Microarrays 2017, 6(2), 9; https://doi.org/10.3390/microarrays6020009 - 29 May 2017
Cited by 9
Abstract
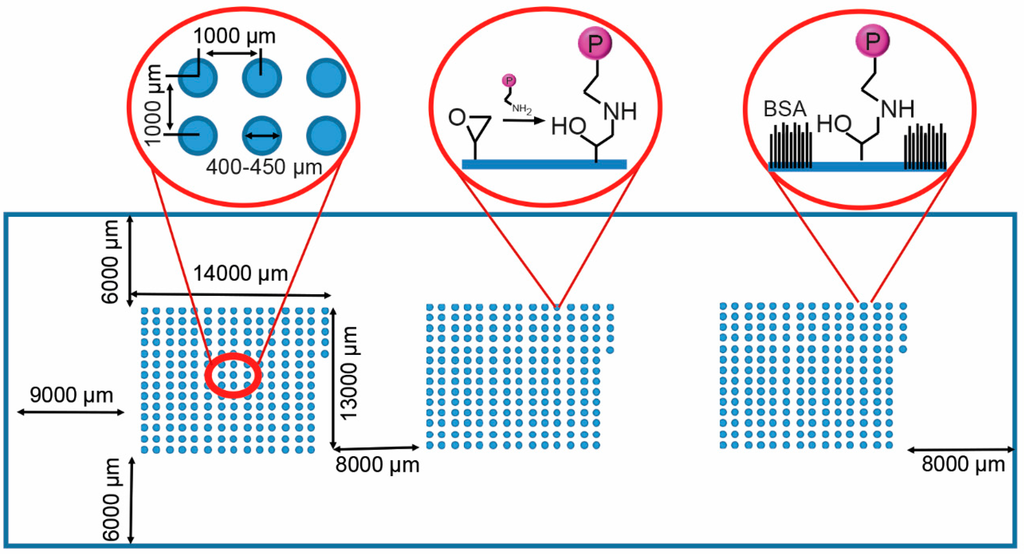
Microfluidic DNA biochips capable of detecting specific DNA sequences are useful in medical diagnostics, drug discovery, food safety monitoring and agriculture. They are used as miniaturized platforms for analysis of nucleic acids-based biomarkers. Binding kinetics between immobilized single stranded DNA on the surface
