Counteracting Effects of Glutathione on the Glutamate-Driven Excitation/Inhibition Imbalance in First-Episode Schizophrenia: A 7T MRS and Dynamic Causal Modeling Study



Abstract
:1. Introduction
2. Materials and Methods
2.1. Participants
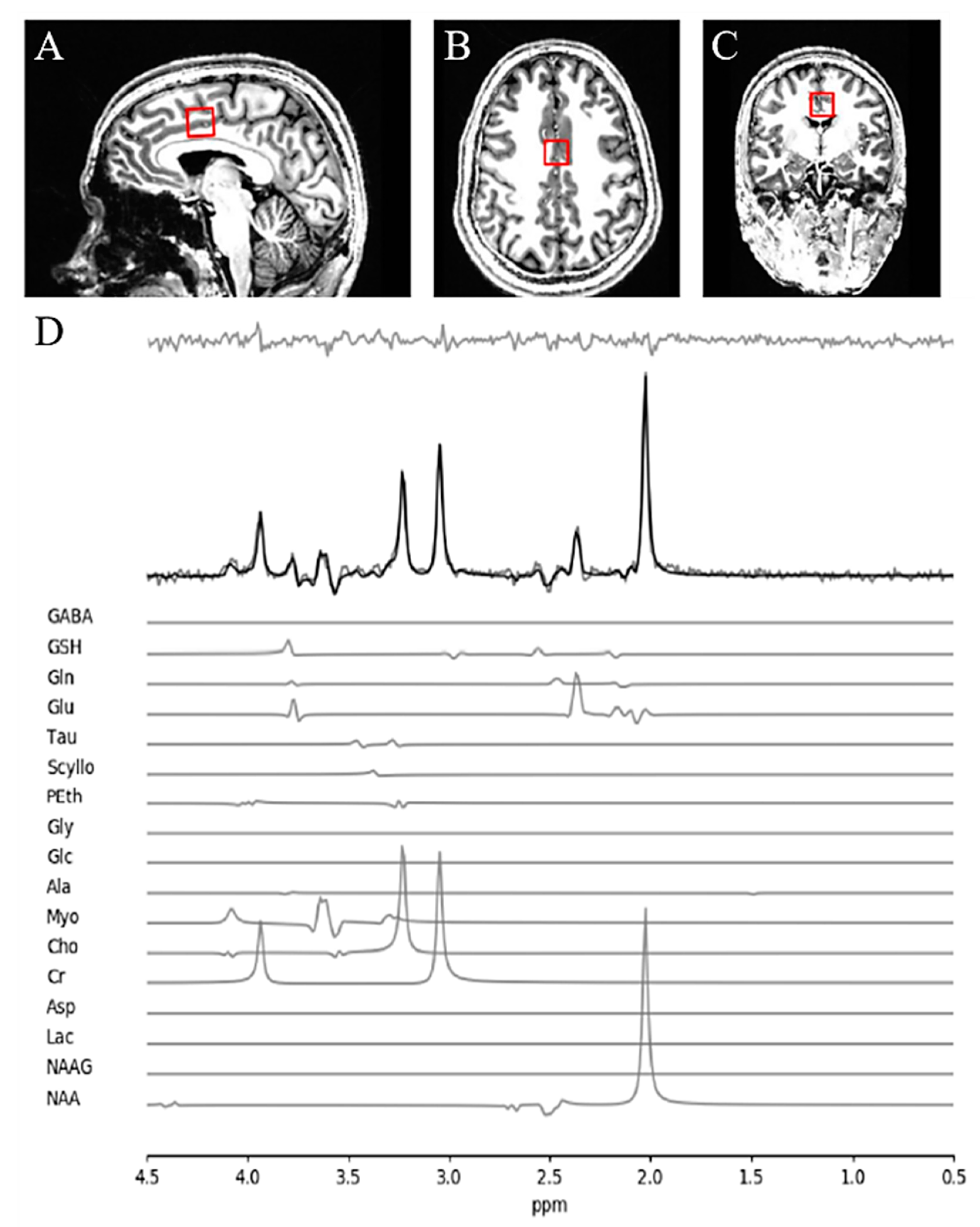
2.2. Magnetic Resonance Spectroscopy (MRS) Acquisition and Analysis
2.3. Bayesian Analysis
2.4. Resting-State fMRI
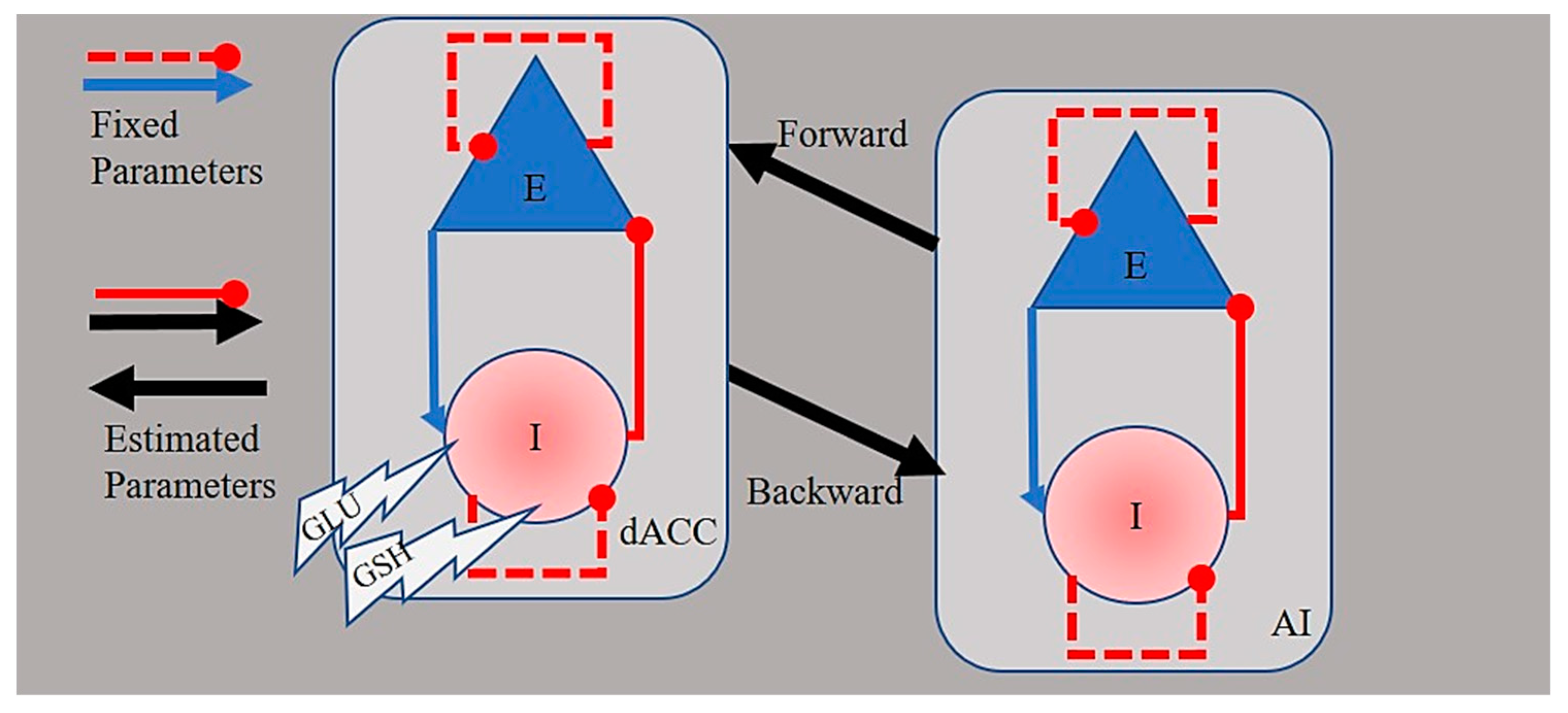
2.5. Spectral Dynamic Causal Modeling of Network Connectivity
3. Results
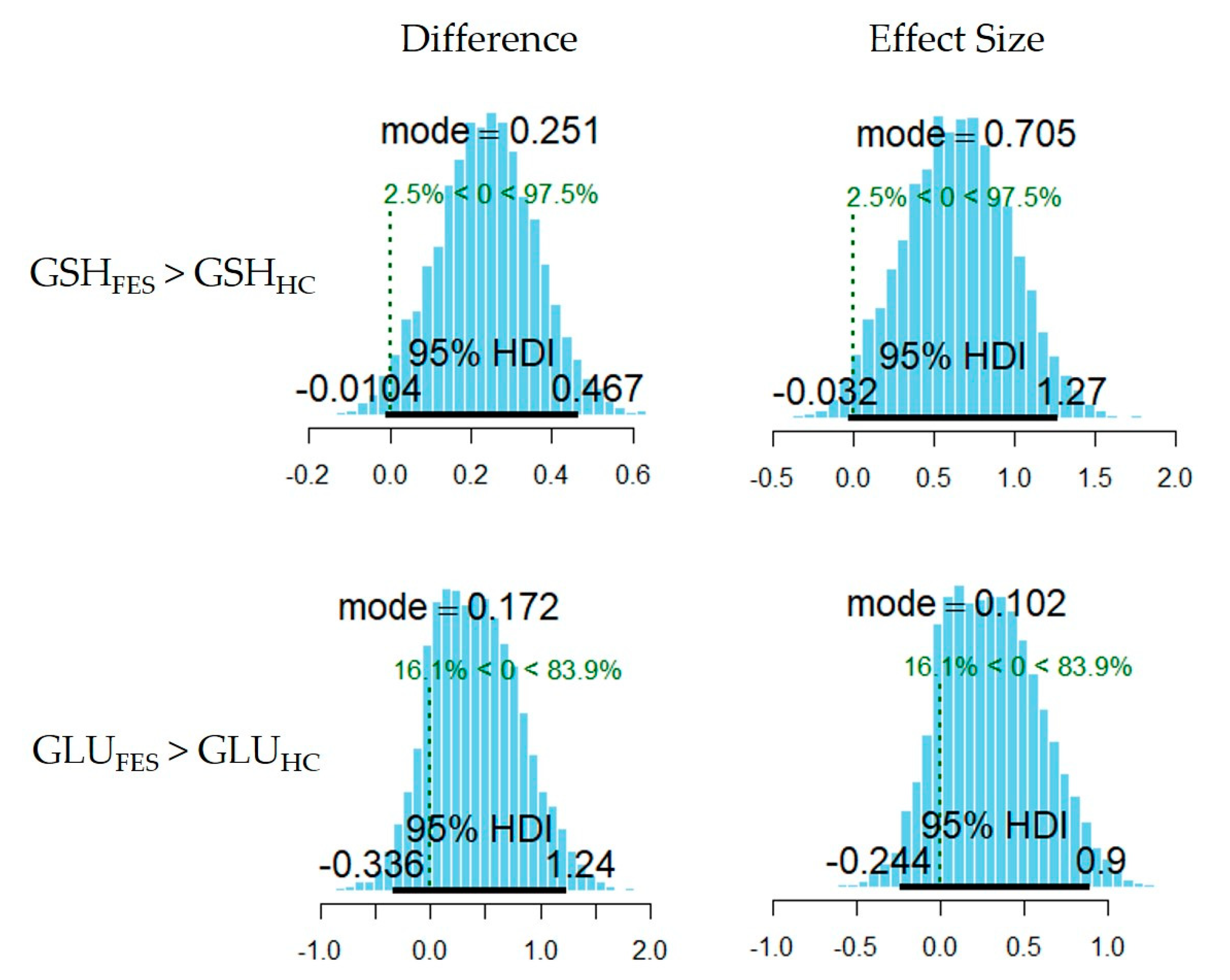
3.1. Between-Group Comparison in Metabolite Levels
3.2. Spectral Dynamic Causal Models of Effective Connectivity
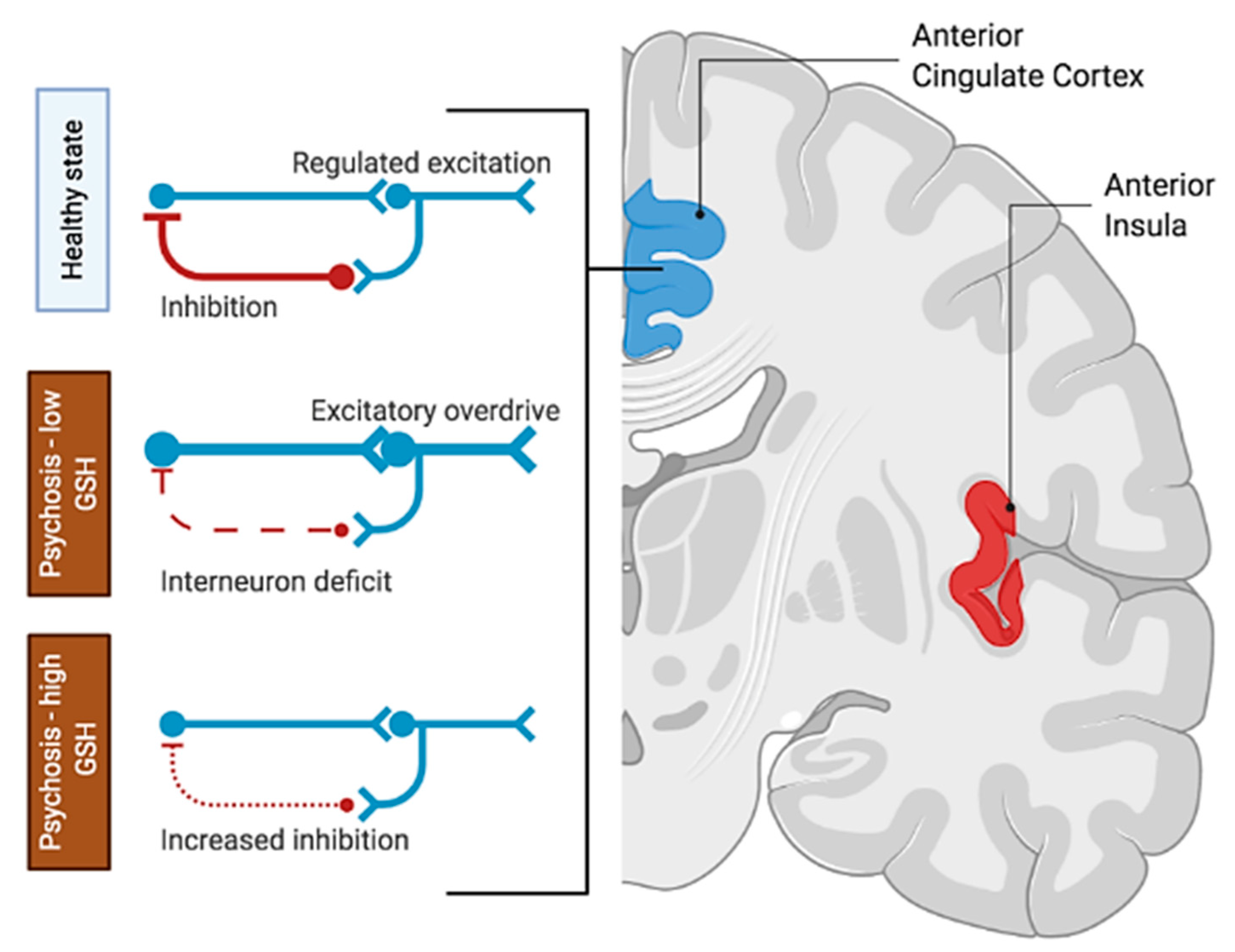
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Emamzadeh, F.N.; Surguchov, A. Parkinson’s Disease: Biomarkers, Treatment, and Risk Factors. Front. Neurosci. 2018, 12. [Google Scholar] [CrossRef] [PubMed]
- Flatow, J.; Buckley, P.; Miller, B.J. Meta-analysis of oxidative stress in schizophrenia. Biol. Psychiatry 2013, 74, 400–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perkins, D.O.; Jeffries, C.D.; Do, K.Q. Potential Roles of Redox Dysregulation in the Development of Schizophrenia. Biol. Psychiatry 2020, 88, 326–336. [Google Scholar] [CrossRef]
- Do, K.Q.; Trabesinger, A.H.; Kirsten-Krüger, M.; Lauer, C.J.; Dydak, U.; Hell, D.; Holsboer, F.; Boesiger, P.; Cuénod, M. Schizophrenia: Glutathione deficit in cerebrospinal fluid and prefrontal cortex in vivo. Eur. J. Neurosci. 2000, 12, 3721–3728. [Google Scholar] [CrossRef] [PubMed]
- Gawryluk, J.W.; Wang, J.F.; Andreazza, A.C.; Shao, L.; Young, L.T. Decreased levels of glutathione, the major brain antioxidant, in post-mortem prefrontal cortex from patients with psychiatric disorders. Int. J. Neuropsychopharmacol. 2011, 14, 123–130. [Google Scholar] [CrossRef] [Green Version]
- Yao, J.K.; Leonard, S.; Reddy, R. Altered Glutathione Redox State in Schizophrenia. Dis. Mark. 2006, 22. [Google Scholar] [CrossRef] [Green Version]
