, F.; Di Stefano, A. Synthesis and study of l-dopa-glutathione codrugs as new anti-Parkinson agents with free radical scavenging properties. J. Med. Chem. 2007, 50, 2506–2515. [Google Scholar] [CrossRef] [PubMed]
- Sozio, P.; Iannitelli, A.; Cerasa, L.S.; Cacciatore, I.; Cornacchia, C.; Giorgioni, G.; Ricciutelli, M.; Nasuti, C.; Cantalamessa, F.; Di Stefano, A. New L-dopa codrugs as potential antiparkinson agents. Arch. Pharm. 2008, 341, 412–417. [Google Scholar] [CrossRef] [PubMed]
- Sozio, P.; D’Aurizio, E.; Iannitelli, A.; Cataldi, A.; Zara, S.; Cantalamessa, F.; Nasuti, C.; Di Stefano, A. Ibuprofen and lipoic acid diamides as potential codrugs with neuroprotective activity. Arch. Pharm. 2010, 343, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Flores-Santana, W.; Moody, T.; Chen, W.; Gorczynski, M.J.; Shoman, M.E.; Velazquez, C.; Thetford, A.; Mitchell, J.B.; Cherukuri, M.K.; King, S.B.; et al. Nitroxide derivatives of non-steroidal anti-inflammatory drugs exert anti-inflammatory and superoxide dismutase scavenging properties in A459 cells. Br. J. Pharmacol. 2012, 165, 1058–1067. [Google Scholar] [CrossRef] [PubMed]
- Eridani, S.; Sgaramella, V.; Cova, L. Stem cells: From embryology to cellular therapy? An appraisal of the present state of art. Cytotechnology 2004, 44, 125–141. [Google Scholar] [PubMed]
- Phillips, A.W.; Johnston, M.V.; Fatemi, A. The potential for cell-based therapy in perinatal brain injuries. Transl. Stroke Res. 2013, 4, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Daley, G.Q.; Goodell, M.A.; Snyder, E.Y. Realistic prospects for stem cell therapeutics. ASH Educ. Program Book 2003, 2003, 398–418. [Google Scholar] [CrossRef]
- Keller, G. Embryonic stem cell differentiation: Emergence of a new era in biology and medicine. Genes Dev. 2005, 19, 1129–1155. [Google Scholar] [CrossRef] [PubMed]
- Delcroix, G.J.; Schiller, P.C.; Benoit, J.P.; Montero-Menei, C.N. Adult cell therapy for brain neuronal damages and the role of tissue engineering. Biomater. 2010, 31, 2105–2120. [Google Scholar] [CrossRef] [PubMed]
- Blum, B.; Benvenisty, N. The tumorigenicity of human embryonic stem cells. Adv. Cancer Res. 2008, 100, 133–158. [Google Scholar] [PubMed]
- Bacigaluppi, M.; Pluchino, S.; Peruzzotti-Jametti, L.; Kilic, E.; Kilic, U.; Salani, G.; Brambilla, E.; West, M.J.; Comi, G.; Martino, G.; et al. Delayed post-ischaemic neuroprotection following systemic neural stem cell transplantation involves multiple mechanisms. Brain 2009, 132 Pt 8, 2239–2251. [Google Scholar] [CrossRef] [PubMed]
- Uccelli, A.; Laroni, A.; Freedman, M.S. Mesenchymal stem cells for the treatment of multiple sclerosis and other neurological diseases. Lancet Neurol. 2011, 10, 649–656. [Google Scholar] [CrossRef]
- Drago, D.; Cossetti, C.; Iraci, N.; Gaude, E.; Musco, G.; Bachi, A.; Pluchino, S. The stem cell secretome and its role in brain repair. Biochimie 2013, 95, 2271–2285. [Google Scholar] [CrossRef] [PubMed]
- Haas, S.; Weidner, N.; Winkler, J. Adult stem cell therapy in stroke. Curr. Opin. Neurol. 2005, 18, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Ji, K.; Guo, L.; Wu, W.; Lu, H.; Shan, P.; Yan, C. Mesenchymal stem cells rescue injured endothelial cells in an in vitro ischemia-reperfusion model via tunneling nanotube like structure-mediated mitochondrial transfer. Microvasc. Res. 2014, 92, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Hess, D.C.; Borlongan, C.V. Stem cells and neurological diseases. Cell Prolif. 2008, 41 (Suppl. S1), 94–114. [Google Scholar] [CrossRef] [PubMed]
