Mitochondria, Oxidative Stress, cAMP Signalling and Apoptosis: A Crossroads in Lymphocytes of Multiple Sclerosis, a Possible Role of Nutraceutics

 ,
, 
 ,
,
Abstract
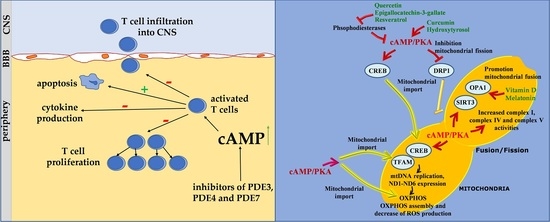
:
1. Introduction
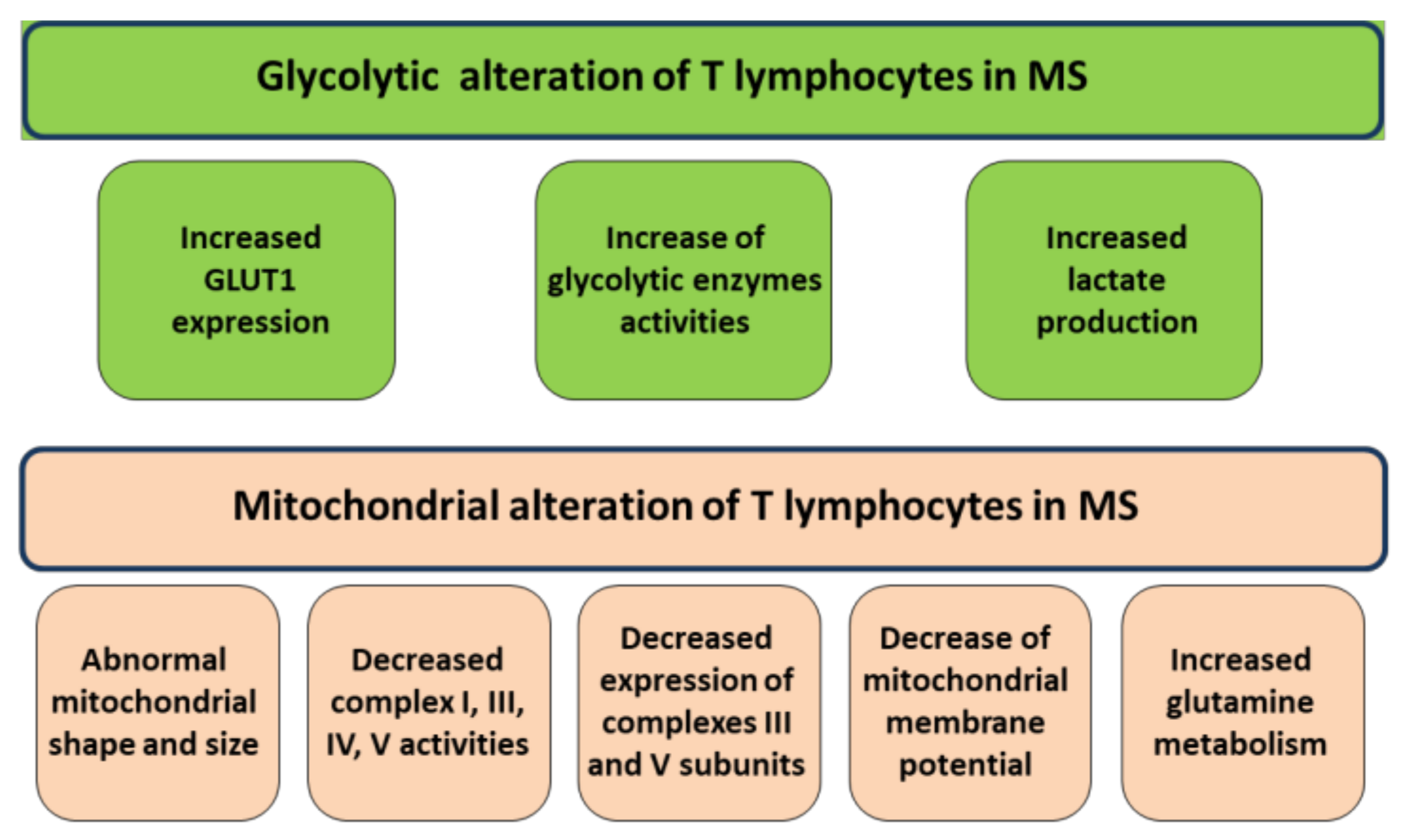
2. Mitochondrial Alterations of T Lymphocytes in MS
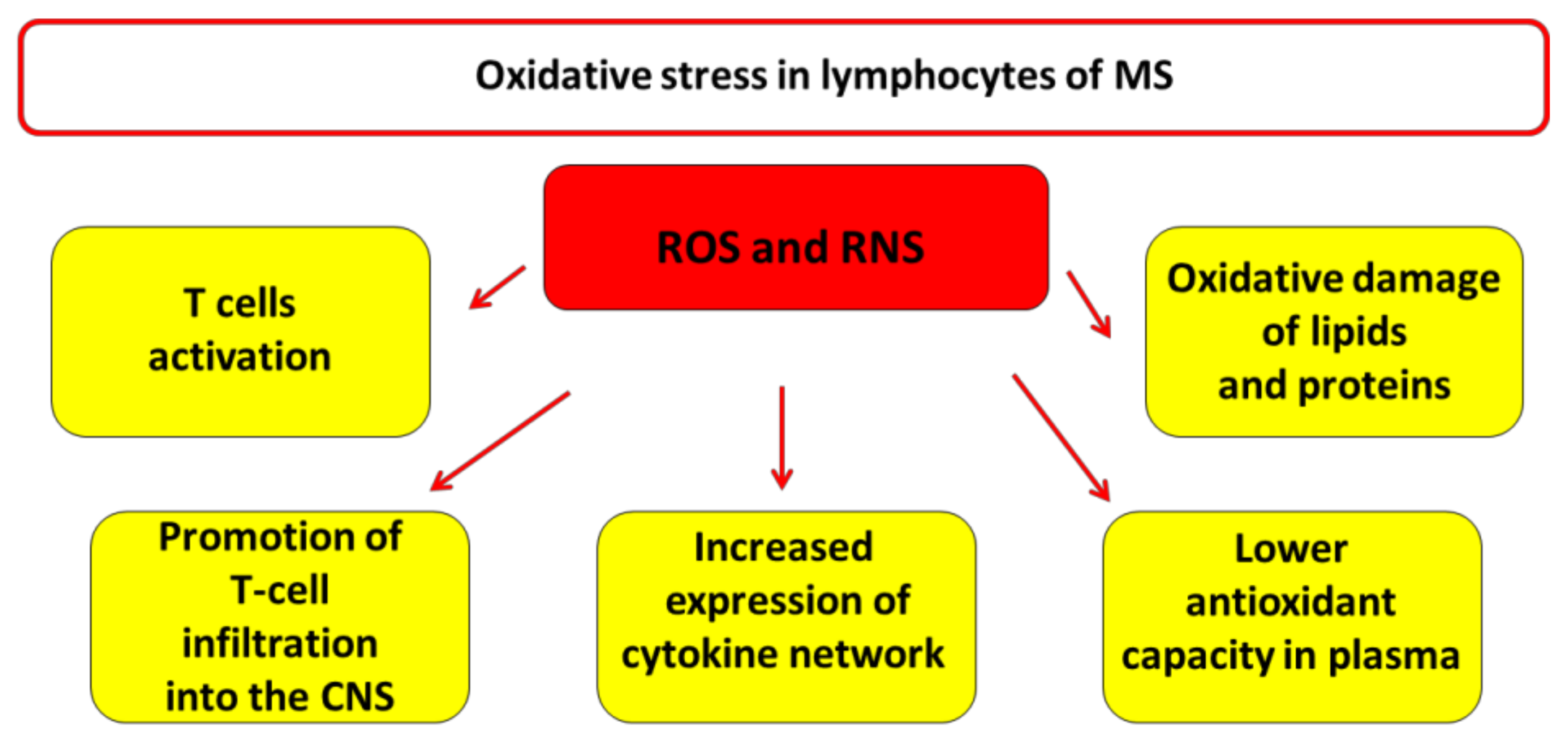
3. Oxidative Stress in Lymphocytes of MS
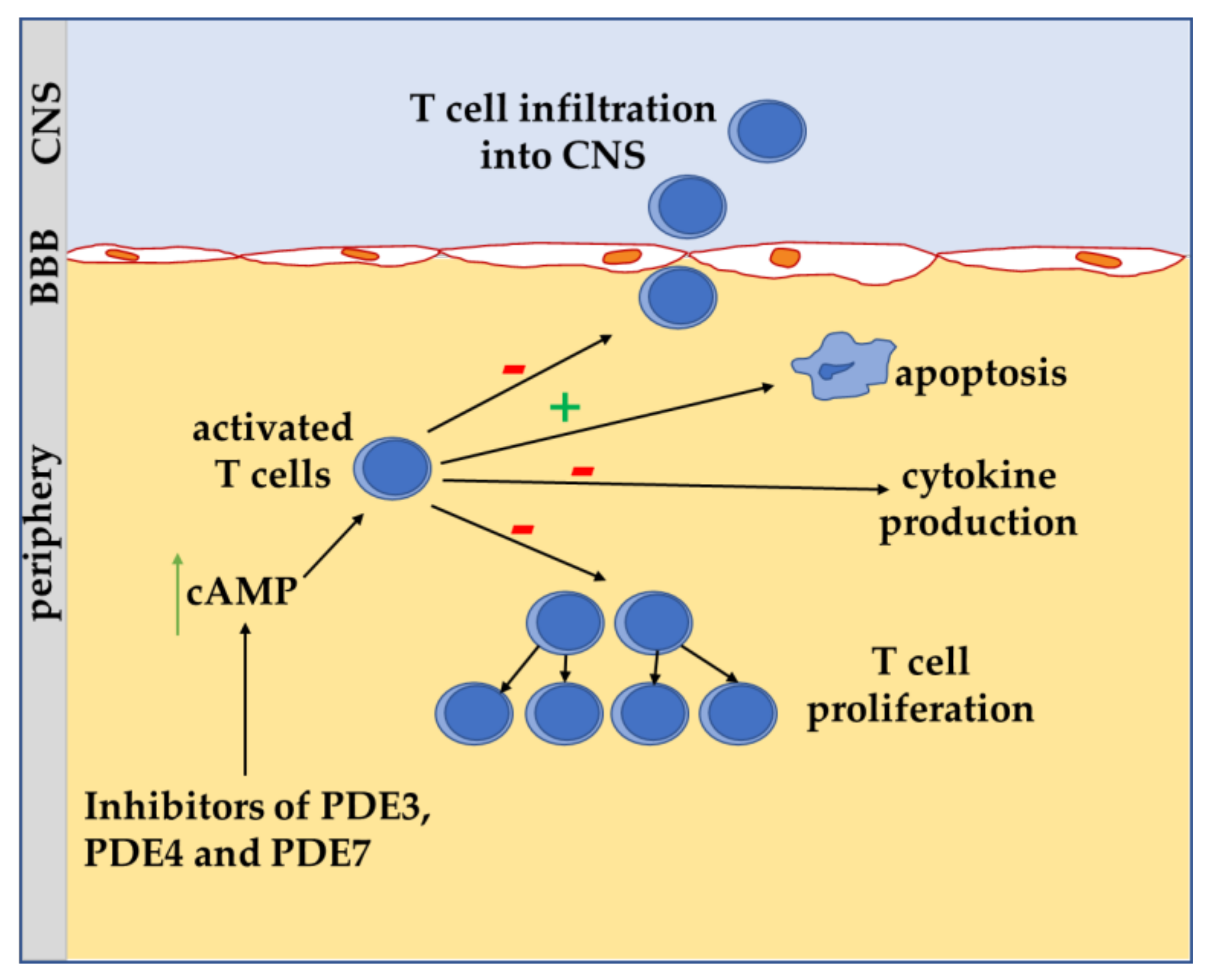
4. Role of cAMP Signaling in T Lymphocyte of MS
5. Apoptosis and Lymphocytes in MS
6. The cAMP Signalling in the Regulation of Mitochondrial Activity, ROS Balance and T Cell Proliferation: Possible Implications of Some Nutraceutics
6.1. cAMP, Mitochondria and ROS Balance
6.2. Nutraceutics
6.2.1. Curcumin
6.2.2. Flavonoids
6.2.3. Resveratrol
6.2.4. Hydroxytyrosol
6.2.5. Vitamin D
6.2.6. Melatonin
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lassmann, H.; Brück, W.; Lucchinetti, C.F. The immunopathology of multiple sclerosis: An overview. Brain Pathol. 2007, 17, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Tobore, T.O. Towards a comprehensive etiopathogenetic and pathophysiological theory of multiple sclerosis. Int. J. Neurosci. 2020, 130, 279–300. [Google Scholar] [CrossRef] [PubMed]
- Lublin, F.D.; Reingold, S.C. Defining the clinical course of multiple sclerosis: Results of an international survey. Neurology 1996, 46, 907–911. [Google Scholar] [CrossRef] [PubMed]
- Klineova, S.; Lublin, F.D. Clinical Course of Multiple Sclerosis. Cold Spring Harb. Perspect. Med. 2018, 8, a028928. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Vécsei, L. Monitoring the Redox Status in Multiple Sclerosis. Biomedicines 2020, 8, 406. [Google Scholar] [CrossRef] [PubMed]
- Sospedra, M.; Martin, R. Immunology of multiple sclerosis. Annu. Rev. Immunol. 2005, 23, 683–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadovnick, A.D.; Armstrong, H.; Rice, G.P.; Bulman, D.; Hashimoto, L.; Party, D.W.; Hashimoto, S.A.; Warren, S.; Hader, W.; Murrary, T.J.; et al. A population-based study of multiple sclerosis in twins: Update. Ann. Neurol. 1993, 33, 281–285. [Google Scholar] [CrossRef]
- Dyment, D.A.; Ebers, G.C.; Sadovnick, A.D. Genetics of multiple sclerosis. Lancet Neurol. 2004, 3, 104–110. [Google Scholar] [CrossRef] [Green Version]
- Hewer, S.; Lucas, R.; van der Mei, I.; Taylor, B.V. Vitamin D and multiple sclerosis. J. Clin. Neurosci. 2013, 20, 634–641. [Google Scholar] [CrossRef]
- Pender, M.P.; Burrows, S.R. Epstein-Barr virus and multiple sclerosis: Potential opportunities for immunotherapy. Clin. Transl. Immunol. 2014, 3, e27. [Google Scholar] [CrossRef]
- Tanaka, M.; Toldi, J.; Vécsei, L. Exploring the Etiological Links behind Neurodegenerative Diseases: Inflammatory Cytokines and Bioactive Kynurenines. J. Mol. Sci. 2020, 21, 2431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lassmann, H. Pathogenic Mechanisms Associated with Different Clinical Courses of Multiple Sclerosis. Front. Immunol. 2019, 9, 3116. [Google Scholar] [CrossRef] [PubMed]
- Van Langelaar, J.; Rijvers, L.; Smolders, J.; van Luijn, M.M. B and T Cells Driving Multiple Sclerosis: Identity, Mechanisms and Potential Triggers. Front. Immunol. 2020, 11, 760. [Google Scholar] [CrossRef]
- Kammer, G.M. The adenylate cyclase-cAMP-protein kinase A pathway and regulation of the immune response. Immunol. Today 1988, 9, 222–229. [Google Scholar] [CrossRef]
- Geltink, R.I.K.; Kyle, R.L.; Pearce, E.L. Unraveling the Complex Interplay Between T Cell Metabolism andFunction. Annu. Rev. Immunol. 2018, 36, 461–488. [Google Scholar] [CrossRef]
- Corthay, A. A three-cell model for activation of naïve T helper cells. Scand. J. Immunol. 2006, 64, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Taghian, K.; Petratos, S. Axonal degeneration in multiple sclerosis: Can we predict and prevent permanent disability? Acta Neuropathol. Commun. 2014, 2, 97. [Google Scholar] [CrossRef]
- Bargiela, D.; Chinnery, P.F. Mitochondria in neuroinflammation - Multiple sclerosis (MS), leber hereditary optic neuropathy (LHON) and LHON-MS. Neurosci. Lett. 2019, 710, 132932. [Google Scholar] [CrossRef]
- Wehbi, V.L.; Taskén, K. Molecular Mechanisms for cAMP-Mediated Immunoregulation in T cells - Role of Anchored Protein Kinase A Signaling Units. Front. Immunol. 2016, 7, 222. [Google Scholar] [CrossRef] [Green Version]
- Frauwirth, K.A.; Thompson, C.B. Activation and inhibition of lymphocytes by costimulation. J. Clin. Investig. 2002, 109, 295–299. [Google Scholar] [CrossRef]
- Schepers, M.; Tiane, A.; Paes, D.; Sanchez, S.; Rombaut, B.; Piccart, E.; Rutten, B.P.F.; Brône, B.; Hellings, N.; Prickaerts, J.; et al. Targeting Phosphodiesterases-Towards a Tailor-Made Approach in Multiple Sclerosis Treatment. Front. Immunol. 2019, 10, 1727. [Google Scholar] [CrossRef] [PubMed]
- Gilgun-Sherki, Y.; Melamed, E.; Offen, D. The role of oxidative stress in the pathogenesis of multiple sclerosis: The need for effective antioxidant therapy. J. Neurol. 2004, 251, 261–268. [Google Scholar] [PubMed]
- Ohl, K.; Tenbrock, K.; Kipp, M. Oxidative stress in multiple sclerosis: Central and peripheral mode of action. Exp. Neurol. 2016, 277, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Gonzalo, H.; Nogueras, L.; Gil-Sánchez, A.; Hervás, J.V.; Valcheva, P.; González-Mingot, C.; Martin-Gari, M.; Canudes, M.; Peralta, S.; Solana, M.J.; et al. Impairment of Mitochondrial Redox Status in Peripheral Lymphocytes of Multiple Sclerosis Patients. Front. Neurosci. 2019, 13, 938. [Google Scholar] [CrossRef] [PubMed]
- De Rasmo, D.; Ferretta, A.; Russo, S.; Ruggieri, M.; Lasorella, P.; Paolicelli, D.; Trojano, M.; Signorile, A. PBMC of Multiple Sclerosis Patients Show Deregulation of OPA1 Processing Associated with Increased ROS and PHB2 Protein Levels. Biomedicines 2020, 8, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McFarland, H.F.; Martin, R. Multiple sclerosis: A complicated picture of autoimmunity. Nat. Immunol. 2007, 8, 913–919. [Google Scholar] [CrossRef]
