
Oxidative Stress and Energy Metabolism in the Brain: Midlife as a Turning Point



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Energy Metabolism of the Brain
3. Mechanisms of ROS Generation
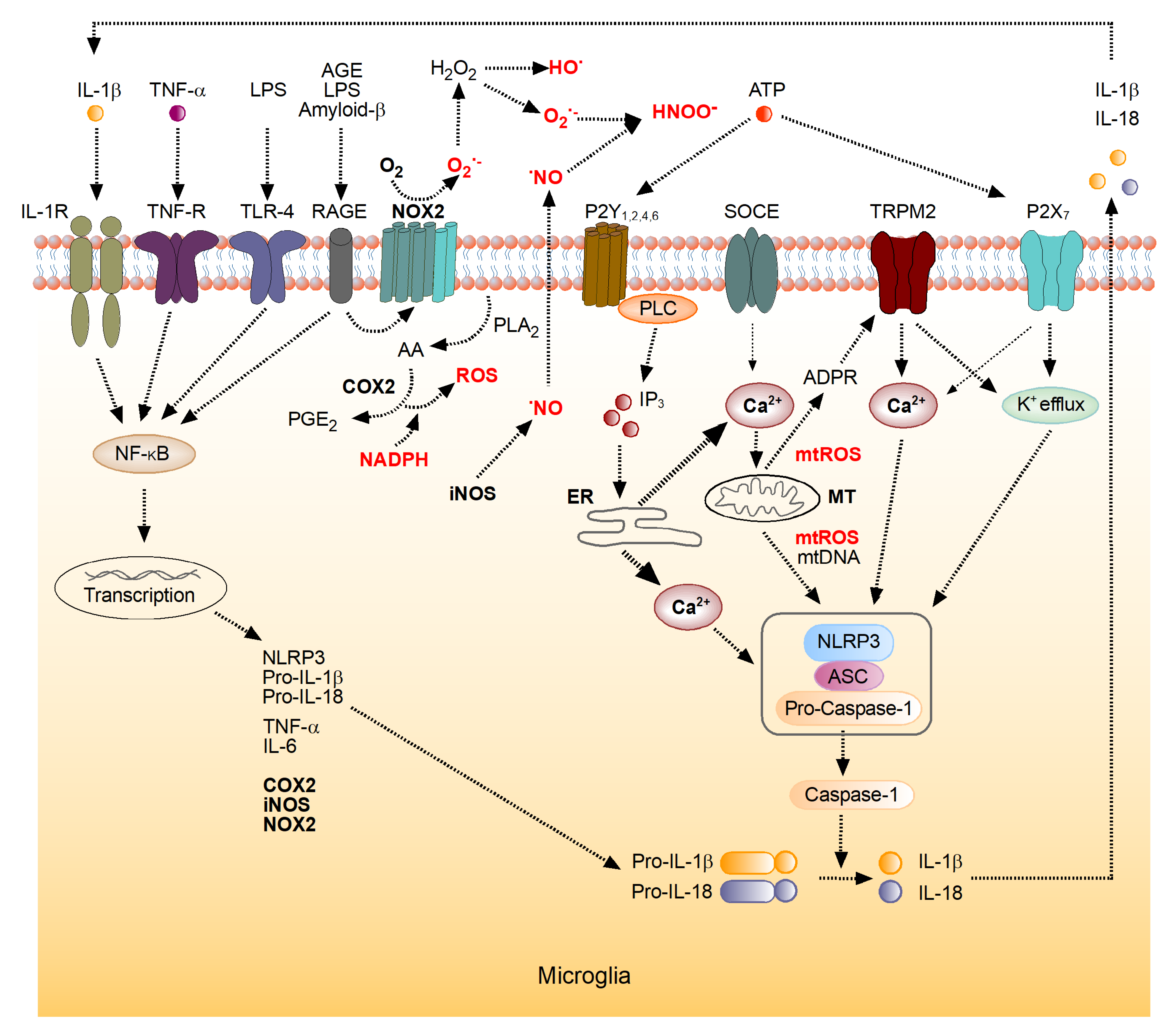
4. The Role of the Brain’s Immune System in the Generation of ROS
5. Midlife Turning Point in Glucose Catabolism: Switch from Glycolysis to Pentose Phosphate Pathway
6. Midlife Increase in Mitochondrial Function Followed by Its Subsequent Decline
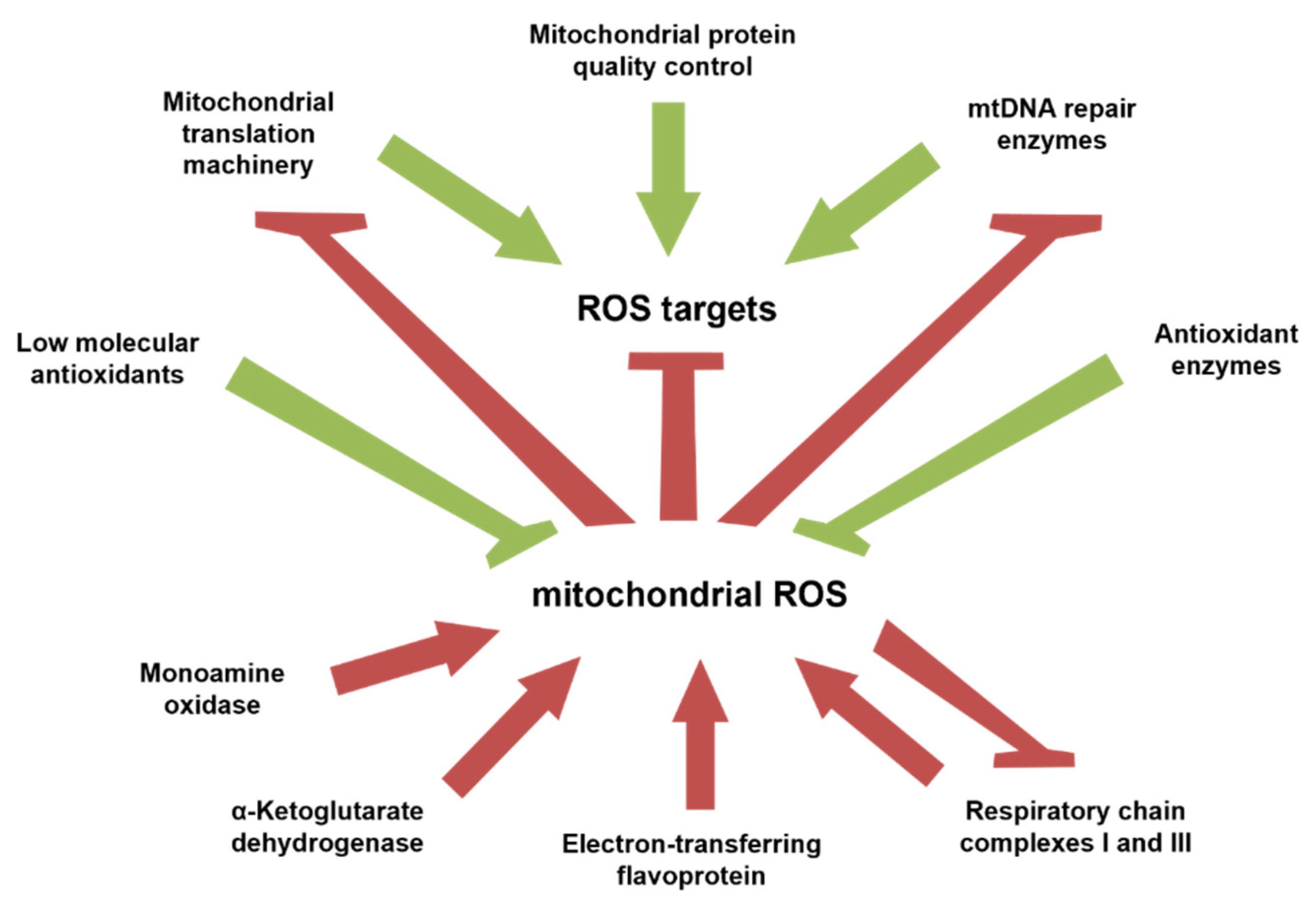
7. Contribution of Mitochondrially Produced ROS to Age-Related Changes in Signaling Pathways
8. Midlife Activation of the Brain’s Immune System and Its Possible Consequences
9. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Adam-Vizi, V. Production of Reactive Oxygen Species in Brain Mitochondria: Contribution by Electron Transport Chain and Non-Electron Transport Chain Sources. Antioxid. Redox Signal. 2005, 7, 1140–1149. [Google Scholar] [CrossRef]
- Lushchak, V.I. Free Radicals, Reactive Oxygen Species, Oxidative Stresses and their Classifications. UKR. Biochem. J. 2015, 87, 11–18. [Google Scholar] [CrossRef] [Green Version]
- Garaschuk, O.; Semchyshyn, H.M.; Lushchak, V.I. Healthy Brain Aging: Interplay between Reactive Species, Inflammation and Energy Supply. Ageing Res. Rev. 2018, 43, 26–45. [Google Scholar] [CrossRef] [PubMed]
- Lushchak, V.I. Interplay between Bioenergetics and Oxidative Stress at Normal Brain Aging. Aging as a Result of Increasing Disbalance in the System Oxidative Stress-Energy Provision. Pflugers Arch. 2021, 473, 713–722. [Google Scholar] [CrossRef]
- Semchyshyn, H. Is Carbonyl/AGE/RAGE Stress a Hallmark of the Brain Aging? Pflugers Arch. 2021, 473, 723–734. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial Reactive Oxygen Species (ROS) and ROS-Induced ROS Release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [Green Version]
- Harman, D. Aging: A Theory Based on Free Radical and Radiation Chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCord, J.M.; Fridovich, I. Superoxide Dismutase. An Enzymic Function for Erythrocuprein (Hemocuprein). J. Biol. Chem. 1969, 244, 6049–6055. [Google Scholar] [CrossRef]
- Reczek, C.R.; Chandel, N.S. ROS-Dependent Signal Transduction. Curr. Opin. Cell Biol. 2015, 33, 8–13. [Google Scholar] [CrossRef] [Green Version]
- Zandalinas, S.I.; Mittler, R. ROS-Induced ROS Release in Plant and Animal Cells. Free Radic. Biol. Med. 2018, 122, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Jones, D.P. Reactive Oxygen Species (ROS) as Pleiotropic Physiological Signalling Agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef] [PubMed]
- Simpson, D.S.A.; Oliver, P.L. ROS Generation in Microglia: Understanding Oxidative Stress and Inflammation in Neurodegenerative Disease. Antioxidants 2020, 9, 743. [Google Scholar] [CrossRef] [PubMed]
- Longo, V.D.; Di Tano, M.; Mattson, M.P.; Guidi, N. Intermittent and Periodic Fasting, Longevity and Disease. Nat. Aging 2021, 1, 47–59. [Google Scholar] [CrossRef]