- Kumar, J.; Liddle, E.B.; Fernandes, C.C.; Palaniyappan, L.; Hall, E.L.; Robson, S.E.; Simmonite, M.; Fiesal, J.; Katshu, M.Z.; Qureshi, A.; et al. Glutathione and glutamate in schizophrenia: A 7T MRS study. Mol. Psychiatry 2020, 25, 873–882. [Google Scholar] [CrossRef] [Green Version]
- Sedlak, T.W.; Nucifora, L.G.; Koga, M.; Shaffer, L.S.; Higgs, C.; Tanaka, T.; Wang, A.M.; Coughlin, J.M.; Barker, P.B.; Fahey, J.W.; et al. Sulforaphane Augments Glutathione and Influences Brain Metabolites in Human Subjects: A Clinical Pilot Study. Mol. Neuropsychiatry 2018, 3, 214–222. [Google Scholar] [CrossRef]
- Baxter, P.S.; Bell, K.F.; Hasel, P.; Kaindl, A.M.; Fricker, M.; Thomson, D.; Cregan, S.P.; Gillingwater, T.H.; Hardingham, G.E. Synaptic NMDA receptor activity is coupled to the transcriptional control of the glutathione system. Nat. Commun. 2015, 6, 6761. [Google Scholar] [CrossRef] [Green Version]
- Sedlak, T.W.; Paul, B.D.; Parker, G.M.; Hester, L.D.; Snowman, A.M.; Taniguchi, Y.; Kamiya, A.; Snyder, S.H.; Sawa, A. The glutathione cycle shapes synaptic glutamate activity. Proc. Natl. Acad. Sci. USA 2019, 116, 2701–2706. [Google Scholar] [CrossRef] [Green Version]
- Janáky, R.; Ogita, K.; Pasqualotto, B.A.; Bains, J.S.; Oja, S.S.; Yoneda, Y.; Shaw, C.A. Glutathione and signal transduction in the mammalian CNS. J. Neurochem. 1999, 73, 889–902. [Google Scholar] [CrossRef] [PubMed]
- Oja, S.S.; Janáky, R.; Varga, V.; Saransaari, P. Modulation of glutamate receptor functions by glutathione. Neurochem. Int. 2000, 37, 299–306. [Google Scholar] [CrossRef]
- Freitas, H.R.; de Melo Reis, R.A. Glutathione induces GABA release through P2X7R activation on Müller glia. Neurogenesis 2017, 4, e1283188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Limongi, R.; Jeon, P.; Mackinley, M.; Das, T.; Dempster, K.; Théberge, J.; Bartha, R.; Wong, D.; Palaniyappan, L. Glutamate and Dysconnection in the Salience Network: Neurochemical, Effective Connectivity, and Computational Evidence in Schizophrenia. Biol. Psychiatry 2020, 88, 273–281. [Google Scholar] [CrossRef]
