3.2. Optimized Antibody Spot Size and Signal Density
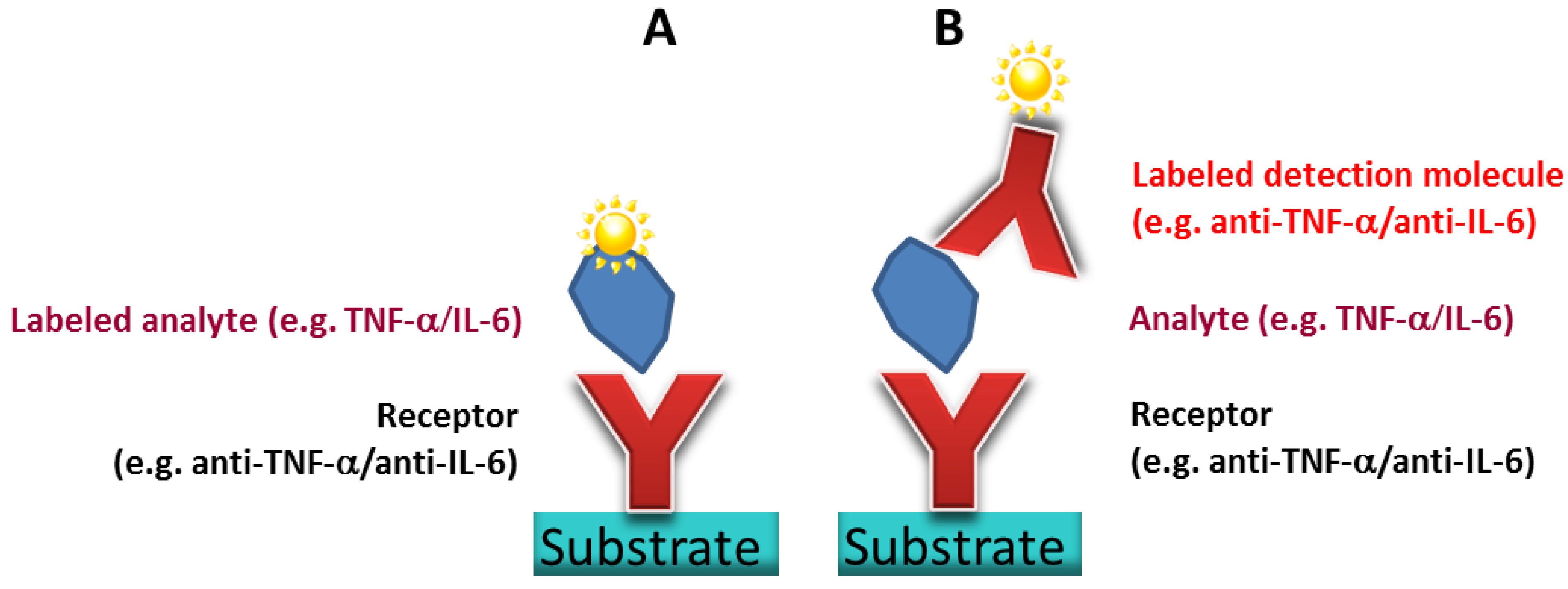
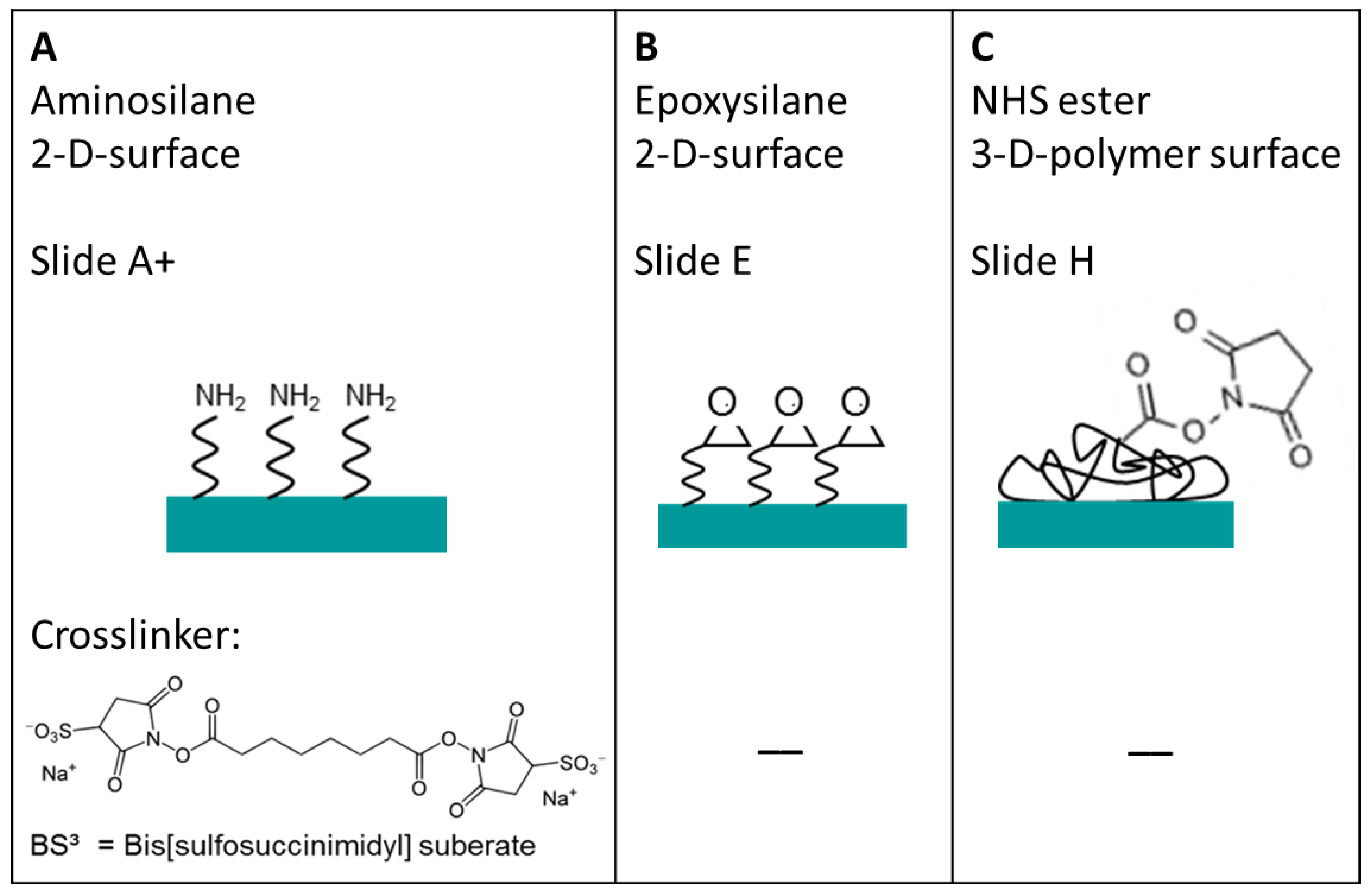
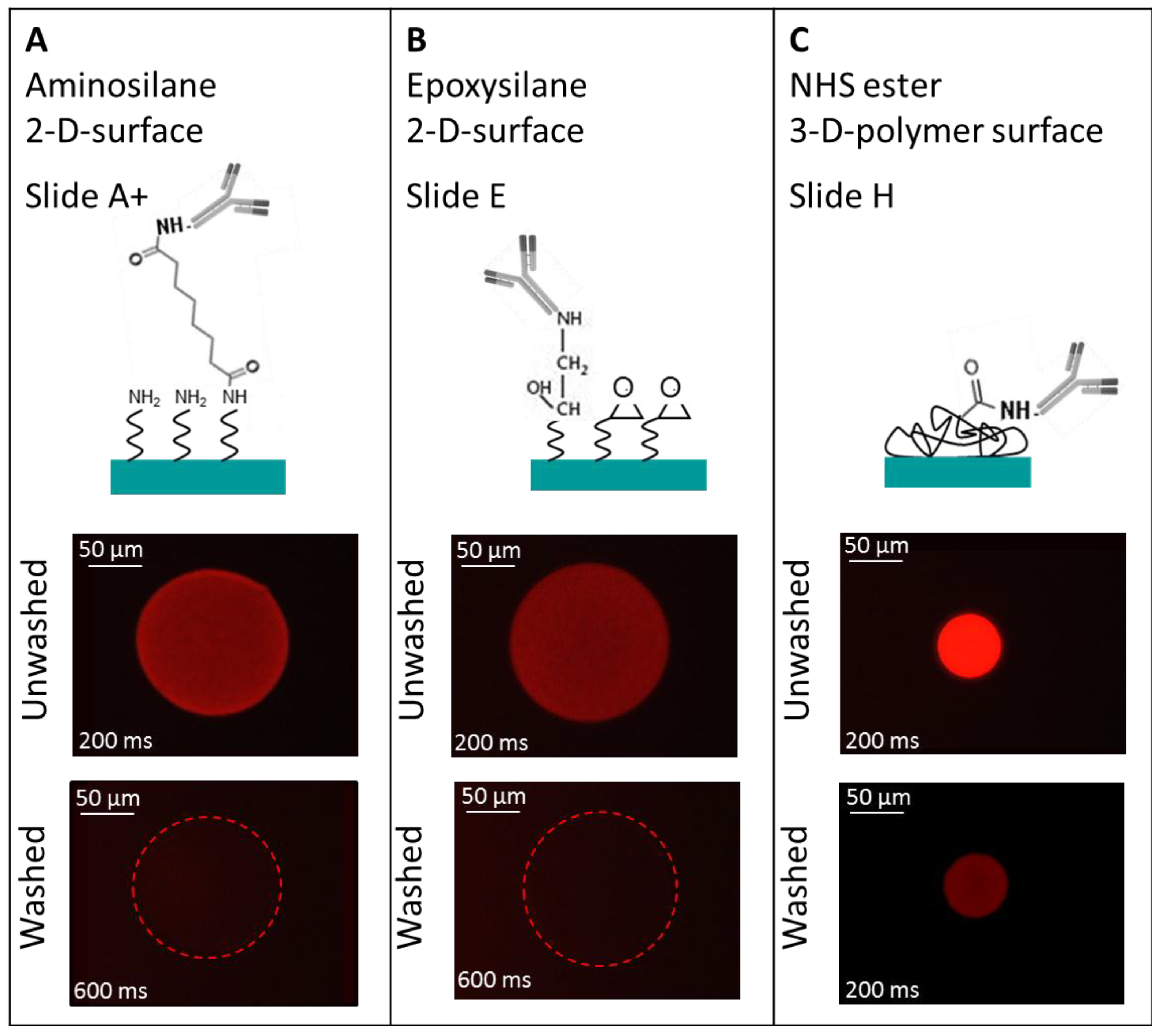
To obtain a high density TNFα or IL-6 antibody microarray with small, homogeneous, reproducible spots and short spot to spot distance for later sensitive cytokine detection, we visualized the spotting of fluorescently labeled antibodies on different surfaces. The three selected surfaces—amine, epoxy, and NHS-3D functionalized hydrogel substrates—were spotted with the Nanoplotter, using a picopipette and the resulting spot patterns were visualized with fluorescence microscopy. A scheme of the resulting immobilized antibodies on the surfaces together with the imaged fluorescence spots before and after washing with PBS buffer of the different slides are shown in
Figure 4. Please note that in the scheme the antibody’s orientation is drawn in the oriented form for simplicity; however this does not reflect the real situation, and the antibodies are arbitrarily immobilized.