- Ferri, A.L.M.; Bersano, A.; Lisini, D.; Boncoraglio, G.; Frigerio, S.; Parati, E. Mesenchymal Stem Cells for Ischemic Stroke: Progress and Possibilities. Curr. Med. Chem. 2016, 23, 1598–1608. [Google Scholar] [CrossRef]
- Keating, A. Mesenchymal stromal cells. Curr. Opin. Hematol. 2006, 13, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Ramos, J.; Song, S.; Cardozo-Pelaez, F.; Hazzi, C.; Stedeford, T.; Willing, A.; Freeman, T.B.; Saporta, S.; Janssen, W.; Patel, N.; et al. Adult bone marrow stromal cells differentiate into neural cells in vitro. Exp. Neurol. 2000, 164, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Caplan, A.I. Mesenchymal stem cells. J. Orthop. Res. 1991, 9, 641–650. [Google Scholar] [CrossRef] [PubMed]
- Rowart, P.; Erpicum, P.; Detry, O.; Weekers, L.; Gregoire, C.; Lechanteur, C.; Briquet, A.; Beguin, Y.; Krzesinski, J.M.; Jouret, F. Mesenchymal Stromal Cell Therapy in Ischemia/Reperfusion Injury. J. Immunol. Res. 2015, 2015, 602597. [Google Scholar] [CrossRef] [PubMed]
- Erpicum, P.; Detry, O.; Weekers, L.; Bonvoisin, C.; Lechanteur, C.; Briquet, A.; Beguin, Y.; Krzesinski, J.M.; Jouret, F. Mesenchymal stromal cell therapy in conditions of renal ischaemia/reperfusion. Nephrol. Dial. Transplant. 2014, 29, 1487–1493. [Google Scholar] [CrossRef] [PubMed]
- Kranz, A.; Wagner, D.C.; Kamprad, M.; Scholz, M.; Schmidt, U.R.; Nitzsche, F.; Aberman, Z.; Emmrich, F.; Riegelsberger, U.M.; Boltze, J. Transplantation of placenta-derived mesenchymal stromal cells upon experimental stroke in rats. Brain Res. 2010, 1315, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Prather, W.R.; Toren, A.; Meiron, M.; Ofir, R.; Tschope, C.; Horwitz, E.M. The role of placental-derived adherent stromal cell (PLX-PAD) in the treatment of critical limb ischemia. Cytotherapy 2009, 11, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Acosta, S.A.; Tajiri, N.; Shinozuka, K.; Ishikawa, H.; Sanberg, P.R.; Sanchez-Ramos, J.; Song, S.; Kaneko, Y.; Borlongan, C.V. Combination therapy of human umbilical cord blood cells and granulocyte colony stimulating factor reduces histopathological and motor impairments in an experimental model of chronic traumatic brain injury. PLoS ONE 2014, 9, e90953. [Google Scholar] [CrossRef] [PubMed]
- Pena, I.D.; Sanberg, P.R.; Acosta, S.; Tajiri, N.; Lin, S.Z.; Borlongan, C.V. Stem cells and G-CSF for treating neuroinflammation in traumatic brain injury: Aging as a comorbidity factor. J. Neurosurg. Sci. 2014, 58, 145–149. [Google Scholar]
- Tajiri, N.; Acosta, S.A.; Shahaduzzaman, M.; Ishikawa, H.; Shinozuka, K.; Pabon, M.; Hernandez-Ontiveros, D.; Kim, D.W.; Metcalf, C.; Staples, M.; et al. Intravenous transplants of human adipose-derived stem cell protect the brain from traumatic brain injury-induced neurodegeneration and motor and cognitive impairments: Cell graft biodistribution and soluble factors in young and aged rats. J. Neurosci. 2014, 34, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Kunioku, H.; Inoue, K.; Tomida, M. Interleukin-6 protects rat PC12 cells from serum deprivation or chemotherapeutic agents through the phosphatidylinositol 3-kinase and STAT3 pathways. Neurosci. Lett. 2001, 309, 13–16. [Google Scholar] [CrossRef]
- Nakajima, A.; Yamada, K.; Zou, L.B.; Yan, Y.; Mizuno, M.; Nabeshima, T. Interleukin-6 protects PC12 cells from 4-hydroxynonenal-induced cytotoxicity by increasing intracellular glutathione levels. Free Radic. Biol. Med. 2002, 32, 1324–1332. [Google Scholar] [CrossRef]
- Yamada, K.; Umegaki, H.; Maezawa, I.; Iguchi, A.; Kameyama, T.; Nabeshima, T. Possible involvement of catalase in the protective effect of interleukin-6 against 6-hydroxydopamine toxicity in PC12 cells. Brain Res. Bull. 1997, 43, 573–577. [Google Scholar] [CrossRef]