- García-Revilla, J.; Alonso-Bellido, I.M.; Burguillos, M.A.; Herrera, A.J.; Espinosa-Oliva, A.M.; Ruiz, R.; Cruz-Hernández, L.; García-Domínguez, I.; Roca-Ceballos, M.A.; Santiago, M.; et al. Reformulating Pro-Oxidant Microglia in Neurodegeneration. J. Clin. Med. 2019, 8, 1719. [Google Scholar] [CrossRef] [Green Version]
- Cepas, V.; Collino, M.; Mayo, J.C.; Sainz, R.M. Redox Signaling and Advanced Glycation Endproducts (AGEs) in Diet-Related Diseases. Antioxidants 2020, 9, 142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garaschuk, O. The Role of NLRP3 Inflammasome for Microglial Response to Peripheral Inflammation. Neural Regen. Res. 2021, 16, 294–295. [Google Scholar] [CrossRef]
- Tschopp, J.; Schroder, K. NLRP3 Inflammasome Activation: The Convergence of Multiple Signalling Pathways on ROS Production? Nat. Rev. Immunol. 2010, 10, 210–215. [Google Scholar] [CrossRef]
- Yang, S.; Qin, C.; Hu, Z.-W.; Zhou, L.-Q.; Yu, H.-H.; Chen, M.; Bosco, D.B.; Wang, W.; Wu, L.-J.; Tian, D.-S. Microglia Reprogram Metabolic Profiles for Phenotype and Function Changes in Central Nervous System. Neurobiol. Dis. 2021, 152, 105290. [Google Scholar] [CrossRef]
- Metcalfe, N.B.; Alonso-Alvarez, C. Oxidative Stress as a Life-History Constraint: The Role of Reactive Oxygen Species in Shaping Phenotypes from Conception to Death: Oxidative Stress as a Life-History Constraint. Funct. Ecol. 2010, 24, 984–996. [Google Scholar] [CrossRef] [Green Version]
- Sikora, E.; Bielak-Zmijewska, A.; Dudkowska, M.; Krzystyniak, A.; Mosieniak, G.; Wesierska, M.; Wlodarczyk, J. Cellular Senescence in Brain Aging. Front. Aging Neurosci. 2021, 13, 646924. [Google Scholar] [CrossRef]
- Olmedillas Del Moral, M.; Asavapanumas, N.; Uzcátegui, N.L.; Garaschuk, O. Healthy Brain Aging Modifies Microglial Calcium Signaling In Vivo. Int. J. Mol. Sci. 2019, 20, 589. [Google Scholar] [CrossRef] [Green Version]
- Yanar, K.; Simsek, B.; Çaylı, N.; Övül Bozkır, H.; Mengi, M.; Belce, A.; Aydin, S.; Çakatay, U. Caloric Restriction and Redox Homeostasis in Various Regions of Aging Male Rat Brain: Is Caloric Restriction Still Worth Trying Even after Early-Adulthood? Redox Homeostasis and Caloric Restriction in Brain. J. Food Biochem. 2019, 43, e12740. [Google Scholar] [CrossRef]
- Olmedillas Del Moral, M.; Fröhlich, N.; Figarella, K.; Mojtahedi, N.; Garaschuk, O. Effect of Caloric Restriction on the In Vivo Functional Properties of Aging Microglia. Front. Immunol. 2020, 11, 750. [Google Scholar] [CrossRef]
- Bayliak, M.M.; Sorochynska, O.M.; Kuzniak, O.V.; Gospodaryov, D.V.; Demianchuk, O.I.; Vasylyk, Y.V.; Mosiichuk, N.M.; Storey, K.B.; Garaschuk, O.; Lushchak, V.I. Middle Age as a Turning Point in Mouse Cerebral Cortex Energy and Redox Metabolism: Modulation by Every-Other-Day Fasting. Exp. Gerontol. 2021, 145, 111182. [Google Scholar] [CrossRef]
- Bayliak, M.M.; Mosiichuk, N.M.; Sorochynska, O.M.; Kuzniak, O.V.; Sishchuk, L.O.; Hrushchenko, A.O.; Semchuk, A.O.; Pryimak, T.V.; Vasylyk, Y.V.; Gospodaryov, D.V.; et al. Middle Aged Turn Point in Parameters of Oxidative Stress and Glucose Catabolism in Mouse Cerebellum during Lifespan: Minor Effects of Every-Other-Day Fasting. Biogerontology 2021, 22, 315–328. [Google Scholar] [CrossRef]