[...] Read more.
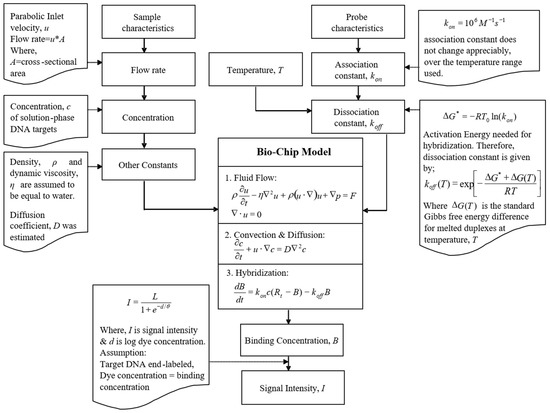
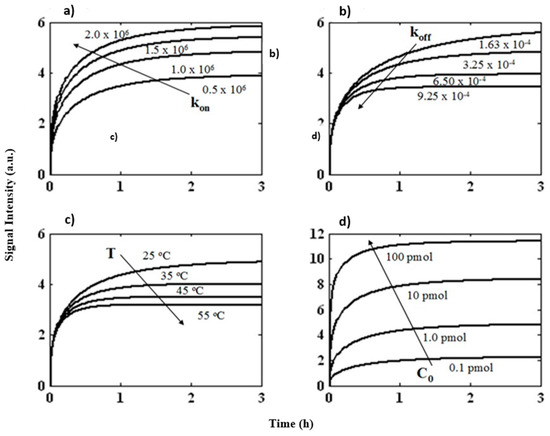
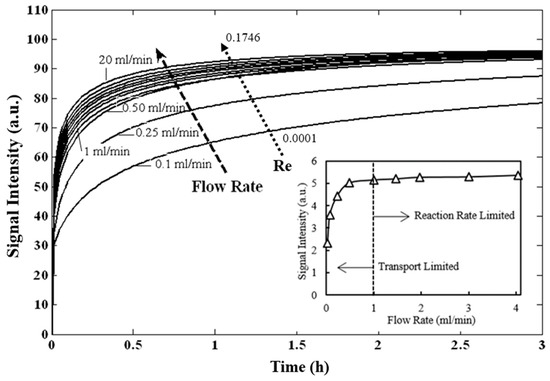
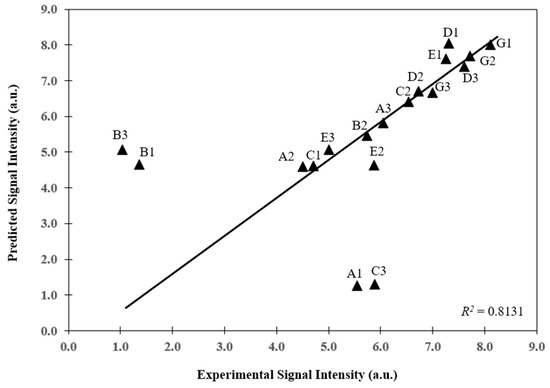
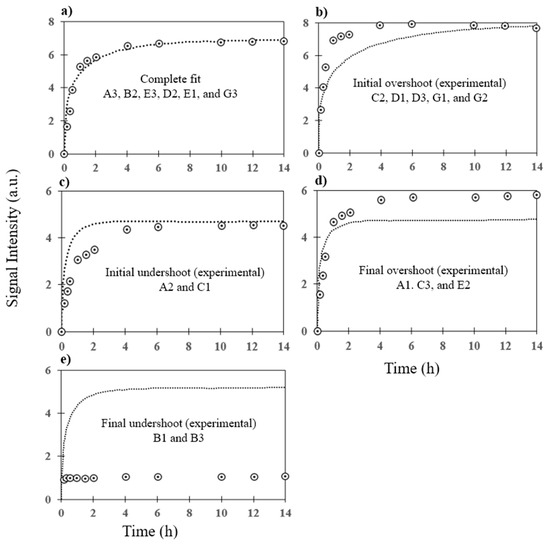
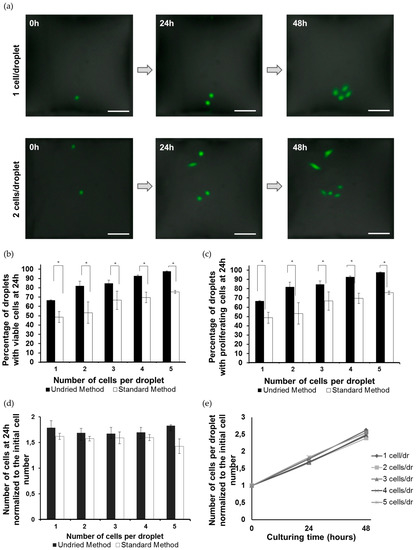
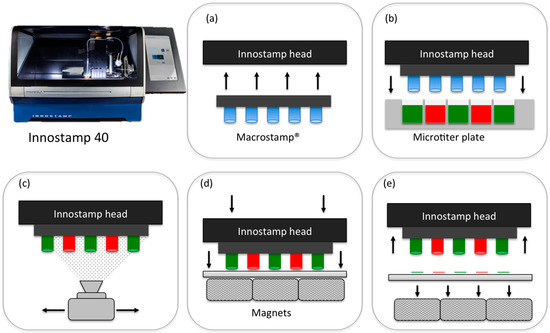
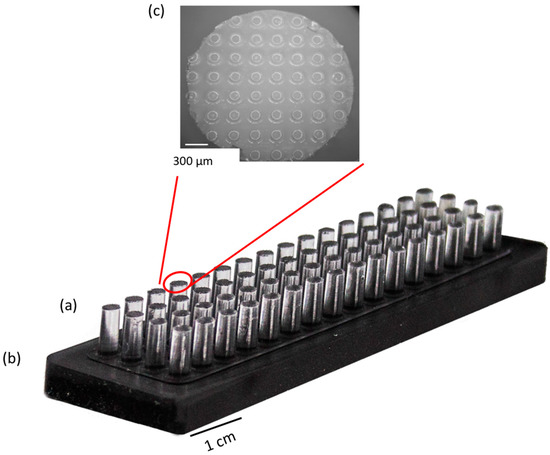
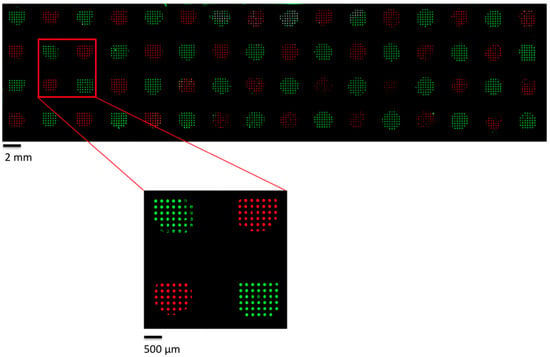