- Comabella, M.; Khoury, S.J. Immunopathogenesis of multiple sclerosis. Clin. Immunol. 2012, 142, 2–8. [Google Scholar] [CrossRef]
- Segal, B.M.; Cross, A.H. Fas(t) track to apoptosis in MS: TNF receptors may suppress or potentiate CNS demyelination. Neurology 2000, 55, 906–907. [Google Scholar] [CrossRef]
- Ruggieri, M.; Avolio, C.; Scacco, S.; Pica, C.; Lia, A.; Zimatore, G.B.; Papa, S.; Livrea, P.; Trojano, M. Glatiramer acetate induces pro-apoptotic mechanisms involving Bcl-2, Bax and Cyt-c in peripheral lymphocytes from multiple sclerosis patients. J. Neurol. 2006, 253, 231–236. [Google Scholar] [CrossRef]
- Julià, E.; Edo, M.C.; Horga, A.; Montalban, X.; Comabella, M. Differential susceptibility to apoptosis of CD4+T cells expressing CCR5 and CXCR3 in patients with MS. Clin. Immunol. 2009, 133, 364–374. [Google Scholar] [CrossRef]
- La Rocca, C.; Carbone, F.; De Rosa, V.; Colamatteo, A.; Galgani, M.; Perna, F.; Lanzillo, R.; Brescia Morra, V.; Orefice, G.; Cerillo, I.; et al. Immunometabolic profiling of T cells from patients with relapsing-remitting multiple sclerosis reveals an impairment in glycolysis and mitochondrial respiration. Metabolism 2017, 77, 39–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shyer, J.A.; Flavell, R.A.; Bailis, W. Metabolic signaling in T cells. Cell Res. 2020, 30, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.G.; Thompson, C.B. Revving the engine: Signal transduction fuels T cell activation. Immunity 2007, 27, 173–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Previte, D.M.; O’Connor, E.C.; Novak, E.A.; Martins, C.P.; Mollen, K.P.; Piganelli, J.D. Reactive oxygen species are required for driving efficient and sustained aerobic glycolysis during CD4+ T cell activation. PLoS ONE 2017, 12, e0175549. [Google Scholar] [CrossRef]
- Johnson, M.O.; Wolf, M.M.; Madden, M.Z.; Andrejeva, G.; Sugiura, A.; Contreras, D.C.; Maseda, D.; Liberti, M.V.; Paz, K.; Kishton, R.J.; et al. Distinct regulation of Th17 and Th1 cell differentiation by glutaminase-dependent metabolism. Cell 2018, 175, 1780–1795. [Google Scholar] [CrossRef] [Green Version]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef] [Green Version]
- Seyfried, T.N.; Arismendi-Morillo, G.; Mukherjee, P.; Chinopoulos, C. On the Origin of ATP Synthesis in Cancer. iScience 2020, 23, 101761. [Google Scholar] [CrossRef]
- Sena, L.A.; Li, S.; Jairaman, A.; Prakriya, M.; Ezponda, T.; Hildeman, D.A.; Wang, C.R.; Schumacker, P.T.; Licht, J.D.; Perlman, H.; et al. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity 2013, 38, 225–236. [Google Scholar] [CrossRef] [Green Version]
- Djaldetti, R.; Achiron, A.; Ziv, I.; Djaldetti, M. Lymphocyte ultrastructure in patients with multiple sclerosis. Biomed. Pharmacother. 1995, 49, 300–303. [Google Scholar] [CrossRef]
- De Riccardis, L.; Rizzello, A.; Ferramosca, A.; Urso, E.; De Robertis, F.; Danieli, A.; Giudetti, A.M.; Trianni, G.; Zara, V.; Maffia, M. Bioenergetics profile of CD4+T cells in relapsing remittingmultiplesclerosissubjects. J. Biotechnol. 2015, 202, 31–39. [Google Scholar] [CrossRef]
- Armon-Omer, A.; Neuman, H.; Sharabi-Nov, A.; Shahien, R. Mitochondrial activity is impaired in lymphocytes of MS patients in correlation with disease severity. Mult. Scler. Relat. Disord. 2020, 41, 102025. [Google Scholar] [CrossRef] [PubMed]
- Salim, S. Oxidative stress and the central nervous system. J. Pharmacol. Exp. Ther. 2017, 360, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Miljković, D.; Spasojević, I. Multiple sclerosis: Molecular mechanisms and therapeutic opportunities. Antioxid. Redox Signal. 2013, 19, 2286–2334. [Google Scholar] [CrossRef] [PubMed]
- Török, N.; Tanaka, M.; Vécsei, L. Searching for Peripheral Biomarkers in Neurodegenerative Diseases: The Tryptophan-Kynurenine Metabolic Pathway. Int. J. Mol. Sci. 2020, 21, 9338. [Google Scholar] [CrossRef] [PubMed]
- Miller, E.; Walczak, A.; Saluk, J.; Ponczek, M.B.; Majsterek, I. Oxidative modification of patient’s plasma proteins andits role in pathogenesis of multiple sclerosis. Clin. Biochem. 2012, 45, 26–30. [Google Scholar] [CrossRef] [PubMed]
- Padureanu, R.; Albu, C.V.; Mititelu, R.R.; Bacanoiu, M.V.; Docea, A.O.; Calina, D.; Padureanu, V.; Olaru, G.; Sandu, R.E.; Malin, R.D.; et al. Oxidative Stress and Inflammation Interdependence in Multiple Sclerosis. J. Clin. Med. 2019, 8, 1815. [Google Scholar] [CrossRef] [Green Version]
- Siotto, M.; Filippi, M.M.; Simonelli, I.; Landi, D.; Ghazaryan, A.; Vollaro, S.; Ventriglia, M.; Pasqualetti, P.; Rongioletti, M.C.A.; Squitti, R.; et al. Oxidative Stress Related to Iron Metabolism in Relapsing Remitting Multiple Sclerosis Patients with Low Disability. Front. Neurosci. 2019, 13, 86. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.; Tsirka, S.E. Endothelial NOS-deficient mice reveal dual roles for nitric oxide during experimental autoimmune encephalomyelitis. Glia 2009, 57, 1204–1215. [Google Scholar] [CrossRef] [Green Version]
- Marui, N.; Offermann, M.K.; Swerlick, R.; Kunsch, C.; Rosen, C.A.; Ahmad, M.; Alexander, R.W.; Medford, R.M. Vascular cell adhesion molecule-1 (VCAM-1) gene transcription and expression are regulated through an antioxidant-sensitive mechanism in human vascular endothelial cells. J. Clin. Investig. 1993, 92, 1866–1874. [Google Scholar] [CrossRef]
- Jäger, A.; Dardalhon, V.; Sobel, R.A.; Bettelli, E.; Kuchroo, V.K. Th1, Th17, and Th9 effector cells induce experimental autoimmune encephalomyelitis with different pathological phenotypes. J. Immunol. 2009, 183, 7169–7177. [Google Scholar]
- Munro, D.; Treberg, J.R. A radical shift in perspective: Mitochondria as regulators of reactive oxygen species. J. Exp. Biol. 2017, 220, 1170–1180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hampton, M.B.; Kettle, A.J.; Winterbourn, C.C. Inside the neutrophil phagosome: Oxidants, myeloperoxidase, and bacterial killing. Blood 1998, 92, 3007–3017. [Google Scholar] [CrossRef] [PubMed]