- Clarke, D.D.; Sokoloff, L. Circulation and Energy Metabolism of the Brain. In Basic Neurochemistry, 4th ed.; Siegel, G., Agrano, B.V., Albers, R.W., Molino, P.V., Eds.; Raven Press: New York, NY, USA, 1999; pp. 565–590. [Google Scholar]
- Hyder, F.; Rothman, D.L.; Bennett, M.R. Cortical Energy Demands of Signaling and Nonsignaling Components in Brain Are Conserved across Mammalian Species and Activity Levels. Proc. Natl. Acad. Sci. USA 2013, 110, 3549–3554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, C.S.; Sherrington, C.S. On the Regulation of the Blood-Supply of the Brain. J. Physiol. 1890, 11, 85–158. [Google Scholar] [CrossRef] [PubMed]
- Cipolla, M.J. The Cerebral Circulation. Colloq. Ser. Integr. Syst. Physiol. Mol. Funct. 2009, 1, 1–59. [Google Scholar] [CrossRef]
- Moore, S.A. Polyunsaturated Fatty Acid Synthesis and Release by Brain-Derived Cells In Vitro. J. Mol. Neurosci. MN 2001, 16, 195–200. [Google Scholar] [CrossRef]
- Halim, N.D.; Mcfate, T.; Mohyeldin, A.; Okagaki, P.; Korotchkina, L.G.; Patel, M.S.; Jeoung, N.H.; Harris, R.A.; Schell, M.J.; Verma, A. Phosphorylation Status of Pyruvate Dehydrogenase Distinguishes Metabolic Phenotypes of Cultured Rat Brain Astrocytes and Neurons. Glia 2010, 58, 1168–1176. [Google Scholar] [CrossRef] [Green Version]
- Di Loreto, S.; Zimmitti, V.; Sebastiani, P.; Cervelli, C.; Falone, S.; Amicarelli, F. Methylglyoxal Causes Strong Weakening of Detoxifying Capacity and Apoptotic Cell Death in Rat Hippocampal Neurons. Int. J. Biochem. Cell Biol. 2008, 40, 245–257. [Google Scholar] [CrossRef]
- Rose, J.; Brian, C.; Pappa, A.; Panayiotidis, M.I.; Franco, R. Mitochondrial Metabolism in Astrocytes Regulates Brain Bioenergetics, Neurotransmission and Redox Balance. Front. Neurosci. 2020, 14, 536682. [Google Scholar] [CrossRef]
- Debernardi, R.; Pierre, K.; Lengacher, S.; Magistretti, P.J.; Pellerin, L. Cell-Specific Expression Pattern of Monocarboxylate Transporters in Astrocytes and Neurons Observed in Different Mouse Brain Cortical Cell Cultures. J. Neurosci. Res. 2003, 73, 141–155. [Google Scholar] [CrossRef] [PubMed]
- Rafiki, A.; Boulland, J.L.; Halestrap, A.P.; Ottersen, O.P.; Bergersen, L. Highly Differential Expression of the Monocarboxylate Transporters MCT2 and MCT4 in the Developing Rat Brain. Neuroscience 2003, 122, 677–688. [Google Scholar] [CrossRef] [PubMed]
- Bröer, S.; Rahman, B.; Pellegri, G.; Pellerin, L.; Martin, J.L.; Verleysdonk, S.; Hamprecht, B.; Magistretti, P.J. Comparison of Lactate Transport in Astroglial Cells and Monocarboxylate Transporter 1 (MCT 1) Expressing Xenopus laevis Oocytes. Expression of Two Different Monocarboxylate Transporters in Astroglial Cells and Neurons. J. Biol. Chem. 1997, 272, 30096–30102. [Google Scholar] [CrossRef] [Green Version]