- Association, A.P. Diagnostic and Statistical Manual of Mental Disorders; American Psychiatric Publishing, Inc.: Arlington, VA, USA, 2013. [Google Scholar]
- Leckman, J.F.; Sholomskas, D.; Thompson, W.D.; Belanger, A.; Weissman, M.M. Best estimate of lifetime psychiatric diagnosis: A methodological study. Arch. Gen. Psychiatry 1982, 39, 879–883. [Google Scholar] [CrossRef] [PubMed]
- Andreasen, N.C.; Carpenter, W.T.J.; Kane, J.M.; Lasser, R.A.; Marder, S.R.; Weinberger, D.R. Remission in schizophrenia: Proposed criteria and rationale for consensus. Am. J. Psychiatry 2005, 162, 441–449. [Google Scholar] [CrossRef]
- Tkáč, I.; Gruetter, R. Methodology of H NMR Spectroscopy of the Human Brain at Very High Magnetic Fields. Appl. Magn. Reson. 2005, 29, 139–157. [Google Scholar] [CrossRef]
- Near, J.; Edden, R.; Evans, C.J.; Paquin, R.; Harris, A.; Jezzard, P. Frequency and phase drift correction of magnetic resonance spectroscopy data by spectral registration in the time domain. Magn. Reson. Med. 2015, 73, 44–50. [Google Scholar] [CrossRef] [Green Version]
- Bartha, R.; Drost, D.J.; Menon, R.S.; Williamson, P.C. Spectroscopic lineshape correction by QUECC: Combined QUALITY deconvolution and eddy current correction. Magn. Reson. Med. 2000, 44, 641–645. [Google Scholar] [CrossRef]
- Van den Boogaart, A.; Ala-Korpela, M.; Jokisaari, J.; Griffiths, J.R. Time and frequency domain analysis of NMR data compared: An application to 1D 1H spectra of lipoproteins. Magn. Reson. Med. 1994, 31, 347–358. [Google Scholar] [CrossRef]
- Bartha, R.; Drost, D.J.; Williamson, P.C. Factors affecting the quantification of short echo in-vivo 1H MR spectra: Prior knowledge, peak elimination, and filtering. NMR. Biomed. 1999, 12, 205–216. [Google Scholar] [CrossRef]