- Pacelli, C.; De Rasmo, D.; Signorile, A.; Grattagliano, I.; di Tullio, G.; D’Orazio, A.; Nico, B.; Comi, G.P.; Ronchi, D.; Ferranini, E.; et al. Mitochondrial defect and PGC-1α dysfunction in parkin-associated familial Parkinson’s disease. Biochim. Biophys. Acta 2011, 1812, 1041–1053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarafdar, A.; Pula, G. The Role of NADPH Oxidases and Oxidative Stress in Neurodegenerative Disorders. Int. J. Mol. Sci. 2018, 19, 3824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahl, K.G.; Zielasek, J.; Uttenthal, L.O.; Rodrigo, J.; Toyka, K.V.; Schmidt, H.H. Protective role of the cytokine-inducible isoform of nitric oxide synthase induction and nitrosative stress in experimental autoimmune encephalomyelitis of the DA rat. J. Neurosci. Res. 2003, 73, 198–205. [Google Scholar] [CrossRef]
- Zehntner, S.; Bourbonniere, L.; Hassan-Zahraee, M.; Tran, E.; Owens, T. Bone marrow-derived versus parenchymal sources of inducible nitric oxide synthase in experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2004, 150, 70–79. [Google Scholar] [CrossRef]
- Dalton, D.; Wittmer, S. Nitric-oxide-dependent and independent mechanisms of protection from CNS inflammation during Th1-mediated autoimmunity: Evidence from EAE in iNOS KO mice. J. Neuroimmunol. 2005, 160, 110–121. [Google Scholar] [CrossRef]
- Thiel, V.E.; Audus, K.L. Nitric oxide and blood-brain barrier integrity. Antioxid. Redox Signal. 2001, 3, 273–278. [Google Scholar] [CrossRef]
- Lassmann, H.; van Horssen, J. Oxidative stress and its impact on neurons and glia in multiple sclerosis lesions. Biochim. Biophys. Acta 2016, 1862, 506–510. [Google Scholar] [CrossRef]
- Wang, Y.; Branicky, R.; Noë, A.; Hekimi, S. Superoxide dismutases: Dual roles in controlling ROS damage and regulating ROS signaling. J. Cell Biol. 2018, 217, 1915–1928. [Google Scholar] [CrossRef]
- Karlík, M.; Valkovič, P.; Hančinová, V.; Krížová, L.; Tóthová, L.; Celec, P. Markers of oxidative stress in plasma and saliva in patients with multiple sclerosis. Clin. Biochem. 2015, 48, 24–28. [Google Scholar]
- Morel, A.; Bijak, M.; Niwald, M.; Miller, E.; Saluk, J. Markers of oxidative/nitrative damage of plasma proteins correlated with EDSS and BDI scores in patients with secondary progressive multiple sclerosis. Redox Rep. 2017, 22, 547–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Benedetto, G.; Gerbino, A.; Lefkimmiatis, K. Shaping mitochondrial dynamics: The role of cAMP signalling. Biochem. Biophys. Res. Commun. 2018, 27, 65–74. [Google Scholar]
- Mosenden, R.; Taskén, K. Cyclic AMP-mediated immune regulation--overview of mechanisms of action in T cells. Cell. Signal. 2011, 23, 1009–1016. [Google Scholar] [CrossRef] [PubMed]
- Raskovalova, T.; Lokshin, A.; Huang, X.; Jackson, E.K.; Gorelik, E. Adenosine-mediated inhibition of cytotoxic activity and cytokine production by IL-2/NKp46-activated NK cells: Involvement of protein kinase A isozyme I (PKA I). Immunol. Res. 2006, 36, 91–99. [Google Scholar] [CrossRef]
- Raker, V.K.; Becker, C.; Steinbrink, K. The cAMP Pathway as Therapeutic Target in Autoimmune and Inflammatory Diseases. Front. Immunol. 2016, 7, 123. [Google Scholar] [CrossRef] [Green Version]
- Kammer, G.M. High prevalence of T cell type I protein kinase A deficiency in systemic lupus erythematosus. Arthritis Rheum. 1999, 42, 1458–1465. [Google Scholar] [CrossRef]
- Salinthone, S.; Yadav, V.; Schillace, R.V.; Bourdette, D.N.; Carr, D.W. Lipoic acid attenuates inflammation via cAMP and protein kinase A signaling. PLoS ONE 2010, 5, e13058. [Google Scholar] [CrossRef] [Green Version]
- Fiedler, S.E.; Yadav, V.; Kerns, A.R.; Tsang, C.; Markwardt, S.; Kim, E.; Spain, R.; Bourdette, D.; Salinthone, S. Lipoic Acid Stimulates cAMP Production in Healthy Control and Secondary Progressive MS Subjects. Mol. Neurobiol. 2018, 55, 6037–6049. [Google Scholar] [CrossRef]
- Lacour, M.; Arrighi, J.F.; Müller, K.M.; Carlberg, C.; Saurat, J.H.; Hauser, C. cAMP up-regulates IL-4 and IL-5 production from activated CD4+ T cells while decreasing IL-2 release and NF-AT induction. Int. Immunol. 1994, 6, 1333–1343. [Google Scholar] [CrossRef] [Green Version]
- Sharifzadeh, M.; Zamanian, A.R.; Gholizadeh, S.; Tabrizian, K.; Etminani, M.; Khalaj, S.; Zarrindast, M.R.; Roghani, A. Post-training intrahippocampal infusion of nicotine-bucladesine combination causes a synergistic enhancement effect on spatial memory retention in rats. Eur. J. Pharmacol. 2007, 562, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Khezri, S.; Javan, M.; Goudarzvand, M.; Semnanian, S.; Baharvand, H. Dibutyryl cyclic AMP inhibits the progression of experimental autoimmune encephalomyelitis and potentiates recruitment of endogenous neural stem cells. J. Mol. Neurosci. 2013, 51, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Vakilzadeh, G.; Khodagholi, F.; Ghadiri, T.; Darvishi, M.; Ghaemi, A.; Noorbakhsh, F.; Gorji, A.; Sharifzadeh, M. Protective Effect of a cAMP Analogue on Behavioral Deficits and Neuropathological Changes in Cuprizone Model of Demyelination. Mol. Neurobiol. 2015, 52, 130–141. [Google Scholar] [CrossRef] [PubMed]
- Szczypka, M. Role of Phosphodiesterase 7 (PDE7) in T Cell Activity: Effects of Selective PDE7 Inhibitors and Dual PDE4/7 Inhibitors on T Cell Functions. Int. J. Mol. Sci. 2020, 21, 6118. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; McIntyre, K.W.; Townsend, R.M.; Shen, H.H.; Pitts, W.J.; Dodd, J.H.; Nadler, S.G.; McKinnon, M.; Watson, A.J. Phosphodiesterase 7A-deficient mice have functional T cells. J. Immunol. 2003, 171, 6414–6420. [Google Scholar] [CrossRef] [Green Version]
- Peter, D.; Jin, S.L.; Conti, M.; Hatzelmann, A.; Zitt, C. Differential expression and function of phosphodiesterase 4 (PDE4) subtypes in human primary CD4+ T cells: Predominant role of PDE4D. J. Immunol. 2007, 178, 4820–4831. [Google Scholar] [CrossRef] [Green Version]