- van Hall, G.; Strømstad, M.; Rasmussen, P.; Jans, O.; Zaar, M.; Gam, C.; Quistorff, B.; Secher, N.H.; Nielsen, H.B. Blood Lactate Is an Important Energy Source for the Human Brain. J. Cereb. Blood Flow Metab. 2009, 29, 1121–1129. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P.; Gleichmann, M.; Cheng, A. Mitochondria in Neuroplasticity and Neurological Disorders. Neuron 2008, 60, 748–766. [Google Scholar] [CrossRef] [Green Version]
- Grimm, A.; Eckert, A. Brain Aging and Neurodegeneration: From a Mitochondrial Point of View. J. Neurochem. 2017, 143, 418–431. [Google Scholar] [CrossRef] [Green Version]
- De Miranda, B.R.; Rocha, E.M.; Castro, S.L.; Greenamyre, J.T. Protection from α-Synuclein Induced Dopaminergic Neurodegeneration by Overexpression of the Mitochondrial Import Receptor TOM20. NPJ Park. Dis. 2020, 6, 38. [Google Scholar] [CrossRef] [PubMed]
- Herrero-Mendez, A.; Almeida, A.; Fernández, E.; Maestre, C.; Moncada, S.; Bolaños, J.P. The Bioenergetic and Antioxidant Status of Neurons Is Controlled by Continuous Degradation of a Key Glycolytic Enzyme by APC/C-Cdh1. Nat. Cell Biol. 2009, 11, 747–752. [Google Scholar] [CrossRef]
- Longden, T.A.; Hill-Eubanks, D.C.; Nelson, M.T. Ion Channel Networks in the Control of Cerebral Blood Flow. J. Cereb. Blood Flow Metab. 2016, 36, 492–512. [Google Scholar] [CrossRef] [Green Version]
- Garthwaite, J.; Charles, S.L.; Chess-Williams, R. Endothelium-Derived Relaxing Factor Release on Activation of NMDA Receptors Suggests Role as Intercellular Messenger in the Brain. Nature 1988, 336, 385–388. [Google Scholar] [CrossRef]
- Roman, R.J. P-450 Metabolites of Arachidonic Acid in the Control of Cardiovascular Function. Physiol. Rev. 2002, 82, 131–185. [Google Scholar] [CrossRef] [Green Version]
- Hall, C.N.; Reynell, C.; Gesslein, B.; Hamilton, N.B.; Mishra, A.; Sutherland, B.A.; O’Farrell, F.M.; Buchan, A.M.; Lauritzen, M.; Attwell, D. Capillary Pericytes Regulate Cerebral Blood Flow in Health and Disease. Nature 2014, 508, 55–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, R.M.; Lourenço, C.F.; Pomerleau, F.; Huettl, P.; Gerhardt, G.A.; Laranjinha, J.; Barbosa, R.M. Brain Nitric Oxide Inactivation Is Governed by the Vasculature. Antioxid. Redox Signal. 2011, 14, 1011–1021. [Google Scholar] [CrossRef]
- Rolett, E.L.; Azzawi, A.; Liu, K.J.; Yongbi, M.N.; Swartz, H.M.; Dunn, J.F. Critical Oxygen Tension in Rat Brain: A Combined (31)P-NMR and EPR Oximetry Study. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2000, 279, R9–R16. [Google Scholar] [CrossRef] [Green Version]
- Wagenführ, L.; Meyer, A.K.; Marrone, L.; Storch, A. Oxygen Tension Within the Neurogenic Niche Regulates Dopaminergic Neurogenesis in the Developing Midbrain. Stem Cells Dev. 2016, 25, 227–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martí-Fàbregas, J.; Romaguera-Ros, M.; Gómez-Pinedo, U.; Martínez-Ramírez, S.; Jiménez-Xarrié, E.; Marín, R.; Martí-Vilalta, J.-L.; García-Verdugo, J.-M. Proliferation in the Human Ipsilateral Subventricular Zone after Ischemic Stroke. Neurology 2010, 74, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Ashok, B.S.; Ajith, T.A.; Sivanesan, S. Hypoxia-Inducible Factors as Neuroprotective Agent in Alzheimer’s Disease. Clin. Exp. Pharmacol. Physiol. 2017, 44, 327–334. [Google Scholar] [CrossRef] [Green Version]