Figure 4.
Three different selected surfaces after protein immobilization. (A) shows the antibody, which is immobilized via the BS3 linker to the amine-modified surface. (B) resembles the direct binding of antibodies’ amine group to the epoxy group on the surface. (C) the antibody binds to the NHS ester activated polymer within the 3D layer. For all surfaces there is a measured spot of unwashed and washed PE labeled anti-TNFα. Note that the fluorescence images also indicate the exposure time, showing the need to increase it from 200 ms to 600 ms for the amine and epoxy slide after washing in order to visualize the spot with the fluorescence microscope.
Figure 4.
Three different selected surfaces after protein immobilization. (A) shows the antibody, which is immobilized via the BS3 linker to the amine-modified surface. (B) resembles the direct binding of antibodies’ amine group to the epoxy group on the surface. (C) the antibody binds to the NHS ester activated polymer within the 3D layer. For all surfaces there is a measured spot of unwashed and washed PE labeled anti-TNFα. Note that the fluorescence images also indicate the exposure time, showing the need to increase it from 200 ms to 600 ms for the amine and epoxy slide after washing in order to visualize the spot with the fluorescence microscope.
It is remarkable that the resulting spot sizes are smallest on NHS-3D slides (~50 µm) compared to the BS
3 activated amine slide and the epoxy slide, which both have about the same diameter of ~150 µm (
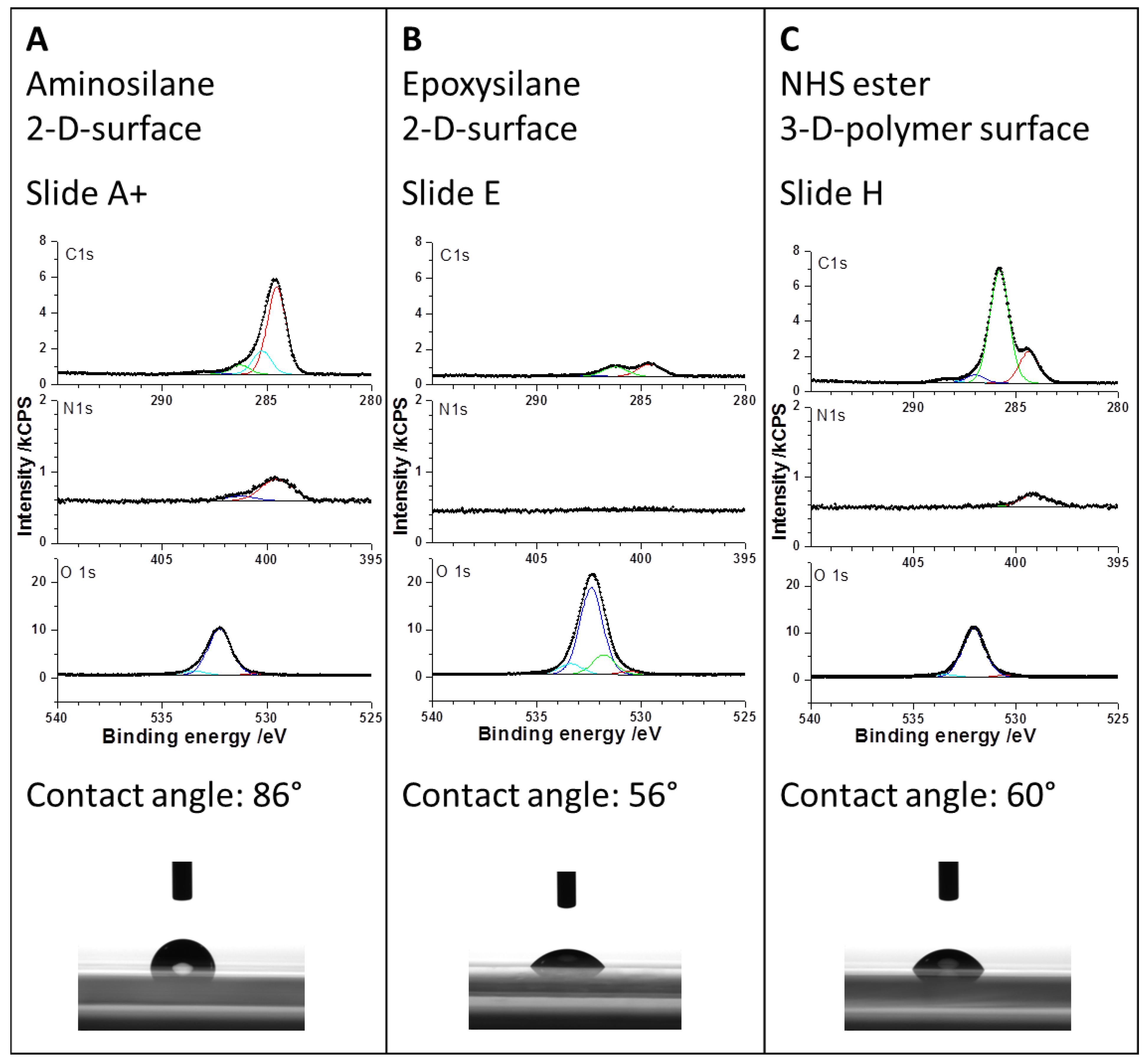
Figure 4). From the contact angle measurements of virgin slides, realized with water droplets (
Figure 3), one would expect a slightly different result. The highest contact angle implicating a small spot size is given in case of the amino slide (86°). At the beginning, the amine terminated surface showed low hydrophilicity; it can be assumed that during the conditioning of the surface with BS
3 in PBS buffer it becomes more hydrophilic due to the protonation of the amine groups and the interaction with the ionic BS
3 linker. This could result in a more hydrophilic surface and hence bigger spots after antibody spotting. On the other side, the epoxy slide and the NHS-3D slide show similar contact angles of 56° and 60°, respectively, but the spot on the NHS-3D slide is much smaller with the same spotting buffer and solution. This can result from the difference in 2D and 3D surfaces, meaning that due to the 3D surface of the NHS modified slide, the layer thickness is bigger, and hence same volume of spotting solution is distributed in three dimensions spotted on a smaller area, resulting in smaller spot sizes but more intense signals compared to the 2D epoxy slide. Another explanation can be the influence of the antibody/dye solution or more likely the influence of our self-optimized buffer system depending on the functional surface. The presence of 1% glycerol possibly changes the contact angle compared to water. It is possible that the epoxy surface is more glycerol-philic compared to the NHS-3D slide with unknown polymer composition and the spotting buffer on the NHS modified slide results in a higher contact angle than with water.