- Sanabra, C.; Johansson, E.M.; Mengod, G. Critical role for PDE4 subfamilies in the development of experimental autoimmune encephalomyelitis. J. Chem. Neuroanat. 2013, 47, 96–105. [Google Scholar] [CrossRef] [Green Version]
- Reyes-Irisarri, E.; Sanchez, A.J.; Garcia-Merino, J.A.; Mengod, G. Selective induction of cAMP phosphodiesterase PDE4B2 expression in experimental autoimmune encephalomyelitis. J. Neuropathol. Exp. Neurol. 2007, 66, 923–931. [Google Scholar] [CrossRef] [Green Version]
- Kureshiro, J.; Miyamoto, K.; Tanaka, N.; Kusunoki, S. Selective phosphodiesterase-3 inhibitor cilostazol ameliorates experimental autoimmune encephalomyelitis. NeuroReport 2009, 20, 718–722. [Google Scholar] [CrossRef]
- De Medeiros, A.S.; Wyman, A.R.; Alaamery, M.A.; Allain, C.; Ivey, F.D.; Wang, L.; Le, H.; Morken, J.P.; Habara, A.; Le, C.; et al. Identification and characterization of a potent and biologically-active PDE4/7 inhibitor via fission yeast-based assays. Cell. Signal. 2017, 40, 73–80. [Google Scholar] [CrossRef]
- Ebrahimiyan, H.; Aslani, S.; Rezaei, N.; Jamshidi, A.; Mahmoudi, M. Survivin and autoimmunity; the ins and outs. Immunol. Lett. 2018, 193, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Rommer, P.S.; Milo, R.; Han, M.H.; Satyanarayan, S.; Sellner, J.; Hauer, L.; Illes, Z.; Warnke, C.; Laurent, S.; Weber, M.S.; et al. Immunological Aspects of Approved MS Therapeutics. Front. Immunol. 2019, 10, 1564. [Google Scholar] [CrossRef] [PubMed]
- Volpe, E.; Sambucci, M.; Battistini, L.; Borsellino, G. Fas-Fas Ligand: Checkpoint of T Cell Functions in Multiple Sclerosis. Front. Immunol. 2016, 7, 382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Oliveira, G.L.; Ferreira, A.F.; Gasparotto, E.P.; Kashima, S.; Covas, D.T.; Guerreiro, C.T.; Brum, D.G.; Barreira, A.A.; Voltarelli, J.C.; Simões, B.P.; et al. Defective expression of apoptosis-related molecules in multiple sclerosis patients is normalized early after autologous haematopoietic stem cell transplantation. Clin. Exp. Immunol. 2017, 187, 383–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balashov, K.E.; Rottman, J.B.; Weiner, H.L.; Hancock, W.W. CCR5(+) and CXCR3(+) T cells are increased in multiple sclerosis and their ligands MIP-1alpha and IP-10 are expressed in demyelinating brain lesions. Proc. Natl. Acad. Sci. USA 1999, 96, 6873–6878. [Google Scholar] [CrossRef] [Green Version]
- Fricker, M.; Tolkovsky, A.M.; Borutaite, V.; Coleman, M.; Brown, G.C. Neuronal Cell Death. Physiol. Rev. 2018, 98, 813–880. [Google Scholar] [CrossRef]
- Naim, S.; Kaufmann, T. The Multifaceted Roles of the BCL-2 Family Member BOK. Front. Cell Dev. Biol. 2020, 8, 574338. [Google Scholar] [CrossRef]
- Martinou, J.C.; Green, D.R. Breaking the mitochondrial barrier. Nat. Rev. Mol. Cell Biol. 2001, 2, 63–67. [Google Scholar] [CrossRef]
- Vaux, D.L.; Flavell, R.A. Apoptosis genes and autoimmunity. Curr. Opin. Immunol. 2000, 12, 719–724. [Google Scholar] [CrossRef]
- Zettl, U.K.; Kuhlmann, T.; Brück, W. Bcl-2 expressing T lymphocytes in multiple sclerosis lesions. Neuropathol. Appl. Neurobiol. 1998, 24, 202–208. [Google Scholar] [CrossRef]
- Schmidt, J.; Gold, R.; Schonrock, L.; Zettl, U.K.; Hartung, H.P.; Toyka, K. T-cell apoptosis in situ inexperimental autoimmune encephalomyelitis following methylprednisolone pulse therapy. Brain 2000, 123, 1431–1441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharief, M.K. Increased cellular expression of the caspase inhibitor FLIP in intrathecal lymphocytes from patients with multiple sclerosis. J. Neuroimmunol. 2000, 111, 203–209. [Google Scholar] [CrossRef]
- Semra, Y.K.; Seidi, O.A.; Sharief, M.K. Overexpression of the apoptosis inhibitor FLIP in T cells correlates with disease activity in multiple sclerosis. J. Neuroimmunol. 2001, 113, 268–274. [Google Scholar] [CrossRef]
- Wai, T.; Langer, T. Mitochondrial Dynamics and Metabolic Regulation. Trends Endocrinol. Metab. 2016, 27, 105–117. [Google Scholar] [CrossRef]
- Signorile, A.; Santeramo, A.; Tamma, G.; Pellegrino, T.; D’Oria, S.; Lattanzio, P.; De Rasmo, D. Mitochondrial cAMP prevents apoptosis modulating Sirt3 protein level and OPA1 processing in cardiac myoblast cells. Biochim. Biophys. Acta Mol. Cell. Res. 2017, 1864, 355–366. [Google Scholar] [CrossRef]
- Kalkavan, H.; Green, D.R. MOMP, cell suicide as a BCL-2 family business. Cell Death Differ. 2018, 25, 46–55. [Google Scholar] [CrossRef]
- Kondadi, A.K.; Anand, R.; Reichert, A.S. Cristae Membrane Dynamics—A Paradigm Change. Trends Cell. Biol. 2020, 30, 923–936. [Google Scholar] [CrossRef]
- Yu-Wai-Man, P.; Spyropoulos, A.; Duncan, H.J.; Guadagno, J.V.; Chinnery, P.F. A multiple sclerosis-like disorder in patients with OPA1 mutations. Ann. Clin. Trans. Neurol. 2016, 3, 723–729. [Google Scholar] [CrossRef]
- MacVicar, T.; Langer, T. OPA1 processing in cell death and disease - the long and short of it. J. Cell Sci. 2016, 129, 2297–2306. [Google Scholar] [CrossRef] [Green Version]
- Rainbolt, T.K.; Lebeau, J.; Puchades, C.; Wiseman, R.L. Reciprocal Degradation of YME1L and OMA1Adapts Mitochondrial Proteolytic Activity during Stress. Cell Rep. 2016, 14, 2041–2049. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.H.; Wei, Y.H. Roles of Mitochondrial Sirtuins in Mitochondrial Function, Redox Homeostasis, Insulin Resistance and Type 2 Diabetes. Int. J. Mol. Sci. 2020, 21, 5266. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Martin-Levilain, J.; Jiménez-Sánchez, C.; Karaca, M.; Foti, M.; Martinou, J.C.; Maechler, P. In vivo stabilization of OPA1 in hepatocytes potentiates mitochondrial respiration and gluconeogenesis in a prohibitin-dependent way. J. Biol. Chem. 2019, 294, 12581–12598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernando-Rodríguez, B.; Artal-Sanz, M. Mitochondrial Quality Control Mechanisms and the PHB (Prohibitin) Complex. Cells 2018, 7, 238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Signorile, A.; Sgaramella, G.; Bellomo, F.; De Rasmo, D. Prohibitins: A Critical Role in Mitochondrial Functions and Implication in Diseases. Cells 2019, 8, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muraguchi, T.; Kawawa, A.; Kubota, S. Prohibitin protects against hypoxia-induced H9c2 cardiomyocyte cell death. Biomed. Res. 2010, 31, 113–122. [Google Scholar] [CrossRef] [Green Version]