- Watts, M.E.; Pocock, R.; Claudianos, C. Brain Energy and Oxygen Metabolism: Emerging Role in Normal Function and Disease. Front. Mol. Neurosci. 2018, 11, 216. [Google Scholar] [CrossRef]
- Papa, S.; Skulachev, V.P. Reactive Oxygen Species, Mitochondria, Apoptosis and Aging. Mol. Cell. Biochem. 1997, 174, 305–319. [Google Scholar] [CrossRef] [PubMed]
- Büeler, H. Mitochondrial and Autophagic Regulation of Adult Neurogenesis in the Healthy and Diseased Brain. Int. J. Mol. Sci. 2021, 22, 3342. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.M.; Geng, L.; Cahill-Smith, S.; Liu, F.; Douglas, G.; Mckenzie, C.-A.; Smith, C.; Brooks, G.; Channon, K.M.; Li, J.-M. Nox2 Contributes to Age-Related Oxidative Damage to Neurons and the Cerebral Vasculature. J. Clin. Investig. 2019, 129, 3374–3386. [Google Scholar] [CrossRef]
- Vignais, P.V. The Superoxide-Generating NADPH Oxidase: Structural Aspects and Activation Mechanism. Cell. Mol. Life Sci. CMLS 2002, 59, 1428–1459. [Google Scholar] [CrossRef]
- Kontos, H.A.; George, E. Brown Memorial Lecture. Oxygen Radicals in Cerebral Vascular Injury. Circ. Res. 1985, 57, 508–516. [Google Scholar] [CrossRef] [Green Version]
- Kukreja, R.C.; Kontos, H.A.; Hess, M.L.; Ellis, E.F. PGH Synthase and Lipoxygenase Generate Superoxide in the Presence of NADH or NADPH. Circ. Res. 1986, 59, 612–619. [Google Scholar] [CrossRef] [Green Version]
- Terada, L.S.; Willingham, I.R.; Rosandich, M.E.; Leff, J.A.; Kindt, G.W.; Repine, J.E. Generation of Superoxide Anion by Brain Endothelial Cell Xanthine Oxidase. J. Cell. Physiol. 1991, 148, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Minaev, B.F. How Cofactor-Free Oxygenases Can Overcome Spin Prohibition in Substrates Oxygenation by Dioxygen. Chem. Phys. 2019, 521, 61–68. [Google Scholar] [CrossRef]
- Hanschmann, E.-M.; Godoy, J.R.; Berndt, C.; Hudemann, C.; Lillig, C.H. Thioredoxins, Glutaredoxins, and Peroxiredoxins—Molecular Mechanisms and Health Significance: From Cofactors to Antioxidants to Redox Signaling. Antioxid. Redox Signal. 2013, 19, 1539–1605. [Google Scholar] [CrossRef]
- Jones, D.P. Redox Theory of Aging. Redox Biol. 2015, 5, 71–79. [Google Scholar] [CrossRef] [Green Version]
- Asiimwe, N.; Yeo, S.G.; Kim, M.-S.; Jung, J.; Jeong, N.Y. Nitric Oxide: Exploring the Contextual Link with Alzheimer’s Disease. Oxid. Med. Cell. Longev. 2016, 2016, 7205747. [Google Scholar] [CrossRef]
- Brawek, B.; Garaschuk, O. Microglial Calcium Signaling in the Adult, Aged and Diseased Brain. Cell Calcium 2013, 53, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Brawek, B.; Garaschuk, O. Monitoring In Vivo Function of Cortical Microglia. Cell Calcium 2017, 64, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Garaschuk, O.; Verkhratsky, A. Physiology of Microglia. Methods Mol. Biol. 2019, 2034, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Brookes, P.S.; Yoon, Y.; Robotham, J.L.; Anders, M.W.; Sheu, S.-S. Calcium, ATP, and ROS: A Mitochondrial Love-Hate Triangle. Am. J. Physiol. Cell Physiol. 2004, 287, C817–C833. [Google Scholar] [CrossRef]