- Shimma, N.; Akiyama, N.; Umezawa, M.; Okuma, Y.; Nomura, Y.; Saito, T.; Horie, S.; Murayama, T. Possible role of interleukin-6 in PC12 cell death induced by MPP+ and tetrahydroisoquinoline. J. Pharmacol. Sci. 2003, 93, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Liu, Y.; Tang, P.; Liu, P.; Hou, C.; Zhang, X.; Chen, L.; Zhang, L.; Gu, C. Hydrogen sulfide prevents OGD/R-induced apoptosis by suppressing the phosphorylation of p38 and secretion of IL-6 in PC12 cells. Neuroreport 2016, 27, 230–234. [Google Scholar] [CrossRef] [PubMed]
- Lazarovici, P.; Marcinkiewicz, C.; Lelkes, P.I. Cross talk between the cardiovascular and nervous systems: Neurotrophic effects of vascular endothelial growth factor (VEGF) and angiogenic effects of nerve growth factor (NGF)-implications in drug development. Curr. Pharm. Des. 2006, 12, 2609–2622. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.P.; Kwon, B.O.; Gho, Y.S.; Chae, C.B. Specific interaction of VEGF165 with beta-amyloid, and its protective effect on beta-amyloid-induced neurotoxicity. J. Neurochem. 2005, 93, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Lu, C.; Han, Q.; Li, J.; Du, Z.; Liao, L.; Zhao, R.C. Adipose-derived mesenchymal stem cells protect PC12 cells from glutamate excitotoxicity-induced apoptosis by upregulation of XIAP through PI3-K/Akt activation. Toxicology 2011, 279, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Q.; Zhou, Y.; Ye, W.; Cai, T.; Zhang, X.; Deng, D.Y. Hypoxia-inducible factor 1-alpha-AA-modified bone marrow stem cells protect PC12 cells from hypoxia-induced apoptosis, partially through VEGF/PI3K/Akt/FoxO1 pathway. Stem Cells Dev. 2012, 21, 2703–2717. [Google Scholar] [CrossRef] [PubMed]
- Borlongan, C.V.; Kaneko, Y.; Maki, M.; Yu, S.J.; Ali, M.; Allickson, J.G.; Sanberg, C.D.; Kuzmin-Nichols, N.; Sanberg, P.R. Menstrual blood cells display stem cell-like phenotypic markers and exert neuroprotection following transplantation in experimental stroke. Stem Cells Dev. 2010, 19, 439–452. [Google Scholar] [CrossRef] [PubMed]
- Nishijima, K.; Ng, Y.S.; Zhong, L.; Bradley, J.; Schubert, W.; Jo, N.; Akita, J.; Samuelsson, S.J.; Robinson, G.S.; Adamis, A.P.; et al. Vascular endothelial growth factor-A is a survival factor for retinal neurons and a critical neuroprotectant during the adaptive response to ischemic injury. Am. J. Pathol. 2007, 171, 53–67. [Google Scholar] [CrossRef] [PubMed]
- Emerich, D.F.; Silva, E.; Ali, O.; Mooney, D.; Bell, W.; Yu, S.J.; Kaneko, Y.; Borlongan, C. Injectable VEGF hydrogels produce near complete neurological and anatomical protection following cerebral ischemia in rats. Cell Transplant. 2010, 19, 1063–1071. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, K.; Nagai, A.; Sheikh, A.M.; Shiota, Y.; Narantuya, D.; Watanabe, T.; Masuda, J.; Kobayashi, S.; Kim, S.U.; Yamaguchi, S. Transplantation of human mesenchymal stem cells promotes functional improvement and increased expression of neurotrophic factors in a rat focal cerebral ischemia model. J. Neurosci. Res. 2010, 88, 1017–1025. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.S.; Jeong, S.Y.; Yang, J.; Kim, S.D.; Zhang, B.; Yoo, H.S.; Song, S.U.; Jeon, M.S.; Song, Y.S. Neuroprotective effect of mesenchymal stem cell through complement component 3 downregulation after transient focal cerebral ischemia in mice. Neurosci. Lett. 2016, 633, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Gebhart, N.; Richelson, E.; Brott, T.G.; Meschia, J.F.; Zubair, A.C. Mechanism of mesenchymal stem cell-induced neuron recovery and anti-inflammation. Cytotherapy 2014, 16, 1336–1344. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Liu, Q.; Jiang, Y.; Wu, L.; Xu, G.; Liu, X. Enhanced angiogenesis promoted by human umbilical mesenchymal stem cell transplantation in stroked mouse is Notch1 signaling associated. Neuroscience 2015, 290, 288–299. [Google Scholar] [CrossRef] [PubMed]