- Kumar, M.K.S.; Nair, S.; Mony, U.; Kalingavarman, S.; Venkat, R.; Sivanarayanan, T.B.; Unni, A.K.K.; Rajeshkannan, R.; Anandakuttan, A.; Radhakrishnan, S.; et al. Significance of elevated Prohibitin 1 levels in Multiple Sclerosis patients lymphocytes towards the assessment of subclinical disease activity and its role in the central nervous system pathology of disease. Int. J. Biol. Macromol. 2018, 110, 573–581. [Google Scholar] [CrossRef]
- Kozin, M.S.; Kulakova, O.G.; Favorova, O.O. Involvement of Mitochondria in Neurodegeneration in Multiple Sclerosis. Biochemistry 2018, 83, 813–830. [Google Scholar] [CrossRef]
- Wei, Y.; Chiang, W.C.; Sumpter, R., Jr.; Mishra, P.; Levine, B. Prohibitin 2 Is an Inner Mitochondrial Membrane. Mitophagy Recept. Cell 2017, 168, 224–238. [Google Scholar]
- Plaza-Zabala, A.; Sierra-Torre, V.; Sierra, A. Autophagy and Microglia: Novel Partners in Neurodegeneration and Aging. Int. J. Mol. Sci. 2017, 18, 598. [Google Scholar] [CrossRef] [Green Version]
- Pua, H.H.; He, Y.W. Maintaining T lymphocyte homeostasis: Another duty of autophagy. Autophagy 2007, 3, 266–267. [Google Scholar] [CrossRef] [Green Version]
- Yousefi, S.; Perozzo, R.; Schmid, I.; Ziemiecki, A.; Schaffner, T.; Scapozza, L.; Brunner, T.; Simon, H.U. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat. Cell Biol 2006, 8, 1124–1132. [Google Scholar] [CrossRef] [PubMed]
- Alirezaei, M.; Fox, H.S.; Flynn, C.T.; Moore, C.S.; Hebb, A.L.; Frausto, R.F.; Bhan, V.; Kiosses, W.B.; Whitton, J.L.; Robertson, G.S.; et al. Elevated ATG5 expression in autoimmune demyelination and multiple sclerosis. Autophagy 2009, 5, 152–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Gambero, A.J.; Sanjuan, C.; Serrano-Castro, P.J.; Suárez, J.; de Fonseca, F.R. The Biomedical Uses of Inositols: A Nutraceutical Approach to Metabolic Dysfunction in Aging and Neurodegenerative Diseases. Biomedicines 2020, 8, 295. [Google Scholar]
- Bergien, S.O.; Petersen, C.M.; Lynning, M.; Kristiansen, M.; Skovgaard, L. Use of natural medicine and dietary supplements concomitant with conventional medicine among people with Multiple Sclerosis. Mult. Scler. Relat. Disord. 2020, 44, 102197. [Google Scholar] [CrossRef] [PubMed]
- Papa, S.; Scacco, S.; De Rasmo, D.; Signorile, A.; Papa, F.; Panelli, D.; Nicastro, A.; Scaringi, R.; Santeramo, A.; Roca, E.; et al. cAMP-dependent protein kinase regulates post-translational processing and expression of complex I subunits in mammalian cells. Biochim. Biophys. Acta 2010, 1797, 649–658. [Google Scholar] [CrossRef] [Green Version]
- Ryu, H.; Lee, J.; Impey, S.; Ratan, R.R.; Ferrante, R.J. Antioxidants modulate mitochondrial PKA and increase CREB binding to D-loop DNA of the mitochondrial genome in neurons. Proc. Natl. Acad. Sci. USA 2005, 102, 13915–13920. [Google Scholar] [CrossRef] [Green Version]
- De Rasmo, D.; Signorile, A.; Roca, E.; Papa, S. cAMP response element-binding protein (CREB) is imported into mitochondria and promotes protein synthesis. FEBS J. 2009, 276, 4325–4333. [Google Scholar] [CrossRef]
- De Rasmo, D.; Signorile, A.; Larizza, M.; Pacelli, C.; Cocco, T.; Papa, S. Activation of the cAMP cascade in human fibroblast cultures rescues the activity of oxidatively damaged complex I. Free Radic. Biol. Med. 2012, 52, 757–764. [Google Scholar] [CrossRef]
- De Rasmo, D.; Palmisano, G.; Scacco, S.; Technikova-Dobrova, Z.; Panelli, D.; Cocco, T.; Sardanelli, A.M.; Gnoni, A.; Micelli, L.; Trani, A.; et al. Phosphorylation pattern of the NDUFS4 subunit of complex I of the mammalian respiratory chain. Mitochondrion 2010, 10, 464–471. [Google Scholar] [CrossRef]
- De Rasmo, D.; Panelli, D.; Sardanelli, A.M.; Papa, S. cAMP-dependent protein kinase regulates the mitochondrial import of the nuclear encoded NDUFS4 subunit of complex I. Cell. Signal. 2008, 20, 989–997. [Google Scholar] [CrossRef]
- Acin-Perez, R.; Salazar, E.; Brosel, S.; Yang, H.; Schon, E.A.; Manfredi, G. Modulation of mitochondrial protein phosphorylation by soluble adenylyl cyclase ameliorates cytochrome oxidase defects. EMBO Mol. Med. 2009, 1, 392–406. [Google Scholar] [CrossRef] [PubMed]
- Acin-Perez, R.; Salazar, E.; Kamenetsky, M.; Buck, J.; Levin, L.R.; Manfredi, G. Cyclic AMP produced inside mitochondria regulates oxidative phosphorylation. Cell. Metab. 2009, 9, 265–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Rasmo, D.; Signorile, A.; Santeramo, A.; Larizza, M.; Lattanzio, P.; Capitanio, G.; Papa, S. Intramitochondrial adenylyl cyclase controls the turnover of nuclear-encoded subunits and activity of mammalian complex I of the respiratory chain. Biochim. Biophys. Acta 2015, 1853, 183–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Rasmo, D.; Micelli, L.; Santeramo, A.; Signorile, A.; Lattanzio, P.; Papa, S. cAMP regulates the functional activity, coupling efficiency and structural organization of mammalian FOF1 ATP synthase. Biochim. Biophys. Acta 2016, 1857, 350–358. [Google Scholar] [CrossRef]
- Trotta, A.P.; Chipuk, J.E. Mitochondrial Dynamics as Regulators of Cancer Biology. Cell. Mol. Life Sci. 2017, 74, 1999–2017. [Google Scholar] [CrossRef]
- Qi, Z.; Huang, Z.; Xie, F.; Chen, L. Dynamin-related protein 1: A critical protein in the pathogenesis of neural system dysfunctions and neurodegenerative diseases. J. Cell. Physiol. 2019, 234, 10032–10046. [Google Scholar] [CrossRef]
- Piccoli, C.; Scacco, S.; Bellomo, F.; Signorile, A.; Iuso, A.; Boffoli, D.; Scrima, R.; Capitanio, N.; Papa, S. cAMP controls oxygen metabolism in mammalian cells. FEBS Lett. 2006, 580, 4539–4543. [Google Scholar] [CrossRef] [Green Version]