- Wong, D. MRI Investigations of Metabolic and Structural Brain Changes in Alzheimer’s Disease and Vitamin D Deprivation. Ph.D. Thesis, The University of Western Ontario, London, ON, Canada, 2019; p. 6611. [Google Scholar]
- Dempster, K.; Jeon, P.; MacKinley, M.; Williamson, P.; Théberge, J.; Palaniyappan, L. Early treatment response in first episode psychosis: A 7-T magnetic resonance spectroscopic study of glutathione and glutamate. Mol. Psychiatry 2020, 25, 1640–1650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kruschke, J.K. Doing Bayesian Data Analysis: A Tutorial with R, JAGS, and STAN; Elsevier: London, UK, 2015. [Google Scholar]
- Razi, A.; Kahan, J.; Rees, G.; Friston, K.J. Construct validation of a DCM for resting state fMRI. Neuroimage 2015, 106, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friston, K.J.; Preller, K.H.; Mathys, C.; Cagnan, H.; Heinzle, J.; Razi, A.; Zeidman, P. Dynamic causal modelling revisited. NeuroImage 2019, 199, 730–744. [Google Scholar] [CrossRef] [PubMed]
- Friston, K.J.; Kahan, J.; Biswal, B.; Razi, A. A DCM for resting state fMRI. NeuroImage 2014, 94, 396–407. [Google Scholar] [CrossRef] [Green Version]
- Kahan, J.; Foltynie, T. Understanding DCM: Ten simple rules for the clinician. Neuroimage 2013, 83, 542–549. [Google Scholar] [CrossRef] [Green Version]
- Marreiros, A.C.; Kiebel, S.J.; Friston, K.J. Dynamic causal modelling for fMRI: A two-state model. Neuroimage 2008, 39, 269–278. [Google Scholar] [CrossRef]
- Kahan, J.; Urner, M.; Moran, R.; Flandin, G.; Marreiros, A.; Mancini, L.; White, M.; Thornton, J.; Yousry, T.; Zrinzo, L.; et al. Resting state functional MRI in Parkinson’s disease: The impact of deep brain stimulation on ‘effective’ connectivity. Brain A J. Neurol. 2014, 137, 1130–1144. [Google Scholar] [CrossRef]