- Wang, L.; Negro, R.; Wu, H. TRPM2, Linking Oxidative Stress and Ca2+ Permeation to NLRP3 Inflammasome Activation. Curr. Opin. Immunol. 2020, 62, 131–135. [Google Scholar] [CrossRef]
- Sorochynska, O.M.; Bayliak, M.M.; Gospodaryov, D.V.; Vasylyk, Y.V.; Kuzniak, O.V.; Pankiv, T.M.; Garaschuk, O.; Storey, K.B.; Lushchak, V.I. Every-Other-Day Feeding Decreases Glycolytic and Mitochondrial Energy-Producing Potentials in the Brain and Liver of Young Mice. Front. Physiol. 2019, 10, 1432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calabrese, V.; Cornelius, C.; Trovato, A.; Cavallaro, M.; Mancuso, C.; Di Rienzo, L.; Condorelli, D.; De Lorenzo, A.; Calabrese, E.J. The Hormetic Role of Dietary Antioxidants in Free Radical-Related Diseases. Curr. Pharm. Des. 2010, 16, 877–883. [Google Scholar] [CrossRef] [PubMed]
- Bayliak, M.M.; Abrat, O.B. Role of Nrf2 in Oxidative and Inflammatory Processes in Obesity and Metabolic Diseases. In Nrf2 and Its Modulation in Inflammation; Deng, H., Ed.; Progress in Inflammation Research; Springer International Publishing: Cham, Switzerland, 2020; pp. 153–187. ISBN 978-3-030-44599-7. [Google Scholar]
- Chorley, B.N.; Campbell, M.R.; Wang, X.; Karaca, M.; Sambandan, D.; Bangura, F.; Xue, P.; Pi, J.; Kleeberger, S.R.; Bell, D.A. Identification of Novel NRF2-Regulated Genes by ChIP-Seq: Influence on Retinoid X Receptor Alpha. Nucleic Acids Res. 2012, 40, 7416–7429. [Google Scholar] [CrossRef] [Green Version]
- Turell, L.; Zeida, A.; Trujillo, M. Mechanisms and Consequences of Protein Cysteine Oxidation: The Role of the Initial Short-Lived Intermediates. Essays Biochem. 2020, 64, 55–66. [Google Scholar] [CrossRef]
- Forman, H.J.; Davies, M.J.; Krämer, A.C.; Miotto, G.; Zaccarin, M.; Zhang, H.; Ursini, F. Protein Cysteine Oxidation in Redox Signaling: Caveats on Sulfenic Acid Detection and Quantification. Arch. Biochem. Biophys. 2017, 617, 26–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alcock, L.J.; Perkins, M.V.; Chalker, J.M. Chemical Methods for Mapping Cysteine Oxidation. Chem. Soc. Rev. 2018, 47, 231–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pomatto, L.C.D.; Davies, K.J.A. The Role of Declining Adaptive Homeostasis in Ageing. J. Physiol. 2017, 595, 7275–7309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, J.; Lv, Z.; Qiao, X.; Li, X.; Li, Y.; Zhang, Y.; Chen, C. The Decay of Redox-Stress Response Capacity Is a Substantive Characteristic of Aging: Revising the Redox Theory of Aging. Redox Biol. 2017, 11, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Peleg, S. Biphasic Modeling of Mitochondrial Metabolism Dysregulation during Aging. Trends Biochem. Sci. 2017, 42, 702–711. [Google Scholar] [CrossRef] [Green Version]
- Navarro, A.; Boveris, A. Rat Brain and Liver Mitochondria Develop Oxidative Stress and Lose Enzymatic Activities on Aging. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004, 287, R1244–R1249. [Google Scholar] [CrossRef] [Green Version]