- Park, H.W.; Moon, H.E.; Kim, H.S.; Paek, S.L.; Kim, Y.; Chang, J.W.; Yang, Y.S.; Kim, K.; Oh, W.; Hwang, J.H.; et al. Human umbilical cord blood-derived mesenchymal stem cells improve functional recovery through thrombospondin1, pantraxin3, and vascular endothelial growth factor in the ischemic rat brain. J. Neurosci. Res. 2015, 93, 1814–1825. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Fernandez, M.; Rodriguez-Frutos, B.; Ramos-Cejudo, J.; Otero-Ortega, L.; Fuentes, B.; Vallejo-Cremades, M.T.; Sanz-Cuesta, B.E.; Diez-Tejedor, E. Comparison between xenogeneic and allogeneic adipose mesenchymal stem cells in the treatment of acute cerebral infarct: Proof of concept in rats. J. Transl. Med. 2015, 13, 46. [Google Scholar] [CrossRef] [PubMed]
- Diez-Tejedor, E.; Gutierrez-Fernandez, M.; Martinez-Sanchez, P.; Rodriguez-Frutos, B.; Ruiz-Ares, G.; Lara, M.L.; Gimeno, B.F. Reparative therapy for acute ischemic stroke with allogeneic mesenchymal stem cells from adipose tissue: A safety assessment: A phase II randomized, double-blind, placebo-controlled, single-center, pilot clinical trial. J. Stroke Cerebrovasc. Dis. 2014, 23, 2694–2700. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.M.; Weng, S.J.; Tsai, T.H.; Li, I.H.; Lu, P.H.; Ma, K.H.; Tai, M.C.; Chen, J.T.; Cheng, C.Y.; Huang, Y.S. Neurotrophic and neuroprotective potential of human limbus-derived mesenchymal stromal cells. Cytotherapy 2014, 16, 1371–1383. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Liu, X.; Zhang, N.; Zhu, X.; Liang, H.; Sun, L.; Cheng, Y. Neuroprotective Effects of Brain-Derived Neurotrophic Factor and Noggin-Modified Bone Mesenchymal Stem Cells in Focal Cerebral Ischemia in Rats. J. Stroke Cerebrovasc. Dis. 2016, 25, 410–418. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Yao, Q.; Liang, Z.; Lin, J.; Xie, Y.; Li, S.; Wu, G.; Yang, Z.; Xu, P. The Dose of Intravenously Transplanted Bone Marrow Stromal Cells Determines the Therapeutic Effect on Vascular Remodeling in a Rat Model of Ischemic Stroke. Cell Transplant. 2016, 25, 2173–2185. [Google Scholar] [CrossRef] [PubMed]
- Lapi, D.; Vagnani, S.; Sapio, D.; Mastantuono, T.; Boscia, F.; Pignataro, G.; Penna, C.; Pagliaro, P.; Colantuoni, A. Effects of bone marrow mesenchymal stem cells (BM-MSCs) on rat pial microvascular remodeling after transient middle cerebral artery occlusion. Front. Cell. Neurosci. 2015, 9, 329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gincberg, G.; Arien-Zakay, H.; Lazarovici, P.; Lelkes, P.I. Neural stem cells: Therapeutic potential for neurodegenerative diseases. Br. Med. Bull. 2012, 104, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Hao, L.; Zou, Z.; Tian, H.; Zhang, Y.; Zhou, H.; Liu, L. Stem cell-based therapies for ischemic stroke. Biomed. Res. Int. 2014, 2014, 468748. [Google Scholar] [CrossRef] [PubMed]
- Kopen, G.C.; Prockop, D.J.; Phinney, D.G. Marrow stromal cells migrate throughout forebrain and cerebellum, and they differentiate into astrocytes after injection into neonatal mouse brains. Proc. Natl. Acad. Sci. USA 1999, 96, 10711–10716. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, J.; Chen, X.G.; Wang, L.; Gautam, S.C.; Xu, Y.X.; Katakowski, M.; Zhang, L.J.; Lu, M.; Janakiraman, N.; et al. Human marrow stromal cell therapy for stroke in rat: Neurotrophins and functional recovery. Neurology 2002, 59, 514–523. [Google Scholar] [CrossRef] [PubMed]