The incubation time of one hour after antibody spotting and before washing was found to be enough for all three different surfaces. This could be observed by varying the incubation time from 1–8 hours on all solid surfaces. All spots on one kind of slide time-independently showed the same intensities after washing, meaning the same grey values, regardless of whether they were incubated for one or 8 hours. It can be assumed that saturation, and hence equilibrium, is reached within 1 hour of antibody incubation time.
The fluorescent signal intensity before washing of the samples revealed the following: The PE-labeled TNFα antibody was spotted at a concentration of 25 µg/mL, meaning one single spot (V = 60 pl) contains 1.5 pg of antibody before washing. Assuming an antibody size of 150 kDa, about 6 × 106 TNFα antibody molecules would be deposited within one spot before washing. On the NHS-3D slide the spots are smallest and hence show a more intense fluorescent signal compared to the epoxy and amine slide, as the 6 × 106 available antibodies are more densely packed. Assuming an antibody-PE diameter dAntibody-PE ~26 nm, under the consideration of dAntibody ~10 nm with ~150 kDa and further dPE ~16 nm for the ~240 kDa PE label, the effective surface area of a PE-labeled antibody would be ~531 nm2. For the densest monolayer, there is theoretically space for ~4 × 106 labeled antibodies in the case of 50 µm spots as we have achieved them on the NHS-3D hydrogel surface and ~33 × 106 spaces in case of 150 µm spots of the amino and epoxy functionalized supports. Admittedly, the label size plays an important role and the results will be different if no label is used for later cytokine detection. However, spotting smaller spots will result in an excess of capture antibodies for the potential immobilization of also the third dimension on the solid support, and thus a more effective binding is possible.
On all three surfaces the spots lost intensity after the PBS washing procedure because non-immobilized labeled antibodies were washed away. Residues of antibodies from the washing process in form of non-specific binding were not observed, leading to the conclusion of an accordingly slow kinetic for the immobilization process under given parameters [
19].
The ~150 µm spot on the amine surface lost its fluorescence intensity after the washing process and could only be observed with the fluorescent microscope by increasing the exposure time from 200 ms to 600 ms. A guide for the eye is given for visualization in
Figure 4. It can be assumed that analyte detection in this case would lead to even weaker signals, due to a low signal to noise ratio. The amine surfaces are known to bind biomolecules, e.g., DNA, by adsorption with its negatively charged phosphate backbone to the positively charged amine surface. Alternatively, biomolecules can bind covalently to amine groups by using crosslinkers e.g., like BS
3. The NHS groups of the BS
3 linker, for example, leave, and the linker forms a stable peptide bond with the amine groups of the biomolecule and surface. The advantage of this relatively easy way of preparation of amine functional groups on glass or glass-like surfaces by silanization is canceled out by the disadvantage of an intermediate step with a crosslinker. At first glance, amino modified slides are not intuitive for antibody arrays, e.g., Schott recommend this surface more for DNA application, however, the contact angle measurements and promising results reported by Seurynck-Servoss
et al. suggested an attempt. They evaluated different surfaces for antibody microarrays and found amine modified surfaces well suited regarding spot size, morphology, signal background, sensitivity and reproducibility, also in comparison with other different epoxy modified surfaces and NHS modified hydrogels [
20]. The proof of antibody activity and binding capacity is not further investigated here, as we aim for small spots and a high antibody density,
i.e., higher signal intensities after the washing procedure.
The epoxy surface showed very similar results compared with the amine surface regarding spot size and fluorescence intensity before and after washing. For signal detection, here also the exposure time had to be increased from 200 ms to 600 ms. Epoxy groups can be attached to glass or glass-like surfaces by silanization similar to amine groups. The advantage of a time efficient direct antibody binding via an amine group of the biomolecule on the epoxy surface did not lead to better results in signal intensity.
In contrast to the amine and epoxy surface, the NHS functionalized hydrogel on a glass slide offers a 3D geometry of polymer with NHS ester groups and reacts directly with the amine group of an antibody. The main advantage of the 3D hydrogel is that it is claimed to be beneficial for protein stability, as the antibody can keep its native form and hence its activity, making it an interesting surface for antibody microarrays [
14]. Additionally, the 3D setup theoretically offers more active binding sites per area, meaning a higher analyte binding capacity per spot of the same size and an increased signal. The antibody density of the ~50 µm spot after washing is still the highest in case of the NHS-3D slide compared to the epoxy slide and the amine/BS
3 slide. This means the antibody loading is maximized for smaller spots even after washing, the final microarray can be spotted more densely, and more tests per slide can be realized. Therefore we decided to fabricate our high-density protein arrays on NHS-modified hydrogel slides. As it is still unclear how many unbound antibodies are washed away during the PBS washing procedure, the binding efficiency of immobilized antibody was estimated to be about 12%. This was done by comparing the fluorescence intensities of different antibody concentrations from 1–100 µg/mL with unwashed and washed spots (see
Supplementary Materials).