- Navarro, A.; López-Cepero, J.M.; Bández, M.J.; Sánchez-Pino, M.-J.; Gómez, C.; Cadenas, E.; Boveris, A. Hippocampal Mitochondrial Dysfunction in Rat Aging. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 294, R501–R509. [Google Scholar] [CrossRef] [Green Version]
- Boveris, A.; Navarro, A. Brain Mitochondrial Dysfunction in Aging. IUBMB Life 2008, 60, 308–314. [Google Scholar] [CrossRef]
- Yao, J.; Hamilton, R.T.; Cadenas, E.; Brinton, R.D. Decline in Mitochondrial Bioenergetics and Shift to Ketogenic Profile in Brain during Reproductive Senescence. Biochim. Biophys. Acta 2010, 1800, 1121–1126. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Kumar Sharma, L.; Li, Y.; Hu, P.; Idowu, A.; Liu, D.; Lu, J.; Bai, Y. Comparative Bioenergetic Study of Neuronal and Muscle Mitochondria during Aging. Free Radic. Biol. Med. 2013, 63, 30–40. [Google Scholar] [CrossRef] [Green Version]
- Gauba, E.; Guo, L.; Du, H. Cyclophilin D Promotes Brain Mitochondrial F1FO ATP Synthase Dysfunction in Aging Mice. J. Alzheimers Dis. 2017, 55, 1351–1362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klosinski, L.P.; Yao, J.; Yin, F.; Fonteh, A.N.; Harrington, M.G.; Christensen, T.A.; Trushina, E.; Brinton, R.D. White Matter Lipids as a Ketogenic Fuel Supply in Aging Female Brain: Implications for Alzheimer’s Disease. EBioMedicine 2015, 2, 1888–1904. [Google Scholar] [CrossRef] [Green Version]
- Mi, Y.; Qi, G.; Brinton, R.D.; Yin, F. Mitochondria-Targeted Therapeutics for Alzheimer’s Disease: The Good, the Bad, the Potential. Antioxid. Redox Signal. 2021, 34, 611–630. [Google Scholar] [CrossRef] [PubMed]
- Norwitz, N.G.; Hu, M.T.; Clarke, K. The Mechanisms by Which the Ketone Body D-β-Hydroxybutyrate May Improve the Multiple Cellular Pathologies of Parkinson’s Disease. Front. Nutr. 2019, 6, 63. [Google Scholar] [CrossRef]
- Kirkwood, T.B.L. Understanding the Odd Science of Aging. Cell 2005, 120, 437–447. [Google Scholar] [CrossRef] [Green Version]
- Diaz, F.; Garcia, S.; Padgett, K.R.; Moraes, C.T. A Defect in the Mitochondrial Complex III, but Not Complex IV, Triggers Early ROS-Dependent Damage in Defined Brain Regions. Hum. Mol. Genet. 2012, 21, 5066–5077. [Google Scholar] [CrossRef] [PubMed]
- Maranzana, E.; Barbero, G.; Falasca, A.I.; Lenaz, G.; Genova, M.L. Mitochondrial Respiratory Supercomplex Association Limits Production of Reactive Oxygen Species from Complex I. Antioxid. Redox Signal. 2013, 19, 1469–1480. [Google Scholar] [CrossRef] [Green Version]
- Pinto, M.; Vempati, U.D.; Diaz, F.; Peralta, S.; Moraes, C.T. Ablation of Cytochrome c in Adult Forebrain Neurons Impairs Oxidative Phosphorylation Without Detectable Apoptosis. Mol. Neurobiol. 2019, 56, 3722–3735. [Google Scholar] [CrossRef]
- Reichart, G.; Mayer, J.; Zehm, C.; Kirschstein, T.; Tokay, T.; Lange, F.; Baltrusch, S.; Tiedge, M.; Fuellen, G.; Ibrahim, S.; et al. Mitochondrial Complex IV Mutation Increases Reactive Oxygen Species Production and Reduces Lifespan in Aged Mice. Acta Physiol. 2019, 225, e13214. [Google Scholar] [CrossRef]