- De Rasmo, D.; Gattoni, G.; Papa, F.; Santeramo, A.; Pacelli, C.; Cocco, T.; Micelli, L.; Sardaro, N.; Larizza, M.; Scivetti, M.; et al. The β-adrenoceptor agonist isoproterenol promotes the activity of respiratory chain complex I and lowers cellular reactive oxygen species in fibroblasts and heart myoblasts. Eur. J. Pharmacol. 2011, 652, 15–22. [Google Scholar] [CrossRef]
- Ramzan, R.; Rhiel, A.; Weber, P.; Kadenbach, B.; Vogt, S. Reversible dimerization of cytochrome c oxidase regulates mitochondrial respiration. Mitochondrion 2019, 49, 149–155. [Google Scholar] [CrossRef]
- Smirnova, E.; Griparic, L.; Shurland, D.L.; van der Bliek, A.M. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol. Biol. Cell 2001, 12, 2245–2256. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Dong, W.; Shan, X.; Hong, H.; Liu, Y.; Liu, X.; Zhang, X.; Zhang, J. The anti-tumor effects of Mfn2 in breast cancer are dependent on promoter DNA methylation, the P21(Ras) motif and PKA phosphorylation site. Oncol. Lett. 2018, 15, 8011–8018. [Google Scholar] [PubMed] [Green Version]
- Bellomo, F.; Signorile, A.; Tamma, G.; Ranieri, M.; Emma, F.; De Rasmo, D. Impact of atypical mitochondrial cyclic AMP level in nephropathiccystinosis. Cell. Mol. Life Sci. 2018, 75, 3411–3422. [Google Scholar] [CrossRef] [PubMed]
- De Rasmo, D.; Signorile, A.; De Leo, E.; Polishchuk, E.V.; Ferretta, A.; Raso, R.; Russo, S.; Polishchuk, R.; Emma, F.; Bellomo, F. Mitochondrial Dynamics of Proximal Tubular Epithelial Cells in Nephropathic Cystinosis. Int. J. Mol. Sci. 2019, 21, 192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Insel, P.A.; Zhang, L.; Murray, F.; Yokouchi, H.; Zambon, A.C. Cyclic AMP is both a pro-apoptotic and anti-apoptotic second messenger. Acta Physiol. 2012, 204, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Porcellini, A.; Messina, S.; De Gregorio, G.; Feliciello, A.; Carlucci, A.; Barone, M.; Picascia, A.; De Blasi, A.; Avvedimento, E.V. The expression of the thyroid-stimulating hormone (TSH) receptor and the cAMP-dependent protein kinase RII beta regulatory subunit confers TSH-cAMP-dependent growth to mouse fibroblasts. J. Biol. Chem. 2003, 278, 40621–40630. [Google Scholar] [CrossRef] [Green Version]
- Miller, E.D.; Dziedzic, A.; Saluk-Bijak, J.; Bijak, M. A Review of Various Antioxidant Compounds and their Potential Utility as Complementary Therapy in Multiple Sclerosis. Nutrients 2019, 11, 1528. [Google Scholar] [CrossRef] [Green Version]
- Bisht, A.; Dickens, M.; Rutherfurd-Markwick, K.; Thota, R.; Mutukumira, A.N.; Singh, H. Chlorogenic Acid Potentiates the Anti-Inflammatory Activity of Curcumin in LPS-Stimulated THP-1 Cells. Nutrients 2020, 12, 2706. [Google Scholar] [CrossRef]
- Liu, J.Q.; Yan, Y.Q.; Liu, J.T.; Wang, Y.R.; Wang, X. Curcumin prevents experimental autoimmune encephalomyelitis by inhibiting proliferation and effector CD4+ T cell activation. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 9108–9116. [Google Scholar]
- Zhang, Y.; Li, L.; Zhang, J. Curcumin in antidepressant treatments: An overview of potential mechanisms, pre-clinical/clinical trials and ongoing challenges. Basic Clin. Pharmacol. Toxicol. 2020, 127, 243–253. [Google Scholar] [CrossRef]
- Zheng, M.; Zhang, Q.; Joe, Y.; Lee, B.H.; Ryu, D.G.; Kwon, K.B.; Ryter, S.W.; Chung, H.T. Curcumin induces apoptotic cell death of activated human CD4+ T cells via increasing endoplasmic reticulum stress and mitochondrial dysfunction. Int. Immunopharmacol. 2013, 15, 517–523. [Google Scholar] [CrossRef]
- Pfeffer, C.M.; Singh, A.T.K. Apoptosis: A Target for Anticancer Therapy. Int. J. Mol. Sci. 2018, 19, 448176–448178. [Google Scholar]
- Pae, H.O.; Jeong, S.O.; Jeong, G.S.; Kim, K.M.; Kim, H.S.; Kim, S.A.; Kim, Y.C.; Kang, S.D.; Kim, B.N.; Chung, H.T. Curcumin induces pro-apoptotic endoplasmic reticulum stress in human leukemia HL-60 cells. Biochem. Biophys. Res. Commun. 2007, 353, 1040–1045. [Google Scholar] [CrossRef] [PubMed]
- Safavifar, F.; Saadat, F.; Jalali, S.Z.; Khoramizadeh, M.R. Augmented cAMP Signaling by Co-Administration of Resveratrol and Curcumin: A Cellular Biosensor Kinetic Assessment. Iran. J. Public. Health 2019, 48, 1310–1316. [Google Scholar] [PubMed]
- Maher, P. The Potential of Flavonoids for the Treatment of Neurodegenerative Diseases. Int. J. Mol. Sci. 2019, 20, 3056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, A.; Ikram, M.; Hahm, J.R.; Kim, M.O. Antioxidant and Anti-Inflammatory Effects of Citrus FlavonoidHesperetin: Special Focus on Neurological Disorders. Antioxidants 2020, 9, 609. [Google Scholar] [CrossRef]
- Karak, P. Biological activities of flavonoids: An overview. Int. J. Pharm. Sci. Res. 2019, 3, 1567–1574. [Google Scholar]
- Maleki, S.J.; Crespo, J.F.; Cabanillas, B. Anti-inflammatory effects of flavonoids. Food Chem. 2019, 299, 125124. [Google Scholar] [CrossRef]
- Wu, D. Green tea EGCG, T-cell function, and T-cell-mediated autoimmune encephalomyelitis. J. Investig. Med. 2016, 64, 1213–1219. [Google Scholar] [CrossRef]
- Meng, Q.; Velalar, C.N.; Ruan, R. Regulating the age-related oxidative damage, mitochondrial integrity, and antioxidative enzyme activity in Fischer 344 rats by supplementation of the antioxidant epigallocatechin-3-gallate. Rejuvenation Res. 2008, 3, 649–660. [Google Scholar] [CrossRef]
- Valenti, D.; De Bari, L.; De Rasmo, D.; Signorile, A.; Henrion-Caude, A.; Contestabile, A.; Vacca, R.A. The polyphenols resveratrol and epigallocatechin-3-gallate restore the severe impairment of mitochondria in hippocampal progenitor cells from a Down syndrome mouse model. Biochim. Biophys. Acta 2016, 1862, 1093–1104. [Google Scholar] [CrossRef]