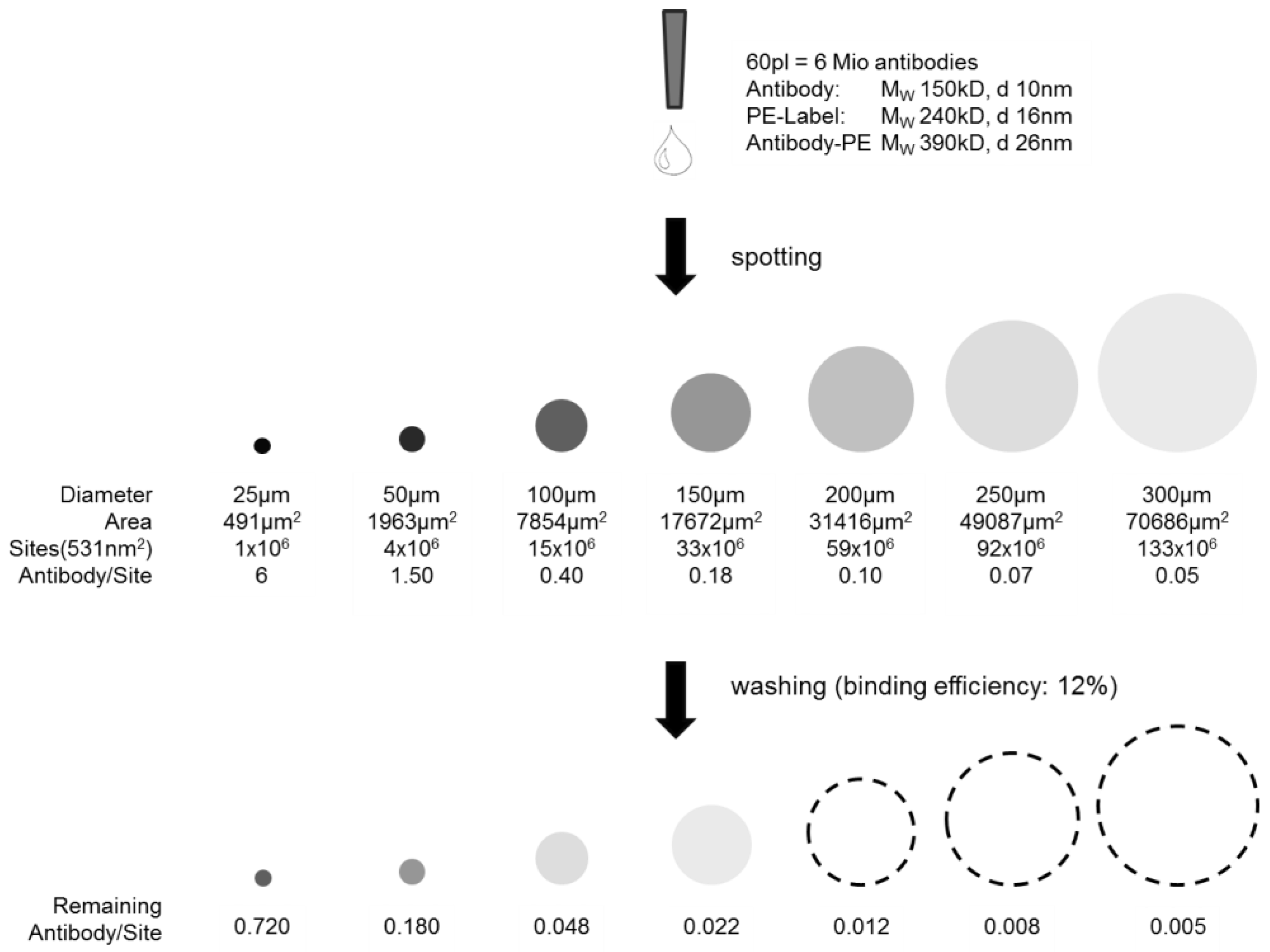
The highest antibody density within one spot would be given if a monolayer, or in case of the 3D hydrogel slide, a multilayer with functional and active antibodies could be achieved. This may later lead to a higher signal density when detecting the analyte. A quantitative evaluation of the spotting results is given below and visualized in
Figure 5. The theoretically possible antibody loading in a 50 µm spot, considering the densest monolayer, was calculated to be 4 × 10
6 PE-labeled anti-TNFα molecules. However, the real antibody loading was estimated from the concentration line given in the
Supplementary Materials: assuming an antibody concentration level of 3 µg/mL within the washed spot by comparing the fluorescent intensities of washed 25 µg/mL concentration with unwashed spots (binding efficiency ~12%), and knowing the drop volume of the nanoplotter (V = 60 pl), there are 0.18 pg antibody within one spot after washing. Assuming a molecular weight of 150 kDa per antibody and taking the Avogadro constant of 6 × 10
23 mol
−1, it can be concluded that the amount of antibody/spot under the given assumptions is ~7.2 × 10
5,
i.e., a loading of ~18% of a theoretical dense antibody monolayer. Taking the PE dye of 240 kDa into consideration, ~47% of a theoretical dense antibody-PE monolayer is achieved. Please note that the term “monolayer” in case of a 3D surface is not appropriate but is taken for comparison and an easy imagination.
Figure 5.
Scheme of capture antibody density per spot. Illustrated before and after washing depending on the spot size. The dashed lines are a guide for the eye, as intensities are not visible anymore.
Figure 5.