- Friston, K.J.; Litvak, V.; Oswal, A.; Razi, A.; Stephan, K.E.; van Wijk, B.C.M.; Ziegler, G.; Zeidman, P. Bayesian model reduction and empirical Bayes for group (DCM) studies. NeuroImage 2016, 128, 413–431. [Google Scholar] [CrossRef] [Green Version]
- Zeidman, P.; Jafarian, A.; Seghier, M.L.; Litvak, V.; Cagnan, H.; Price, C.J.; Friston, K.J. A guide to group effective connectivity analysis, part 2: Second level analysis with PEB. NeuroImage 2019, 200, 12–25. [Google Scholar] [CrossRef]
- Friston, K.; Zeidman, P.; Litvak, V. Empirical Bayes for DCM: A Group Inversion Scheme. Front. Syst. Neurosci. 2015, 9, 164. [Google Scholar] [CrossRef] [Green Version]
- Penny, W.D. Comparing Dynamic Causal Models using AIC, BIC and Free Energy. NeuroImage 2012, 59, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Kass, R.E.; Raftery, A.E. Bayes Factors. J. Am. Stat. Assoc. 1995, 90, 773–795. [Google Scholar] [CrossRef]
- Palaniyappan, L.; Liddle, P.F. Does the salience network play a cardinal role in psychosis? An emerging hypothesis of insular dysfunction. J. Psychiatry Neurosci. 2012, 37, 17–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, T.K.; Javadzadeh, A.; Dey, A.; Sabesan, P.; Théberge, J.; Radua, J.; Palaniyappan, L. Antioxidant defense in schizophrenia and bipolar disorder: A meta-analysis of MRS studies of anterior cingulate glutathione. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2019, 91, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Brandes, M.S.; Gray, N.E. NRF2 as a Therapeutic Target in Neurodegenerative Diseases. ASN Neuro 2020, 12. [Google Scholar] [CrossRef]
- Murray, R.M.; Quattrone, D.; Natesan, S.; van Os, J.; Nordentoft, M.; Howes, O.; Di Forti, M.; Taylor, D. Should psychiatrists be more cautious about the long-term prophylactic use of antipsychotics? Br. J. Psychiatry 2016, 209, 361–365. [Google Scholar] [CrossRef] [Green Version]