- Borlongan, C.V.; Hadman, M.; Sanberg, C.D.; Sanberg, P.R. Central nervous system entry of peripherally injected umbilical cord blood cells is not required for neuroprotection in stroke. Stroke 2004, 35, 2385–2389. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.; Seo, S.; Moon, M.; Park, S. IGF-I inhibition of apoptosis is associated with decreased expression of prostate apoptosis response-4. J. Endocrinol. 2007, 194, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Besner, G.E. Heparin-binding epidermal growth factor-like growth factor is a potent neurotrophic factor for PC12 cells. Neurosignals 2010, 18, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Mang, J.; Mei, C.L.; Wang, J.Q.; Li, Z.S.; Chu, T.T.; He, J.T.; Xu, Z.X. Endogenous protection derived from activin A/Smads transduction loop stimulated via ischemic injury in PC12 cells. Molecules 2013, 18, 12977–12986. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Q.; He, J.T.; Du, Z.W.; Li, Z.S.; Liu, Y.F.; Mang, J.; Xu, Z.X. Effects of SARA on oxygen-glucose deprivation in PC12 cell line. Neurochem. Res. 2013, 38, 961–971. [Google Scholar] [CrossRef] [PubMed]
- Tinhofer, I.; Maly, K.; Dietl, P.; Hochholdinger, F.; Mayr, S.; Obermeier, A.; Grunicke, H.H. Differential Ca2+ signaling induced by activation of the epidermal growth factor and nerve growth factor receptors. J. Biol. Chem. 1996, 271, 30505–30509. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.; Lee, S.B.; Lee, Y.S.; Lee, K.H.; Ahn, J.Y. Neuroprotection by NGF and BDNF against neurotoxin-exerted apoptotic death in neural stem cells are mediated through Trk receptors, activating PI3-kinase and MAPK pathways. Neurochem. Res. 2009, 34, 942–951. [Google Scholar] [CrossRef] [PubMed]
- Franklin, J.L. Redox regulation of the intrinsic pathway in neuronal apoptosis. Antioxid. Redox Signal 2011, 14, 1437–1448. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Wu, D.M.; Hu, B.; Zheng, Y.L.; Zhang, Z.F.; Wang, Y.J. NGF-Dependent activation of TrkA pathway: A mechanism for the neuroprotective effect of troxerutin in D-galactose-treated mice. Brain Pathol. 2010, 20, 952–965. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.H.; Yang, L.Y.; Chen, W.; Li, Y.K.; Yuan, H.B. Fibroblast growth factor 10 protects neuron against oxygen-glucose deprivation injury through inducing heme oxygenase-1. Biochem. Biophys. Res. Commun. 2015, 456, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Shen, X.; Xu, Y.; He, Y.; Hu, W. The effect of activin A on signal transduction pathways in PC12 cells subjected to oxygen and glucose deprivation. Int. J. Mol. Med. 2014, 33, 135–141. [Google Scholar] [CrossRef] [PubMed]
- He, J.T.; Mang, J.; Mei, C.L.; Yang, L.; Wang, J.Q.; Xing, Y.; Yang, H.; Xu, Z.X. Neuroprotective effects of exogenous activin A on oxygen-glucose deprivation in PC12 cells. Molecules 2011, 17, 315–327. [Google Scholar] [CrossRef] [PubMed]
- Souvenir, R.; Flores, J.J.; Ostrowski, R.P.; Manaenko, A.; Duris, K.; Tang, J. Erythropoietin inhibits HIF-1alpha expression via upregulation of PHD-2 transcription and translation in an in vitro model of hypoxia-ischemia. Transl. Stroke Res. 2014, 5, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.; Seger, R. The extracellular signal-regulated kinase: Multiple substrates regulate diverse cellular functions. Growth Factors 2006, 24, 21–44. [Google Scholar] [CrossRef] [PubMed]