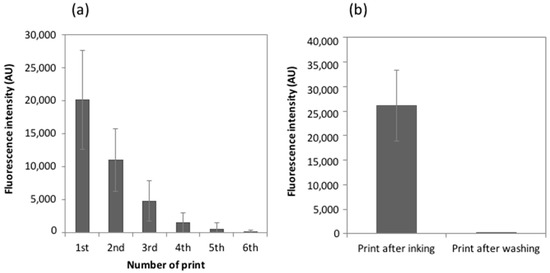
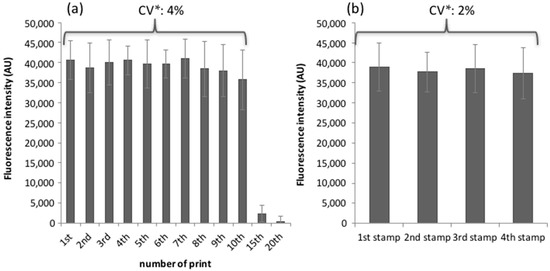
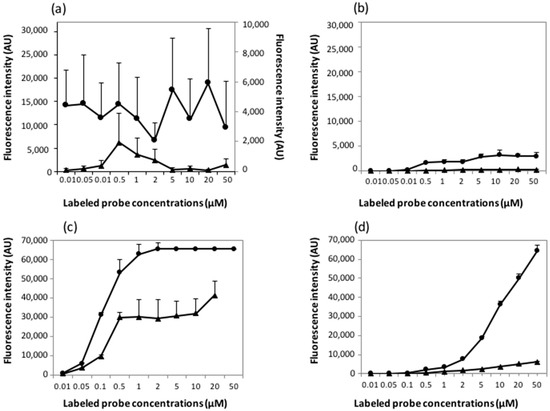
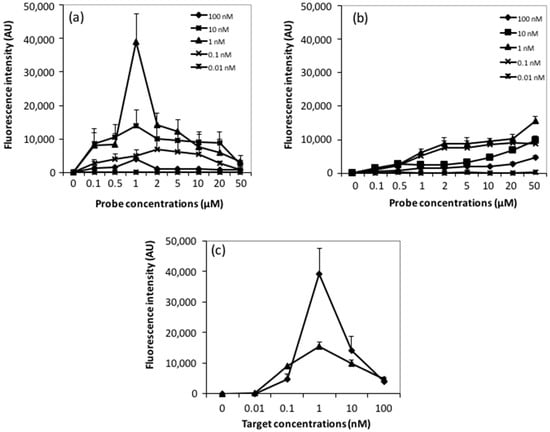
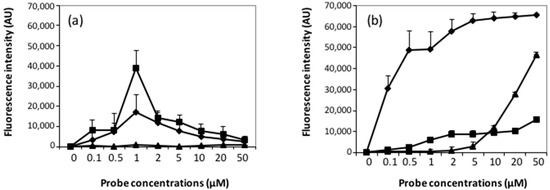
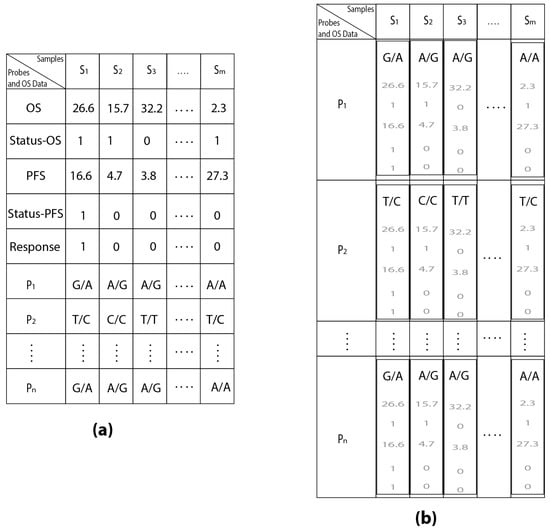
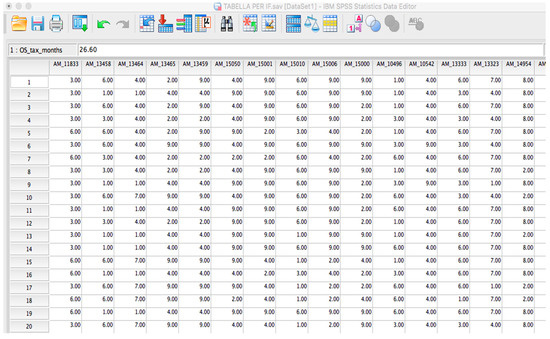
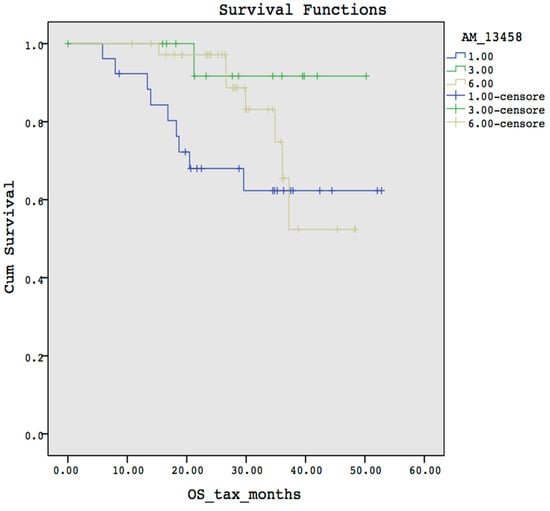
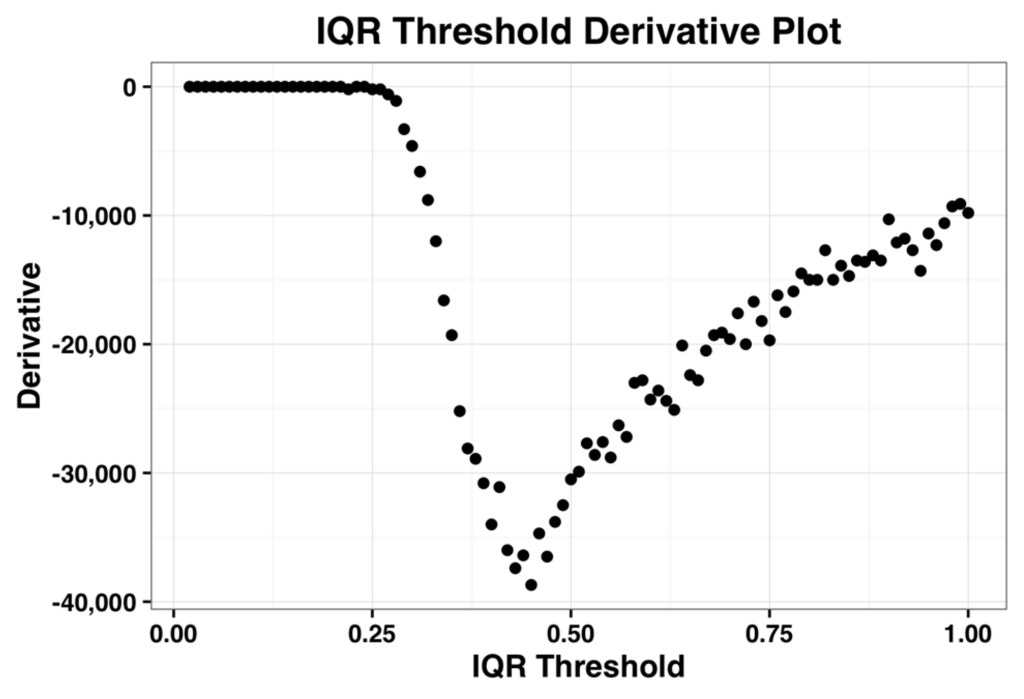
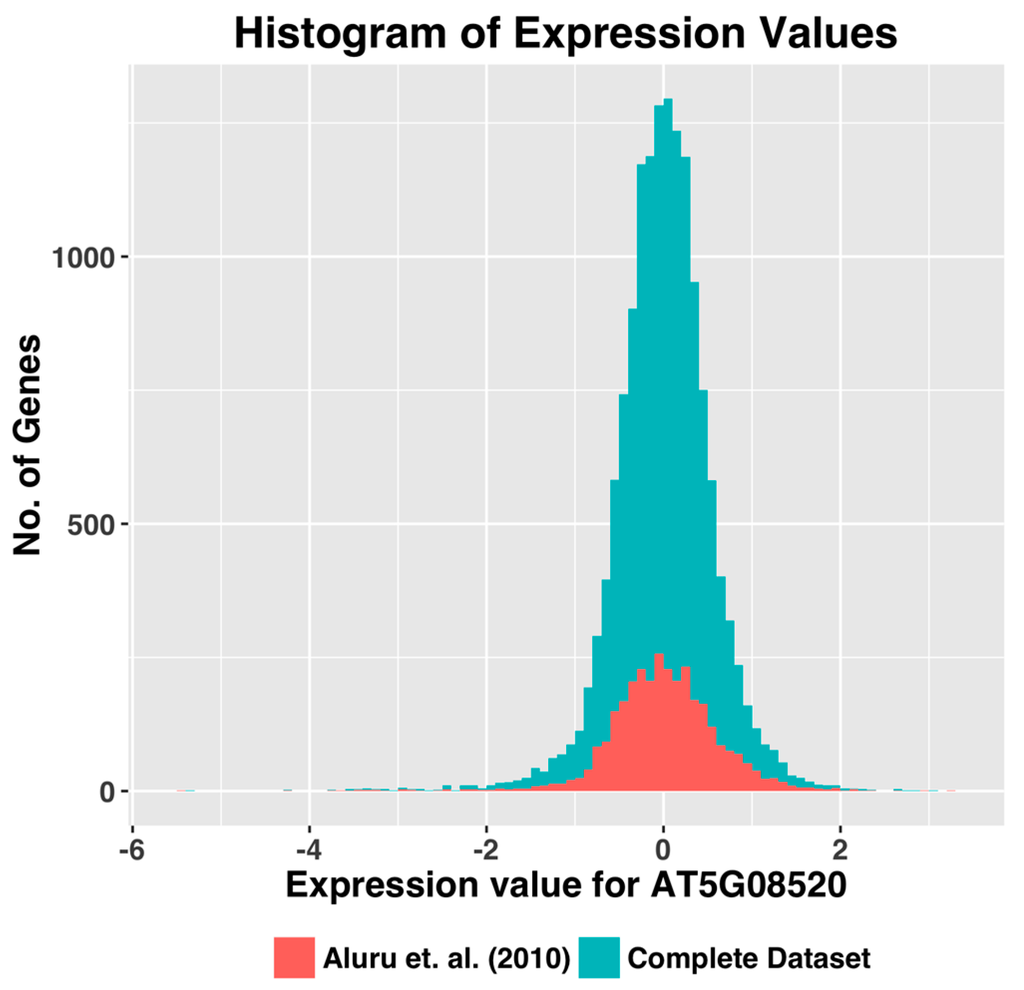
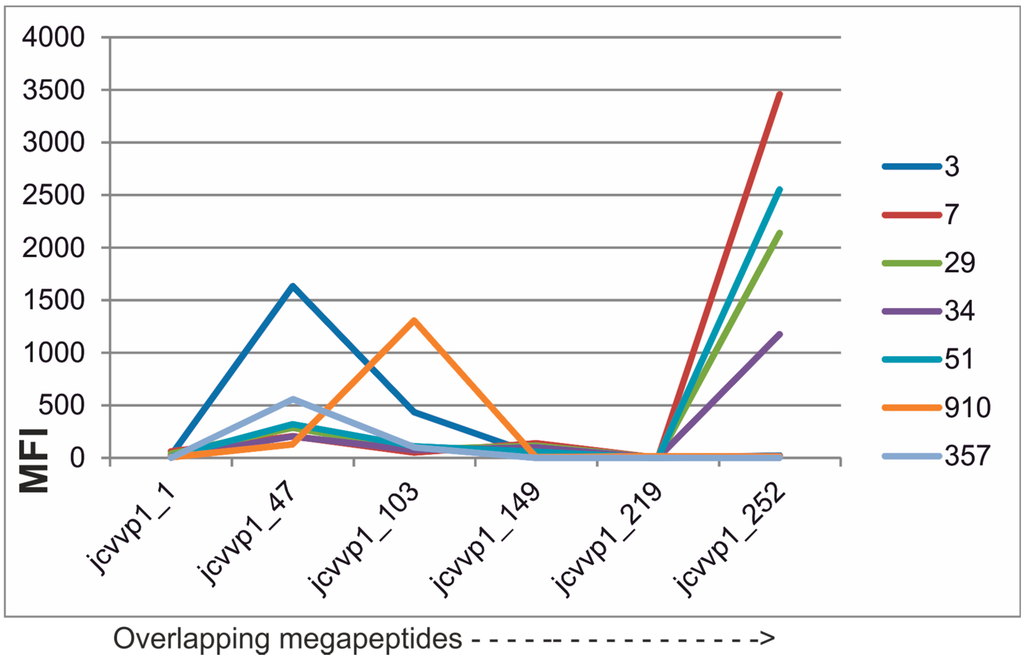