- Valenti, D.; De Rasmo, D.; Signorile, A.; Rossi, L.; De Bari, L.; Scala, I.; Granese, B.; Papa, S.; Vacca, R.A. Epigallocatechin-3-gallate prevents oxidative phosphorylation deficit and promotes mitochondrial biogenesis in human cells from subjects with Down’s syndrome. Biochim. Biophys. Acta 2013, 1832, 542–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valenti, D.; Braidy, N.; De Rasmo, D.; Signorile, A.; Rossi, L.; Atanasov, A.G.; Volpicella, M.; Henrion-Caude, A.; Nabavi, S.M.; Vacca, R.A. Mitochondria as pharmacological targets in Down syndrome. Free Radic. Biol. Med. 2018, 114, 69–83. [Google Scholar] [CrossRef] [PubMed]
- Ok, W.J.; Cho, H.J.; Kim, H.H.; Lee, D.H.; Kang, H.Y.; Kwon, H.W.; Rhee, M.H.; Kim, M.; Park, H.J. Epigallocatechin-3-gallate has an anti-platelet effect in a cyclic AMP-dependent manner. J. Atheroscler. Thromb. 2012, 19, 337–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muthian, G.; Bright, J.J. Quercetin, a flavonoid phytoestrogen, ameliorates experimental allergic encephalomyelitis by blocking IL-12 signaling through JAK-STAT pathway in T lymphocyte. J. Clin. Immunol. 2004, 24, 542–545. [Google Scholar] [CrossRef] [PubMed]
- Ha, E.J.; Kim, K.Y.; Kim, C.E.; Jun, D.Y.; Kim, Y.H. Enhancement of Quercetin-Induced Apoptosis by Cotreatment with Autophagy Inhibitor Is Associated with Augmentation of BAK-Dependent Mitochondrial Pathway in Jurkat T Cells. Oxid. Med. Cell. Longev. 2019, 15, 7989276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, W.C.; Shih, C.M.; Chen, M.C.; Lai, Y.H.; Chen, J.H.; Chen, C.M.; Lin, C.N. Suppressive effects of 3-O-methylquercetin on ovalbumin-induced airway hyperresponsiveness. Planta Med. 2004, 70, 1123–1127. [Google Scholar] [CrossRef]
- Malaguarnera, L. Influence of Resveratrol on the Immune Response. Nutrients 2019, 11, 946. [Google Scholar] [CrossRef] [Green Version]
- Park, S.J.; Ahmad, F.; Philp, A.; Baar, K.; Williams, T.; Luo, H.; Ke, H.; Rehmann, H.; Taussig, R.; Brown, A.L.; et al. Resveratrol ameliorates aging-related metabolic phenotypes by inhibiting cAMP phosphodiesterases. Cell 2012, 148, 421–433. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Tian, Z.; Zhou, G.; Niu, Q.; Chen, J.; Li, P.; Dong, L.; Xia, T.; Zhang, S.; Wang, A. SIRT1-dependent mitochondrial biogenesis supports therapeutic effects of resveratrol against neurodevelopment damage by fluoride. Theranostics 2020, 10, 4822–4838. [Google Scholar] [CrossRef]
- Vergara, D.; Gaballo, A.; Signorile, A.; Ferretta, A.; Tanzarella, P.; Pacelli, C.; Di Paola, M.; Cocco, T.; Maffia, M. Resveratrol Modulation of ProteinExpression in parkin-Mutant Human Skin Fibroblasts: A Proteomic Approach. Oxid. Med. Cell. Longev. 2017, 2017, 2198243. [Google Scholar] [CrossRef] [Green Version]
- Stacchiotti, A.; Corsetti, G. Natural Compounds and Autophagy: Allies Against Neurodegeneration. Front. Cell Dev. Biol. 2020, 8, 555409. [Google Scholar] [CrossRef] [PubMed]
- Aparicio-Soto, M.; Sánchez-Hidalgo, M.; Rosillo, M.A.; Castejón, M.L.; Alarcón-de-la-Lastra, L. Extra virgin olive oil: A key functional food for prevention of immune-inflammatory diseases. Food Funct. 2016, 7, 4492–4505. [Google Scholar] [CrossRef] [PubMed]
- Gallardo-Fernández, M.; Hornedo-Ortega, R.; Cerezo, A.B.; Troncoso, A.M.; García-Parrilla, M.C. Melatonin, protocatechuic acid and hydroxytyrosol effects on vitagenes system against alpha-synuclein toxicity. Food Chem. Toxicol. 2019, 134, 110817. [Google Scholar] [CrossRef] [PubMed]
- Tuck, K.L.; Hayball, P.J.; Stupans, I. Structural characterization of the metabolites of hydroxytyrosol, the principal phenolic component in olive oil, in rats. J. Agric. Food Chem. 2002, 50, 2404–2409. [Google Scholar] [CrossRef]
- De la Torre, R. Bioavailability of olive oil phenolic compounds in humans. Inflammopharmacology 2008, 16, 245–247. [Google Scholar] [CrossRef]
- Richard, N.; Arnold, S.; Kilpert, C.; Wertz, K. Hydroxytyrosol is the major anti-inflammatory compound in aqueous olive extracts and impairs cytokine and chemokine production in macrophages. Planta Med. 2011, 77, 1890–1897. [Google Scholar] [CrossRef] [Green Version]
- Conde, C.; Escribano, B.M.; Luque, E.; Feijóo, M.; Caballero-Villarraso, J.; Valdelvira, M.E.; Ochoa-Sepúlveda, J.J.; Lillo, R.; Paz, E.; Santamaría, A.; et al. Extra-Virgin Olive Oil Modifies the Changes Induced in Non-Nervous Organs and Tissues by Experimental Autoimmune Encephalomyelitis Models. Nutrients 2019, 11, 2448. [Google Scholar] [CrossRef] [Green Version]
- Toteda, G.; Lupinacci, S.; Vizza, D.; Bonofiglio, R.; Perri, E.; Bonofiglio, M.; Lofaro, D.; La Russa, A.; Leone, F.; Gigliotti, P.; et al. High doses of hydroxytyrosol induce apoptosis in papillary and follicular thyroid cancer cells. J. Endocrinol. Investig. 2017, 40, 153–162. [Google Scholar] [CrossRef]
- Echeverría, F.; Ortiz, M.; Valenzuela, R.; Videla, L.A. Hydroxytyrosol and Cytoprotection: A Projection for Clinical Interventions. Int. J. Mol. Sci. 2017, 18, 930. [Google Scholar]
- Hao, J.; Shen, W.; Yu, G.; Jia, H.; Li, X.; Feng, Z.; Wang, Y.; Weber, P.; Wertz, K.; Sharman, E.; et al. Hydroxytyrosol promotes mitochondrial biogenesis and mitochondrial function in 3T3-L1 adipocytes. J. Nutr. Biochem. 2010, 21, 634–644. [Google Scholar] [CrossRef]
- Signorile, A.; Micelli, L.; De Rasmo, D.; Santeramo, A.; Papa, F.; Ficarella, R.; Gattoni, G.; Scacco, S.; Papa, S. Regulation of the biogenesis of OXPHOS complexes in cell transition from replicating to quiescent state: Involvement of PKA and effect of hydroxytyrosol. Biochim. Biophys. Acta 2014, 1843, 675–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, W.J.; Chen, Y.; Shi, L.X.; Cheng, H.R.; Banda, I.; Ji, Y.H.; Wang, Y.T.; Li, X.M.; Mao, Y.X.; Zhang, D.F.; et al. AKT-GSK3 β Signaling Pathway Regulates Mitochondrial Dysfunction-Associated OPA1 Cleavage Contributing to Osteoblast Apoptosis: Preventative Effects of Hydroxytyrosol. Oxid. Med. Cell. Longev. 2019, 2, 4101738. [Google Scholar]