Scheme of capture antibody density per spot. Illustrated before and after washing depending on the spot size. The dashed lines are a guide for the eye, as intensities are not visible anymore.
One possibility to further increase the loading of capture antibodies on the NHS modified hydrogel would be to further reduce the spot size significantly below 50 µm, which was not possible here with the given slide and instrumentation, or to increase the spotting concentration. The second option has been investigated and neglected, as high concentrations of antibodies are spotted, but also washed off afterwards due to an already good saturation of antibodies on each spot.
Another possibility to increase the antibody loading per spot would be to exchange the fluorescent label to one of a smaller effective size, or even to utilize label-free antibodies. The protein label itself is very big (PE 240 kDa, APC 105 kDa); a small dye, for example Alexa
® dyes or FITC (389.2 g/mol ~0.4 kDa), could lead to higher antibody coverage and a closer approximation. The point is that the label at this stage is only used for visualization and quantification estimations before detecting the cytokine molecules. This is important to consider as density calculations show an error and most probably the pure antibody is packed even more densely on the NHS-3D slide. The theoretical optimum would be a dense layer of capture molecules; however, this is not possible due to steric hindrances and other protein-protein interactions, resulting in variable distances in-between antibodies. Additionally, it has also been reported that a highly dense antibody layer does not automatically mean that more analyte can be detected, again due to steric hindrances and interactions [
14].
Finally, for a highly sensitive cytokine array the first step is the selection of an appropriate antibody-antigen pair with specific and high association constant
K. The second one is, as Roger Ekins claims in his ambient analyte theory, that signal density per spot increases with smaller spot sizes till a constant level of signal density is reached,
i.e., the geometry and spot size reduction is of great importance for a high sensitive cytokine microarray [
16,
17,
21,
22]. Additional to Ekin’s theory, Kusnezow
et al. [
23] experimentally discussed the optimal design of microarray immunoassays, and we adapted the theory to our microarray. As illustrated and calculated in
Figure 5, the estimated remaining antibody density after washing within one spot differs strongly in spot size at a constant antibody concentration spotting. The smallest spots show highest antibody density before and after washing.
Assuming ~7.2 × 105 available cytokine binding sites in a 50 µm spot (see above), an attomolar concentration, i.e., 6 × 105 cytokine molecules, would form a monolayer, which can potentially be detected, knowing that in practice this is difficult to achieve due to different side effects. To achieve this detection in the small spots with the calculated low analyte (antigen) concentration, the diffusion coefficient plays an important role to transport the analyte from the sample solution to the spot. The diffusion coefficient of the analyte must be very high or, as this often cannot be modified, the transport length has to be decreased in order to avoid non-specific binding and to achieve the ambient analyte status and no analyte depletion.
3.3. High-Density Antibody Array
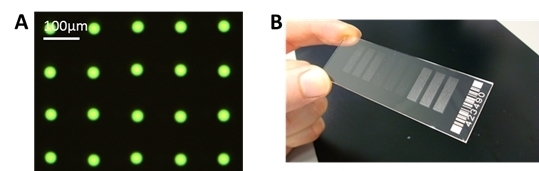
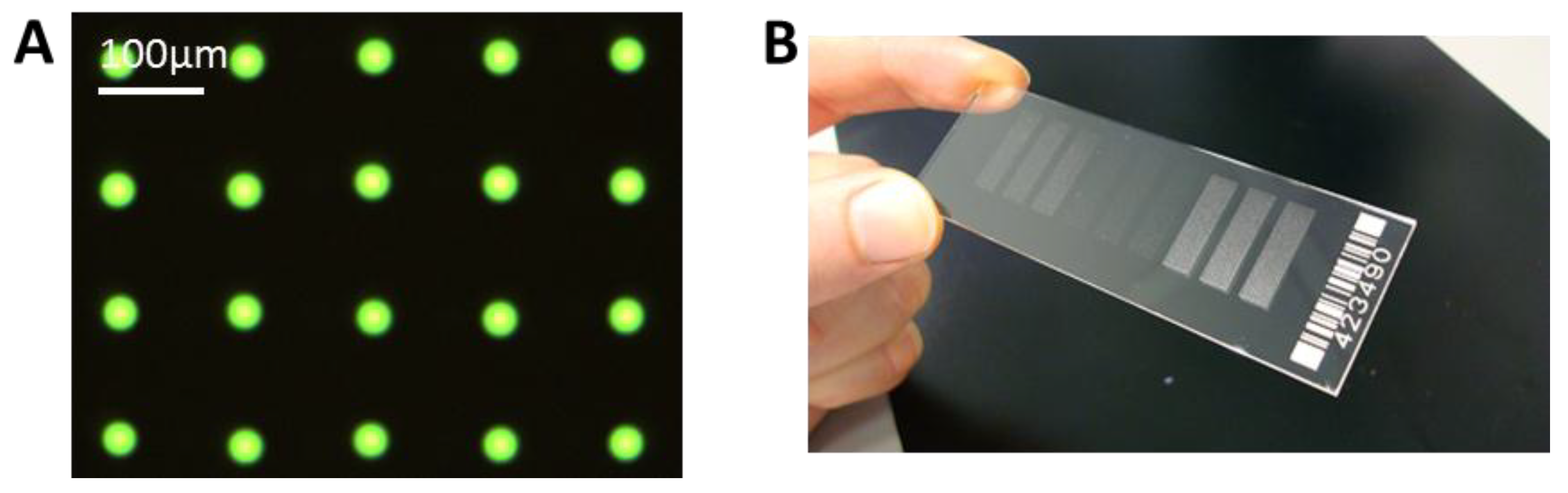
We have achieved a very reproducible spot size with a standard deviation of ± <1 µm for spot sizes around 30 µm. This can be seen in
Figure 6A, where only one type of antibody, FITC labeled anti-TNFα is spotted. With this small and reproducible spot size, a spot to spot distance of 100 µm can be achieved, and it is possible to spot more than 90,000 spots onto a microscopic slide, with 100 spots/mm
2.