- Conus, P.; Seidman, L.J.; Fournier, M.; Xin, L.; Cleusix, M.; Baumann, P.S.; Ferrari, C.; Cousins, A.; Alameda, L.; Gholam-Rezaee, M.; et al. N-acetylcysteine in a Double-Blind Randomized Placebo-Controlled Trial: Toward Biomarker-Guided Treatment in Early Psychosis. Schizophr. Bull. 2018, 44, 317–327. [Google Scholar] [CrossRef] [Green Version]
- Wong, D.; Schranz, A.L.; Bartha, R. Optimized in vivo brain glutamate measurement using long-echo-time semi-LASER at 7 T. NMR Biomed. 2018, 31, e4002. [Google Scholar] [CrossRef]
- Hermens, D.F.; Lagopoulos, J.; Naismith, S.L.; Tobias-Webb, J.; Hickie, I.B. Distinct neurometabolic profiles are evident in the anterior cingulate of young people with major psychiatric disorders. Transl. Psychiatry 2012, 2, e110. [Google Scholar] [CrossRef] [Green Version]
- Xin, L.; Mekle, R.; Fournier, M.; Baumann, P.S.; Ferrari, C.; Alameda, L.; Jenni, R.; Lu, H.; Schaller, B.; Cuenod, M.; et al. Genetic Polymorphism Associated Prefrontal Glutathione and Its Coupling with Brain Glutamate and Peripheral Redox Status in Early Psychosis. Schizophr. Bull. 2016, 42, 1185–1196. [Google Scholar] [CrossRef]
- Krienen, F.M.; Goldman, M.; Zhang, Q.; del Rosario, R.C.H.; Florio, M.; Machold, R.; Saunders, A.; Levandowski, K.; Zaniewski, H.; Schuman, B.; et al. Innovations present in the primate interneuron repertoire. Nature 2020, 586, 262–269. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subject | Group | SOFAS | Parental SES (NSSEC) | Age at Study Date | Gender | PANSS Sub-Scores | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| P1 | P2 | P3 | N1 | N4 | N6 | G5 | G9 | ||||||
| 1 | HC | 73 | 3 | 26 | Male | - | - | - | - | - | - | - | - |
| 2 | 85 | 4 | 23 | Female | - | - | - | - | - | - | - | - | |
| 3 | 79 | 2 | 17 | Male | - | - | - | - | - | - | - | - | |
| 4 | 85 | 5 | 23 | Male | - | - | - | - | - | - | - | - | |
| 5 | 87 | 1 | 16 | Female | - | - | - | - | - | - | - | - | |
| 6 | 81 | 5 | 25 | Male | - | - | - | - | - | - | - | - | |
| 7 | 83 | 2 | 16 | Male | - | - | - | - | - | - | - | - | |