- Bikle, D.D.; Patzek, S.; Wang, Y. Physiologica and pathophysiologic roles of extra renal CYP27b1: Case report. Bone Rep. 2018, 8, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Ložnjak, P.; Jakobsen, J. Stability of vitamin D3 and vitamin D2 in oil, fish and mushrooms after household cooking. Food Chem. 2018, 254, 144–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munger, K.L.; Levin, L.I.; Hollis, B.W.; Howard, N.S.; Ascherio, A. Serum 25-hydroxyvitamin D levels and risk of multiple sclerosis. JAMA 2006, 296, 2832–2838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveira, S.R.; Simão, A.N.C.; Alfieri, D.F.; Flauzino, T.; Kallaur, A.P.; Mezzaroba, L.; Lozovoy, M.A.B.; Sabino, B.S.; Ferreira, K.P.Z.; Pereira, W.L.; et al. Vitamin D deficiency is associated with disability and disease progression in multiple sclerosis patients independently of oxidative and nitrosative stress. J. Neurol. Sci. 2017, 381, 213–219. [Google Scholar] [CrossRef]
- Nashold, F.E.; Hoag, K.A.; Goverman, J.; Hayes, C.E. Rag-1-dependent cells are necessary for 1,25-dihydroxyvitamin D3 prevention of experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2001, 119, 16–29. [Google Scholar] [CrossRef]
- Nashold, F.E.; Miller, D.J.; Hayes, C.E. 1,25-dihydroxyvitamin D3 treatment decreases macrophage accumulation in the CNS of mice with experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2000, 103, 171–179. [Google Scholar] [CrossRef]
- Spach, K.M.; Pedersen, L.B.; Nashold, F.E.; Kayo, T.; Yandell, B.S.; Prolla, T.A.; Hayes, C.E. Gene expressionstimulating inflammatory cell apoptosis. Physiol. Genom. 2004, 18, 141–151. [Google Scholar] [CrossRef] [Green Version]
- Ravid, A.; Koran, R.; Narinsky, R.; Rotem, C.; Novogrodsky, A.; Liberman, U.A. 1,25-Dihydroxyvitamin D3 and agents that increase intracellular adenosine3’,5’-monophosphate synergistically inhibit the mitogenic stimulation of human lymphocytes. J. Clin. Endocrinol. Metab. 1990, 70, 1687–1692. [Google Scholar] [CrossRef]
- Ren, L.; Xuan, L.; Han, F.; Zhang, J.; Gong, L.; Lv, Y.; Zhang, W.; Yang, S.; Xu, B.; Yan, Y.; et al. Vitamin D supplementation rescues simvastatin induced myopathy in mice via improving mitochondrial cristae shape. Toxicol. Appl. Pharmacol. 2020, 401, 115076. [Google Scholar] [CrossRef] [PubMed]
- Hardeland, R.; Poeggeler, B. Non-vertebrate melatonin. J. Pineal Res. 2003, 34, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Sanchez, N.; Cruz-Chamorro, I.; Lopez-Gonzalez, A.; Utrilla, J.C.; Fernández-Santos, J.M.; Martínez-López, A.; Lardone, P.J.; Guerrero, J.M.; Carrillo-Vico, A. Melatonin controls experimental autoimmune encephalomyelitis by altering the T effector/regulatory balance. Brain Behav. Immun. 2015, 50, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Emamgholipour, S.; Hossein-Nezhad, A.; Sahraian, M.A.; Askarisadr, A.; Ansari, M. Evidence for possible role of melatonin in reducing oxidative stress in multiple sclerosis through its effect on SIRT1 and anti-oxidant enzymes. Life Sci. 2016, 145, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Hardeland, R.; Cardinali, D.P.; Srinivasan, V.; Spence, D.W.; Brown, G.M.; Pandi-Perumal, S.R. Melatonin—A pleiotropic, orchestrating regulator molecule. Prog. Neurobiol. 2011, 93, 350–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardeland, R.; Madrid, J.A.; Tan, D.X.; Reiter, R.J. Melatonin, the circadian multioscillator system and health: The need for detailed analyses of peripheral melatonin signaling. J. Pineal Res. 2012, 52, 139–166. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Rosales-Corral, S.; Tan, D.; Jou, M.J.; Galano, A.; Xu, B. Melatonin as a mitochondria-targeted antioxidant: One of evolution’s best ideas. Cell. Mol. Life Sci. 2017, 74, 3863–3881. [Google Scholar] [CrossRef]
- Agil, A.; Chayah, M.; Visiedo, L.; Navarro-Alarcon, M.; Rodríguez Ferrer, J.M.; Tassi, M.; Reiter, R.J.; Fernández-Vázquez, G. Melatonin Improves Mitochondrial Dynamics and Function in the Kidney of Zücker Diabetic Fatty Rats. J. Clin. Med. 2020, 9, 2916. [Google Scholar] [CrossRef]
- Tryfonos, C.; Mantzorou, M.; Fotiou, D.; Vrizas, M.; Vadikolias, K.; Pavlidou, E.; Giaginis, C. Dietary Supplements on Controlling Multiple Sclerosis Symptoms and Relapses: Current Clinical Evidence and Future Perspectives. Medicines 2019, 6, 95. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Deregulation in MS | Reference |
|---|---|---|
| Bcl2 | Increased expression | [29,89,90] |
| OPA1 | Altered proteolytic processing | [25,98] |
| PHB2 | Increased expression | [25,106,107] |
| SIRT3 | No response to oxidative stress | [25] |
| OMA1 | No response to oxidative stress | [25] |
| ATG5 | Upregulation and post-translational modification | [112] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Signorile, A.; Ferretta, A.; Ruggieri, M.; Paolicelli, D.; Lattanzio, P.; Trojano, M.; De Rasmo, D. Mitochondria, Oxidative Stress, cAMP Signalling and Apoptosis: A Crossroads in Lymphocytes of Multiple Sclerosis, a Possible Role of Nutraceutics. Antioxidants 2021, 10, 21. https://doi.org/10.3390/antiox10010021
Signorile A, Ferretta A, Ruggieri M, Paolicelli D, Lattanzio P, Trojano M, De Rasmo D. Mitochondria, Oxidative Stress, cAMP Signalling and Apoptosis: A Crossroads in Lymphocytes of Multiple Sclerosis, a Possible Role of Nutraceutics. Antioxidants. 2021; 10(1):21. https://doi.org/10.3390/antiox10010021
Chicago/Turabian StyleSignorile, Anna, Anna Ferretta, Maddalena Ruggieri, Damiano Paolicelli, Paolo Lattanzio, Maria Trojano, and Domenico De Rasmo. 2021. "Mitochondria, Oxidative Stress, cAMP Signalling and Apoptosis: A Crossroads in Lymphocytes of Multiple Sclerosis, a Possible Role of Nutraceutics" Antioxidants 10, no. 1: 21. https://doi.org/10.3390/antiox10010021
APA StyleSignorile, A., Ferretta, A., Ruggieri, M., Paolicelli, D., Lattanzio, P., Trojano, M., & De Rasmo, D. (2021). Mitochondria, Oxidative Stress, cAMP Signalling and Apoptosis: A Crossroads in Lymphocytes of Multiple Sclerosis, a Possible Role of Nutraceutics. Antioxidants, 10(1), 21. https://doi.org/10.3390/antiox10010021