Figure 6.
(A) Example of spotted microarray (before washing) with optimized parameters, 60 pl of 25 µg/mL anti-TNFα, fluorescein isothiocyanate (FITC) labeled, NHS-3D slide. The resulting spot to spot distance is 100 ± 3 µm. The spot diameter in this case is 27 ± 1 µm. (B) Example of ready to use antibody microarray on NHS-3D slide, nine blocks, 7500 spots each).
Figure 6.
(A) Example of spotted microarray (before washing) with optimized parameters, 60 pl of 25 µg/mL anti-TNFα, fluorescein isothiocyanate (FITC) labeled, NHS-3D slide. The resulting spot to spot distance is 100 ± 3 µm. The spot diameter in this case is 27 ± 1 µm. (B) Example of ready to use antibody microarray on NHS-3D slide, nine blocks, 7500 spots each).
To explore the possibility of preparing high density multiplex arrays with high reproducibility, as well as to verify the label suitability, three different label/antibody complexes—PE-labeled anti-TNFα, APC-labeled anti-TNFα, and FITC-labeled anti-IL-6—were spotted. It can be seen in
Figure 7 that also in the high density multiplex arrays the spots show good reproducibility and homogeneity in morphology and size within one type of spotted antibody.
Figure 7.
Three different fluorescence microscopy images of a fabricated multiplex cytokine array. The antibodies are differently labeled with fluorescence dyes for visualization: PE-labeled anti-TNFα (yellow-orange), allophycocyanin (APC)-labeled anti-TNFα (red), and FITC-labeled anti-IL-6 (green). The final image (D) shows the processed image after merging the pictures taken with different filters (A)–(C).
Figure 7.
Three different fluorescence microscopy images of a fabricated multiplex cytokine array. The antibodies are differently labeled with fluorescence dyes for visualization: PE-labeled anti-TNFα (yellow-orange), allophycocyanin (APC)-labeled anti-TNFα (red), and FITC-labeled anti-IL-6 (green). The final image (D) shows the processed image after merging the pictures taken with different filters (A)–(C).
The unwashed spots appear to be well visible after spotting with an exposure time of 100 ms. In A, using a filter with an excitation of 450–490 nm and an emission of 515 nm (Filter 09), the FITC labeled anti-IL-6 is visible as green spots and the PE-labeled anti-TNFα appears yellow. In B, using a filter with an excitation of 546/12 nm and an emission of 607/80 nm (Filter 20HE), the before yellow PE-labeled anti-TNFα here appears orange-red, and the APC-labeled anti-TNFα is red. The FITC labeled anti-IL-6 is not visible with this filter. In C, using a filter with an excitation of 546/12 nm and an emission of 590 nm (Filter 15) only the APC-labeled anti-TNFα appears as red spots. The three taken images were merged by image processing using the computer software to depict and to differentiate between the three differently labeled cytokines within one image, the initial parameters were not modified.
It is noted that the spot diameter of the green FITC-labeled anti-IL-6 is slightly smaller (28 ± 2 µm) than that of the other labeled TNFα spots (PE 37 ± 2 µm, APC 41 ± 2 µm). One reason for the slight deviation of the spot size could be that the cytokines themselves have different physico-chemical properties and structures, which will result in different ways of interacting with the surface. A further reason could be that FITC is a rather hydrophobic label. Additionally, it could be an optical effect, as FITC bleaches faster than PE or APC and hence the FITC spot appears slightly smaller. In summary, the same spotting pipette, the same substrate, and the same dilution buffer result in very reproducible spots of the same size, only the changing antibody/dye/additives show an influence on spot size, and we could observe differences of about 10–20 µm in diameter. Furthermore, by spotting such a mixed array, it is noted that the position of the spots is more difficult to control compared to single antibody solution, which is limited by the precision of the instrument.
3.4. Analyte Detection
Direct assay: The antibodies are receptor molecules selected to target cytokines (here: TNFα and IL-6) as disease biomarkers. Those cytokines occur in exhaled breath condensate and can potentially give a statement of COPD and asthma levels in patients. The analyte detection clarifies if the antibody microarray can potentially fulfill the detection of low concentrated biomarkers.
To prove the analyte detection with the prepared high-density arrays, the antigen was directly labeled with a fluorescence dye. The labeling efficiency of the used labeling kit was not analyzed, a 1:1 ratio of cytokine to PE label was assumed according to supplier’s information. This means, it has to be considered that if the labeling efficiency is not 100%, not all bound cytokines can be optically detected by fluorescence microscopy, resulting in an effectively even higher sensitivity. The previously labeled cytokine solution (3 µg/mL) reacted in the microarray with a defined volume, given by the LifterSlip
TM geometry. After reaction of the labeled antigen with the unlabeled, spotted antibody array and washing to remove unbound label as well as unbound antigen, we observed the fluorescent signal shown in
Figure 8A. We chose a low µg/mL concentration for the first experiments in order to be able to expect a visible fluorescence result with the microscope. This means here 45 ng of cytokine was available for binding to the 66,000 spots in the array below the 22 × 30 mm LifterSlip
TM. When the geometry of the array is taken into account, as in a conventional antibody array, we will only have e.g., single spots or more likely triplicates of one type of antibody, hence we determined that one single spot has a detection volume of 0.2 nl/spot (100 µm × 100 µm × 20 µm—spot to spot distances and height of the LifterSlip
TM). Thus, if 3 µg/mL IL-6 (M
W = 20 kDa) can be detected with the antibody microarray, one single spot detected ~0.6 pg IL-6 (=30 attomoles or ~1.8 × 10
7 molecules). Considering that if just the spot to spot distance is doubled within the same setup, the detectable mass of a single spot at the same concentration is at ~2.4 pg. As the spot offers ~7.2 × 10
5 binding sites, only 4% of the cytokine molecules are able to find corresponding binding partners. This calculation reveals that with this method,
i.e., by direct analyte labeling, it is possible to detect pure analyte in the low pg range/spot, having a µg/mL sample concentration, and that the cytokine concentration can be potentially further reduced. The incubation overnight showed to be beneficial for signal intensities. Long incubation times were also suggested by Krusnezow
et al. [
23].