- Mo, Z.T.; Li, W.N.; Zhai, Y.R.; Gao, S.Y. The effects of icariin on the expression of HIF-1alpha, HSP-60 and HSP-70 in PC12 cells suffered from oxygen-glucose deprivation-induced injury. Pharm. Biol. 2017, 55, 848–852. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Huang, W.; Wang, J.; Song, H.; Cen, J.; Ji, B. Anthraquinone derivative exerted hormetic effect on the apoptosis in oxygen-glucose deprivation-induced PC12 cells via ERK and Akt activated Nrf2/HO-1 signaling pathway. Chem. Biol. Interact. 2017, 262, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, K.; Kotani, Y.; Nakajima, Y.; Shimazawa, M.; Yoshimura, S.; Nakashima, S.; Iwama, T.; Hara, H. Fasudil, a Rho kinase (ROCK) inhibitor, protects against ischemic neuronal damage in vitro and in vivo by acting directly on neurons. Brain Res. 2007, 1154, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, T.; Wakasa, T.; Okuma, Y.; Inanami, O.; Nomura, Y.; Kuwabara, M.; Kawahara, K. Dual inhibition of protein phosphatase-1/2A and calpain rescues nerve growth factor-differentiated PC12 cells from oxygen-glucose deprivation-induced cell death. J. Neurosci. Res. 2006, 83, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Madineni, A.; Alhadidi, Q.; Shah, Z.A. Cofilin Inhibition Restores Neuronal Cell Death in Oxygen-Glucose Deprivation Model of Ischemia. Mol. Neurobiol. 2016, 53, 867–878. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.S.; Kim, S.W.; Baek, K.J.; Lee, H.S.; Kwon, N.S.; Kim, Y.M.; Yun, H.Y. Involvement of Ras in survival responsiveness to nitric oxide toxicity in pheochromocytoma cells. J. Neurooncol. 2002, 60, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Heo, J.; Campbell, S.L. Ras regulation by reactive oxygen and nitrogen species. Biochemistry 2006, 45, 2200–2210. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.; Dickens, M.; Raingeaud, J.; Davis, R.J.; Greenberg, M.E. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science 1995, 270, 1326–1331. [Google Scholar] [CrossRef] [PubMed]
- Lambeng, N.; Willaime-Morawek, S.; Mariani, J.; Ruberg, M.; Brugg, B. Activation of mitogen-activated protein kinase pathways during the death of PC12 cells is dependent on the state of differentiation. Mol. Brain Res. 2003, 111, 52–60. [Google Scholar] [CrossRef]
- Winter-Vann, A.M.; Johnson, G.L. Integrated activation of MAP3Ks balances cell fate in response to stress. J. Cell. Biochem. 2007, 102, 848–858. [Google Scholar] [CrossRef] [PubMed]
- An, J.M.; Kim, S.S.; Rhie, J.H.; Shin, D.M.; Seo, S.R.; Seo, J.T. Carmustine induces ERK- and JNK-dependent cell death of neuronally-differentiated PC12 cells via generation of reactive oxygen species. Toxicol. In Vitro 2011, 25, 1359–1365. [Google Scholar] [CrossRef] [PubMed]
- Wruck, C.J.; Claussen, M.; Fuhrmann, G.; Romer, L.; Schulz, A.; Pufe, T.; Waetzig, V.; Peipp, M.; Herdegen, T.; Gotz, M.E. Luteolin protects rat PC12 and C6 cells against MPP+ induced toxicity via an ERK dependent Keap1-Nrf2-ARE pathway. In Neuropsychiatric Disorders an Integrative Approach; Springer: Vienna, Austria, 2007; pp. 57–67. [Google Scholar]
- Gu, D.M.; Lu, P.H.; Zhang, K.; Wang, X.; Sun, M.; Chen, G.Q.; Wang, Q. EGFR mediates astragaloside IV-induced Nrf2 activation to protect cortical neurons against in vitro ischemia/reperfusion damages. Biochem. Biophys. Res. Commun. 2015, 457, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Chiarugi, P.; Cirri, P. Redox regulation of protein tyrosine phosphatases during receptor tyrosine kinase signal transduction. Trends Biochem. Sci. 2003, 28, 509–514. [Google Scholar] [CrossRef]