| 8 | 86 | 2 | 16 | Male | - | - | - | - | - | - | - | - | |
| 9 | 80 | 3 | 29 | Male | - | - | - | - | - | - | - | - | |
| 10 | 79 | 2 | 22 | Female | - | - | - | - | - | - | - | - | |
| 11 | 79 | 2 | 23 | Female | - | - | - | - | - | - | - | - | |
| 12 | 83 | 1 | 20 | Male | - | - | - | - | - | - | - | - | |
| 13 | 80 | 4 | 20 | Male | - | - | - | - | - | - | - | - | |
| 14 | 80 | 2 | 20 | Male | - | - | - | - | - | - | - | - | |
| 15 | 85 | 3 | 20 | Male | - | - | - | - | - | - | - | - | |
| 16 | 80 | 2 | 20 | Female | - | - | - | - | - | - | - | - | |
| 17 | 85 | 5 | 27 | Female | - | - | - | - | - | - | - | - | |
| 18 | 85 | 5 | 22 | Female | - | - | - | - | - | - | - | - | |
| 19 | 80 | 5 | 18 | Female | - | - | - | - | - | - | - | - | |
| 20 | 85 | 2 | 22 | Female | - | - | - | - | - | - | - | - | |
| 1 | FES | 40 | 4 | 19 | Male | 4 | 4 | 4 | 4 | 5 | 3 | 2 | 4 |
| 2 | 37 | 5 | 20 | Male | 5 | 4 | 5 | 2 | 3 | 1 | 3 | 4 | |
| 3 | 40 | 2 | 19 | Male | 4 | 5 | 4 | 5 | 4 | 3 | 1 | 3 | |
| 4 | 60 | 2 | 17 | Male | 5 | 1 | 5 | 3 | 4 | 3 | 1 | 4 | |
| 5 | 30 | 4 | 18 | Male | 5 | 3 | 4 | 2 | 3 | 3 | 1 | 3 | |
| 6 | 51 | 4 | 17 | Female | 5 | 1 | 5 | 3 | 5 | 3 | 3 | 2 | |
| 7 | 34 | 5 | 24 | Male | 6 | 5 | 6 | 1 | 1 | 1 | 4 | 5 | |
| 8 | 50 | 2 | 21 | Male | 5 | 4 | 4 | 2 | 4 | 2 | 1 | 4 | |
| 9 | 25 | 2 | 25 | Male | 7 | 3 | 5 | 4 | 3 | 1 | 2 | 6 | |
| 10 | 40 | 2 | 28 | Male | 6 | 4 | 2 | 1 | 1 | 1 | 1 | 3 | |
| 11 | 33 | 2 | 20 | Female | 6 | 3 | 2 | 1 | 1 | 1 | 1 | 4 | |
| 12 | 65 | 3 | 23 | Female | 4 | 2 | 5 | 3 | 1 | 2 | 1 | 3 | |
| 13 | 25 | 2 | 23 | Male | 5 | 6 | 5 | 1 | 3 | 4 | 3 | 5 | |
| 14 | 44 | 3 | 24 | Female | 5 | 4 | 2 | 1 | 1 | 1 | 1 | 5 | |
| 15 | 20 | 2 | 23 | Female | 7 | 4 | 6 | 1 | 1 | 1 | 1 | 6 | |
| 16 | 55 | 4 | 20 | Male | 5 | 1 | 5 | 4 | 3 | 1 | 1 | 3 | |
| 17 | 50 | 1 | 27 | Male | 7 | 3 | 2 | 5 | 4 | 3 | 1 | 3 | |
| 18 | 40 | 5 | 26 | Female | 5 | 1 | 5 | 1 | 5 | 1 | 1 | 3 | |