Sandwich assay: Alternatively, the analyte was detected by a second labeled antibody in the sandwich assay format, even the PE labeled anti-TNFα was premixed with TNFα, which made it possible to minimize the incubation on the microarray to one step. We saw that this procedure showed less non-specific binding and a higher signal to background as if the two steps were realized on the antibody microarray one after the other (data not shown). The ratio of PE: anti-TNFα was 1:1 according to the supplier’s information. The premixed solution with a cytokine concentration of 5 µg/mL reacted with a defined volume on the microarray. This means that 75 ng of cytokine were available for binding to all spots. After reaction with the unlabeled, spotted antibody array and washing, fluorescent signals as shown in
Figure 8B resulted. Taking again the geometry of the array and the 66,000 spots into account, (V = 0.2 nl/spot), it can be calculated, that if 5 µg/mL TNFα (M
W = 17 kDa) can be detected with the antibody microarray, one single spot detected ~1 pg TNFα (=60 amol or ~3.6 × 10
7 molecules). As the spot offers ~7.2 × 10
5 binding sites, only 2% of the cytokine molecules are able to find corresponding binding partners. The calculation reveals again that with this method it is also possible to detect the cytokine in the low pg range/spot in a µg/mL sample concentration, and that the cytokine concentration can be potentially further reduced.
Figure 8.
Original fluorescence images. (A) Direct assay, detection of ~0.6 pg IL-6 per spot on a prepared anti-IL-6 microarray, assuming 100% label efficiency with PE. (B) Sandwich assay, 5 µg/mL TNFα was pre-incubated with 100 µg/mL PE-labeled anti-TNFα. Detection of ~1 pg TNFα per spot on a prepared anti-TNFα microarray. A concentration gradient from border to center becomes visible in this experiment. Note: only a representative part of the whole array is depicted.
Figure 8.
Original fluorescence images. (A) Direct assay, detection of ~0.6 pg IL-6 per spot on a prepared anti-IL-6 microarray, assuming 100% label efficiency with PE. (B) Sandwich assay, 5 µg/mL TNFα was pre-incubated with 100 µg/mL PE-labeled anti-TNFα. Detection of ~1 pg TNFα per spot on a prepared anti-TNFα microarray. A concentration gradient from border to center becomes visible in this experiment. Note: only a representative part of the whole array is depicted.
The blocking of the NHS modified surface by an amine-containing buffer, 1M TRIS, resulted as suitable in all experiments; however, non-specific binding was still present. Alternatively, the blocking buffer recommended by the supplier of the hydrogel slide worked well for given slide, but should be avoided because of carcinogenic, mutagenic and toxic to reproduction (CMR) containing material. Lower TRIS buffer concentrations, like 20 mM or 150 mM showed an increased non-specific binding (data not shown).
It was shown that the antibody microarray works and both ways of detection (direct and sandwich assay) result in similar findings. As we wanted to further illustrate the proof of principle in detection with the fluorescent microscope, we chose to run the experiments at a higher cytokine concentration in the low µg/mL-range; detection was even possible in the ng/mL range (data not shown, weaker fluorescence signals). The determination of the LoD at this stage was not yet targeted. However, during all experiments, it was observed that the signal intensity was not equal over the whole microarray, which is clearly visible in
Figure 8B. The spots at the border of the microarray and LifterSlip
TM, where the solution was inserted manually with the pipette, showed stronger signals than those in the center of the array. This also applied if the incubation was over night, allowing more time for the reaction. One possibility could be the depletion of analyte concentration when applying the sample from the edges of a slide with a pipette. This would mean that the spot in the upper row (left) detects the initial cytokine concentration and towards the bottom (right) we approach the LoD, without knowing this final concentration. In the first instance, the reason for analyte depletion is the amount of antibody because the high spot density array of the same antibody offers many specific binding sites, similar to a larger sized spot, leading to depletion of the analyte in the solution. The observation leads, as expected, to the conclusion that ambient analyte conditions, as described by Ekins do not yet apply for this microarray, as the assay is not independent of sample volume and nor of the present amount of antibodies. However, in a real assay, one would of course not target an array with such a spot density, as more than a triplicate per type of antibody is usually not needed. This layout was only selected to demonstrate the spot density. The second point is that Ekins does not consider the unspecific binding, which always causes analyte depletion, especially for low analyte concentrations. This means, to approximate Ekins theory, higher optimization will be needed, considering for example the measurement of the diffusion constants. Due to the analyte depletion from the edges towards the center of the array, the feeding of the target analyte onto the point of detection is of importance. A promising solution could be the spotting of the analyte spot directly onto the detection spot. Assuming that the sample is not in contact with any material, which unspecifically binds the proteins, this would lead to a minimal loss of sample volume, a minimum of unspecific binding and a minimum in diffusion length for protein-protein binding. Spotting of sample on the antibody microarray using a nanoplotter is unfortunately not user friendly, this is why a design of microfluidics under consideration of gained knowledge becomes interesting. Work in this direction to combine an antibody microarray with microfluidics is ongoing and similar work is reported by different groups [
13,
24,
25,
26].

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