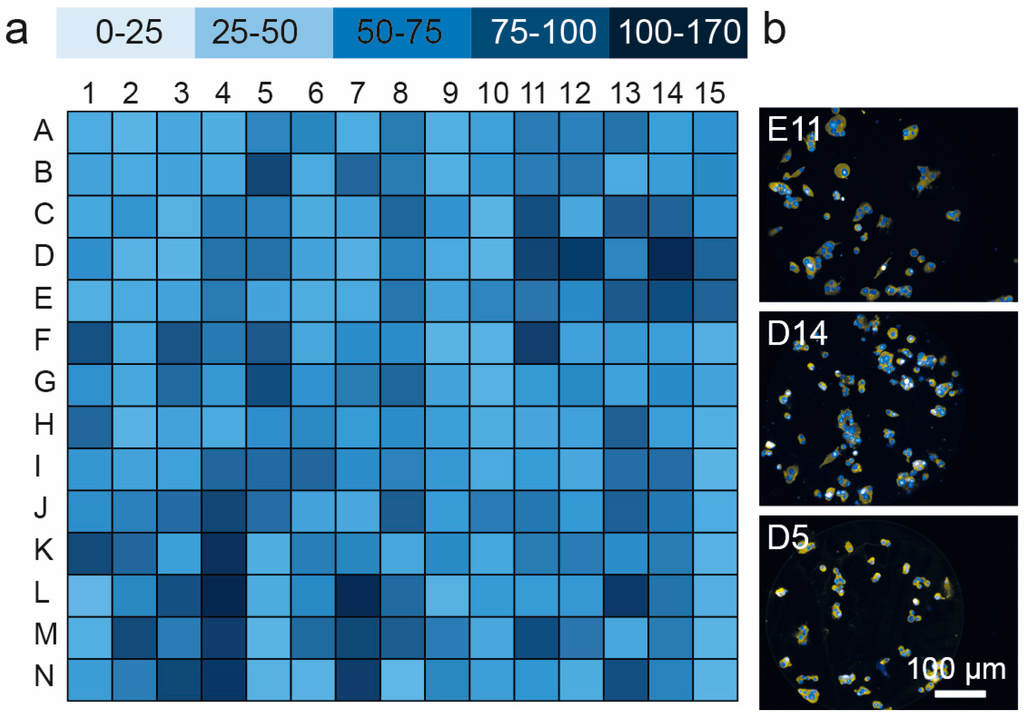
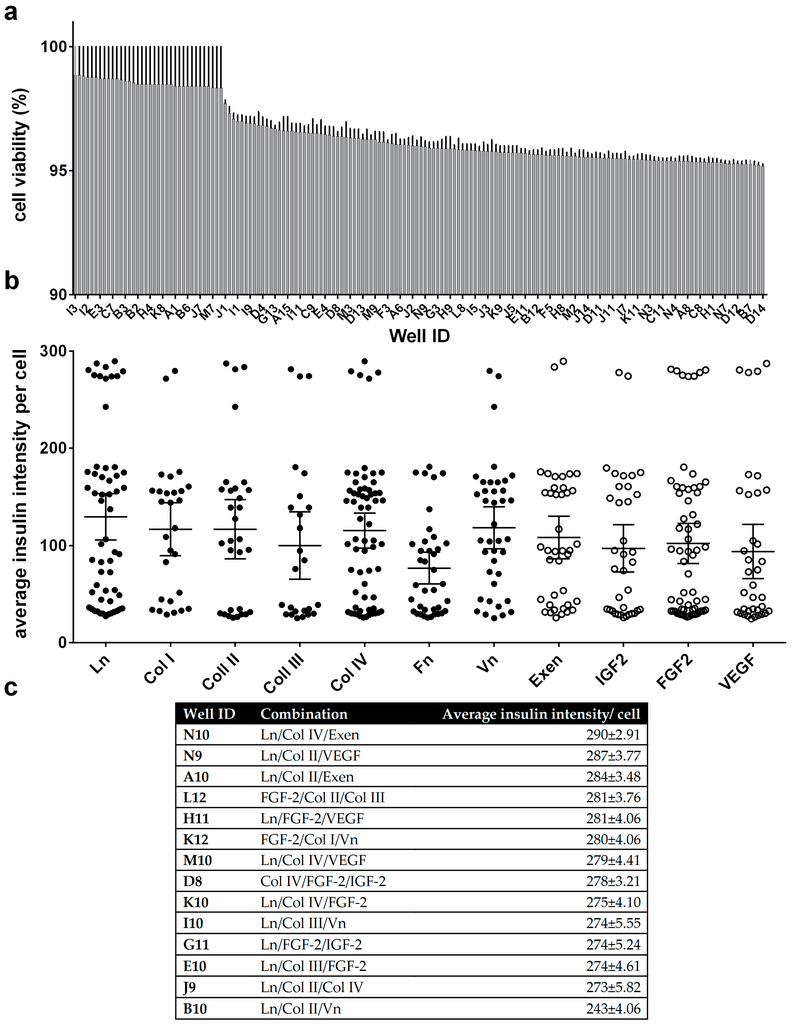
Microfluidic DNA biochips capable of detecting specific DNA sequences are useful in medical diagnostics, drug discovery, food safety monitoring and agriculture. They are used as miniaturized platforms for analysis of nucleic acids-based biomarkers. Binding kinetics between immobilized single stranded DNA on the surface and its complementary