- Torres, M. Mitogen-activated protein kinase pathways in redox signaling. Front. Biosci. 2003, 8, d369–d391. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, B.A.; Minnerup, J.; Balami, J.S.; Arba, F.; Buchan, A.M.; Kleinschnitz, C. Neuroprotection for ischaemic stroke: Translation from the bench to the bedside. Int. J. Stroke 2012, 7, 407–418. [Google Scholar] [CrossRef] [PubMed]


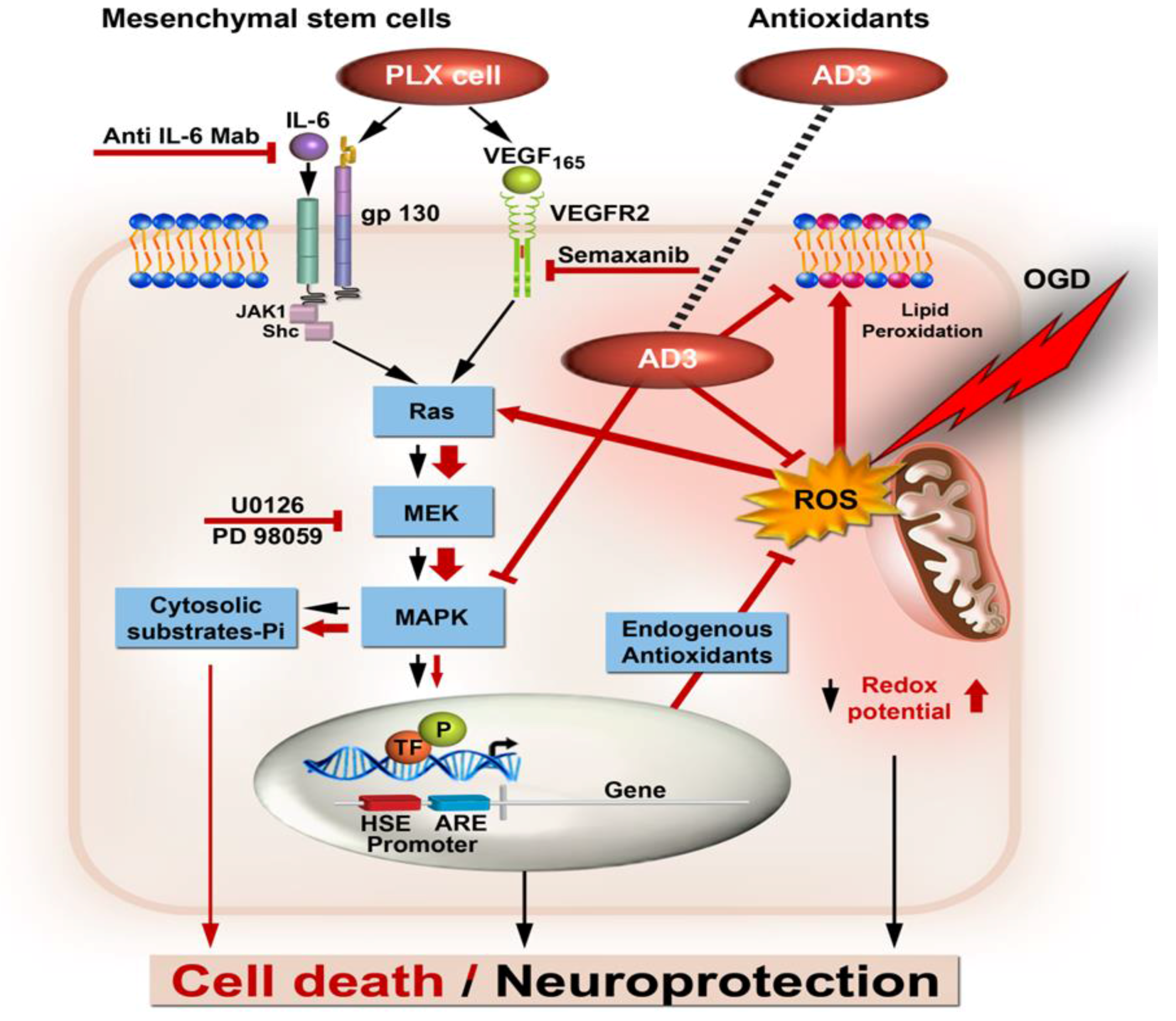{kind=link}
| Drug Classification/Name | Insult Oxygen: O2/CO2/N2 % Glucose: +/− | OGD/Reoxygenation Duration (h/h) a | Differentiation by NGF (Days) b | Neuroprotections Assays c | Mechanism d | Reference |
|---|---|---|---|---|---|---|
| Calcium Channel Blockers: | Reduced Ca overload and redox potential | [90,124,125] | ||||
| Nimodipine and Nifedipine | 1/5/94, − | 18 | Yes (10–14) | LDH, Caspase 3 activity, MMP, MTT, Hoechst/PI | ||
| Mebudipine | 0/5/95, − | 0.5–1.5/24 | No | |||
| Cinepazide maleate | 0/5/95, − | 2.5/24 | No | |||
| Anti-coagulants | 0/5/95, − | MTT, Annexin-PI, MMP | Reduced Ca overload and redox potential | [126,127] | ||
| Heparin | 4 | No | ||||
| Benzopyrone derivative | 24 | No | ||||
| Opiates | 0/5/95, − | 24 | No | LDH | Enhanced activation of ERK1/2 | [128] |
| Naloxone | ||||||
| Tetracycline antibiotic | 1/5/94, − | Hypoxia 2–4 | No | MTT | Reduced activation of ERK1/2 and p-38 | [129] |
| Minocycline | ||||||
| Brain natural histidine dipeptide antioxidants | 0/5/95, − | 24 | No | LDH, Caspase 3 activity | Reduced activation of ERK1/2, JNK1/2 and p-38 | [108] |
| Homocarnosine-antioxidant | ||||||
| Saponins | Annexin-PPI, LDH, Tunnel, MTT | [130,131] | ||||
| Ginsenosides | 0/5/95, − | 6 | No | Enhanced activation of ERK1/2 | ||
| Dioscin | 1/5/94, − | 24 | No | Reduced activation of ERK1/2, p-38, JNK | ||
| Sesquiterpene alkaloid | 0/5/95, − | 24 | No | LDH, Hoechst, MTT, DNA fragmentation | Reduced redox potential and LPO | [132,133] |
| Huperzine A acetylcholinesterase inhibitor | ||||||
| Stilbenoid Phenol antioxidant | 1/5/94, − | 24 | Yes (>7); No | Annexin, Caspase 3,9 activity, MTT, Hoechst/PI, LDH | Reduced redox potential, LPO and Ca overload, Increased antioxidant level | [134,135,136] |
| Resveratrol | ||||||
| Glycosyl anthraquinone antioxidant | 0/5/95, - | 24 | No | LDH, Annexin V, MMP, MTT | Reduced redox potential and LPO | [137] |
| Aloin | ||||||
| Thiol antioxidants | 0/5/95, − | 24 | Yes (5) | LDH, MMP, Caspase activity, MTT | Reduced redox potential, Ca overload and activation of p-38 | [138] |
| s-Methyl cysteine (SMC) | ||||||
| Thiol and NO donors antioxidants | 0/5/95, − | 18 | No | LDH | Reduced redox potential, LPO and activation of ERK1/2, p-38, JNK | [88] |
| α-Lipoic Acid | ||||||
| Tempol | ||||||
| α-Tempol ester-ω-lipo ester PEG-AD3 | ||||||
| Lipid metabolism co-factors | 0/5/95, − | 0.5–2/4 | No | DNA fragmentation, TUNEL, MTT | Reduced redox potential and LPO | [139] |
| l-carnitine , Acetyl-l-carnitine |
| Species | Mesenchymal Stem Cells Origin | Ischemic Insult, Duration (h) and Model | Cytokines Estimation | Neuroprotection Evaluation | Mechanisms | Reference |
|---|---|---|---|---|---|---|
| Human | Bone marrow | Rat MCAO 1 h | Gene expression | Neurological score | Increased expression of rat and human VEGF, IL-6 AND rat bFGF, BDNF, NT-3, GDNF, CCL2 | [200,201,202] |
| Human M17 neuroblastoma cell line-OGD (20–24/72) | Cytometric Bead Array | Infarct volume | ||||
| Umbilical Cord stroma | Mice MCAO 1.5–2 h | ELISA | Neurological score | Increased secretion of human VEGF165, upregulation of IL-4, TGF-β1 and IL-10 | [2,51,203,204] | |