| 19 | 45 | 4 | 19 | Female | 5 | 3 | 1 | 3 | 3 | 3 | 1 | 4 | |
| Metabolite | Parameter | Mean | Median | Mode | HDI (Low) | HDI (High) |
|---|---|---|---|---|---|---|
| GLU | β0 | 6.641 | 6.641 | 6.650 | 6.212 | 7.074 |
| βFES | 0.203 | 0.195 | 0.086 | −0.168 | 0.622 | |
| βHC | −0.203 | −0.195 | −0.086 | −0.622 | 0.168 | |
| σβ | 1.386 | 0.923 | 0.361 | 0.000 | 4.266 | |
| σi | 1.374 | 1.358 | 1.309 | 1.076 | 1.727 | |
| GSH | β0 | 1.604 | 1.604 | 1.610 | 1.482 | 1.719 |
| βFES | 0.118 | 0.117 | 0.118 | −0.003 | 0.236 | |
| βHC | −0.118 | −0.117 | −0.118 | −0.236 | 0.003 | |
| σβ | 0.515 | 0.365 | 0.197 | 0.001 | 1.460 | |
| σi | 0.374 | 0.369 | 0.362 | 0.294 | 0.469 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Limongi, R.; Jeon, P.; Théberge, J.; Palaniyappan, L. Counteracting Effects of Glutathione on the Glutamate-Driven Excitation/Inhibition Imbalance in First-Episode Schizophrenia: A 7T MRS and Dynamic Causal Modeling Study. Antioxidants 2021, 10, 75. https://doi.org/10.3390/antiox10010075
Limongi R, Jeon P, Théberge J, Palaniyappan L. Counteracting Effects of Glutathione on the Glutamate-Driven Excitation/Inhibition Imbalance in First-Episode Schizophrenia: A 7T MRS and Dynamic Causal Modeling Study. Antioxidants. 2021; 10(1):75. https://doi.org/10.3390/antiox10010075
Chicago/Turabian StyleLimongi, Roberto, Peter Jeon, Jean Théberge, and Lena Palaniyappan. 2021. "Counteracting Effects of Glutathione on the Glutamate-Driven Excitation/Inhibition Imbalance in First-Episode Schizophrenia: A 7T MRS and Dynamic Causal Modeling Study" Antioxidants 10, no. 1: 75. https://doi.org/10.3390/antiox10010075
APA StyleLimongi, R., Jeon, P., Théberge, J., & Palaniyappan, L. (2021). Counteracting Effects of Glutathione on the Glutamate-Driven Excitation/Inhibition Imbalance in First-Episode Schizophrenia: A 7T MRS and Dynamic Causal Modeling Study. Antioxidants, 10(1), 75. https://doi.org/10.3390/antiox10010075