strand present in the sample are of interest. To achieve optimal sensitivity with minimum sample size and rapid hybridization, ability to predict the kinetics of hybridization based on the thermodynamic characteristics of the probe is crucial. In this study, a computer aided numerical model for the design and optimization of a flow-through biochip was developed using a finite element technique packaged software tool (FEMLAB; package included in COMSOL Multiphysics) to simulate the transport of DNA through a microfluidic chamber to the reaction surface. The model accounts for fluid flow, convection and diffusion in the channel and on the reaction surface. Concentration, association rate constant, dissociation rate constant, recirculation flow rate, and temperature were key parameters affecting the rate of hybridization. The model predicted the kinetic profile and signal intensities of eighteen 20-mer probes targeting vancomycin resistance genes (VRGs). Predicted signal intensities and hybridization kinetics strongly correlated with experimental data in the biochip (R2 = 0.8131).
Full article
(This article belongs to the Special Issue Microfluidics Technology)
►
Show Figures
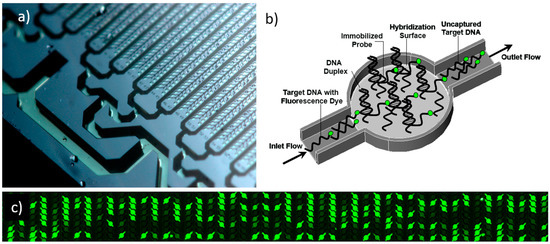
Figure 1



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}