| Mice primary cortical neurons- OGD/R (2/24) | Gene expression | Infarct volume | Decrease secretion of IL-6, IL-1β | |||
| Rat MCAO 2 h | IHC, Angiogenic Microarray | |||||
| Rabbit MCAO 2 h | Western blot | |||||
| Placenta and umbilical cord blood | NGF differentiated PC12 | Gene expression | Necrosis—LDH | Increased secretion of human VEGF and IL-6, | [89,92] | |
| (7 days)—OGD/R (4/18) | ELISA | Neuroprotection Index | Reduced redox potential and LPO | |||
| Adipose tissue | Rat MCAO 1 h | IHC | Neurological score | Increased expression of human VEGF | [205,206] | |
| Phase II clinical trials | Infarct volume | |||||
| Patients with acute ischemic stroke within 24 h of onset | Not applicable | NIH Stroke scale/score (NIHSS) | ||||
| Cornea | Rat MCAO | ELISA, Cytokine array, Gene expression | Infarct volume Open field memory test | Increased expression of human VEGF and BDNF | [207] | |
| Rat | Bone marrow | Rat MCAO 1 h | ELISA, Western Blot, IHC | Neurological score | Increase expression of VEGF and bFGF | [208,209,210] |
| Infarct volume | ||||||
| Adipose tissue | Rat MCAO 1 h | IHC | Neurological score | Increased secretion of rat VEGF | [205] | |
| Infarct volume |
| Drug Classification/Name | Insult Oxygen: O2/CO2/N2 % Glucose: +/− | OGD/Reoxygenation Duration (h/h) a | Differentiation by NGF (Days) b | Neuroprotections Assays c | Mechanism d | Reference |
|---|---|---|---|---|---|---|
| Nerve growth factor (NGF) | 0/5/95, − | 18 | No | LDH, caspase activity | Inhibition of JNK 1, p-38 α, p-38 β activity | [77,111] |
| Insulin-like growth factor-1 (IGF-1) | 0.1/5/94.9, − | 18 | No | MTT | ERK1/2 and PI3K pathways contribute to neuroprotection | [216] |
| Fibroblast growth factor 10 (FGF-10) | 1/5/94, − | 2–4/0 | No | LDH, Annexin | Attenuation of redox potential | [224] |
| TGF-β superfamily (Activin A) | 0/5/95, − | 3–16/0 | Yes (3) | Caspase-3, MTT | Modulation of gene expression | [225] |
| 0/5/95, − | 6/0 | Yes (6) | Annexin, Caspase-3, MTT | Attenuation of redox potential; Modulation of cellular endogenous antioxidants | [226] | |
| 0/5/95, − | 1–24/0 | No/Yes (7) | Morphological changes, Annexin, MTT | Modulation of gene expression | [218,219] | |
| Erythropoietin (EPO) | 1/5/94, − | 18 | Yes (5) | Pro-apoptotic gene, MMP | Attenuation of redox potential | [227] |
| Heparin-binding epidermal growth factor like (HB-EGF) | 0/5/95, − | 21 | No | LDH, Annexin, MTT | Activation of ERK1/2 phosphorylation | [217] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lahiani, A.; Brand-Yavin, A.; Yavin, E.; Lazarovici, P. Neuroprotective Effects of Bioactive Compounds and MAPK Pathway Modulation in “Ischemia”—Stressed PC12 Pheochromocytoma Cells. Brain Sci. 2018, 8, 32. https://doi.org/10.3390/brainsci8020032
Lahiani A, Brand-Yavin A, Yavin E, Lazarovici P. Neuroprotective Effects of Bioactive Compounds and MAPK Pathway Modulation in “Ischemia”—Stressed PC12 Pheochromocytoma Cells. Brain Sciences. 2018; 8(2):32. https://doi.org/10.3390/brainsci8020032
Chicago/Turabian StyleLahiani, Adi, Annette Brand-Yavin, Ephraim Yavin, and Philip Lazarovici. 2018. "Neuroprotective Effects of Bioactive Compounds and MAPK Pathway Modulation in “Ischemia”—Stressed PC12 Pheochromocytoma Cells" Brain Sciences 8, no. 2: 32. https://doi.org/10.3390/brainsci8020032
APA StyleLahiani, A., Brand-Yavin, A., Yavin, E., & Lazarovici, P. (2018). Neuroprotective Effects of Bioactive Compounds and MAPK Pathway Modulation in “Ischemia”—Stressed PC12 Pheochromocytoma Cells. Brain Sciences, 8(2), 32. https://doi.org/10.3390/brainsci8020032