- Lopez-Fabuel, I.; Le Douce, J.; Logan, A.; James, A.M.; Bonvento, G.; Murphy, M.P.; Almeida, A.; Bolaños, J.P. Complex I Assembly into Supercomplexes Determines Differential Mitochondrial ROS Production in Neurons and Astrocytes. Proc. Natl. Acad. Sci. USA 2016, 113, 13063–13068. [Google Scholar] [CrossRef] [Green Version]
- Lushchak, O.V.; Piroddi, M.; Galli, F.; Lushchak, V.I. Aconitase Post-Translational Modification as a Key in Linkage between Krebs Cycle, Iron Homeostasis, Redox Signaling, and Metabolism of Reactive Oxygen Species. Redox Rep. Commun. Free Radic. Res. 2014, 19, 8–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Pinto, V.; Reina, S.; Gupta, A.; Messina, A.; Mahalakshmi, R. Role of Cysteines in Mammalian VDAC Isoforms’ Function. Biochim. Biophys. Acta 2016, 1857, 1219–1227. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Wu, J.; Jing, S.; Forster, M.J.; Yan, L.-J. Mitochondrial Protein Sulfenation during Aging in the Rat Brain. Biophys. Rep. 2018, 4, 104–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, W.-G.; Miranda, C.L.; Maier, C.S. Detection of Carbonyl-Modified Proteins in Interfibrillar Rat Mitochondria Using N’-Aminooxymethylcarbonylhydrazino-D-Biotin as an Aldehyde/Keto-Reactive Probe in Combination with Western Blot Analysis and Tandem Mass Spectrometry. Electrophoresis 2008, 29, 1317–1324. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Luo, X.; Yan, L.-J. Two Dimensional Blue Native/SDS-PAGE to Identify Mitochondrial Complex I Subunits Modified by 4-Hydroxynonenal (HNE). Front. Physiol. 2015, 6, 98. [Google Scholar] [CrossRef]
- Graziewicz, M.A.; Day, B.J.; Copeland, W.C. The Mitochondrial DNA Polymerase as a Target of Oxidative Damage. Nucleic Acids Res. 2002, 30, 2817–2824. [Google Scholar] [CrossRef] [Green Version]
- Niedernhofer, L.J.; Gurkar, A.U.; Wang, Y.; Vijg, J.; Hoeijmakers, J.H.J.; Robbins, P.D. Nuclear Genomic Instability and Aging. Annu. Rev. Biochem. 2018, 87, 295–322. [Google Scholar] [CrossRef]
- Kazak, L.; Reyes, A.; Holt, I.J. Minimizing the Damage: Repair Pathways Keep Mitochondrial DNA Intact. Nat. Rev. Mol. Cell Biol. 2012, 13, 659–671. [Google Scholar] [CrossRef] [PubMed]
- Andziak, B.; O’Connor, T.P.; Qi, W.; DeWaal, E.M.; Pierce, A.; Chaudhuri, A.R.; Van Remmen, H.; Buffenstein, R. High Oxidative Damage Levels in the Longest-Living Rodent, the Naked Mole-Rat. Aging Cell 2006, 5, 463–471. [Google Scholar] [CrossRef]
- Munro, D.; Baldy, C.; Pamenter, M.E.; Treberg, J.R. The Exceptional Longevity of the Naked Mole-Rat May Be Explained by Mitochondrial Antioxidant Defenses. Aging Cell 2019, 18, e12916. [Google Scholar] [CrossRef] [Green Version]
- Lewis, K.N.; Wason, E.; Edrey, Y.H.; Kristan, D.M.; Nevo, E.; Buffenstein, R. Regulation of Nrf2 Signaling and Longevity in Naturally Long-Lived Rodents. Proc. Natl. Acad. Sci. USA 2015, 112, 3722–3727. [Google Scholar] [CrossRef] [Green Version]
- Vyssokikh, M.Y.; Holtze, S.; Averina, O.A.; Lyamzaev, K.G.; Panteleeva, A.A.; Marey, M.V.; Zinovkin, R.A.; Severin, F.F.; Skulachev, M.V.; Fasel, N.; et al. Mild Depolarization of the Inner Mitochondrial Membrane Is a Crucial Component of an Anti-Aging Program. Proc. Natl. Acad. Sci. USA 2020, 117, 6491–6501. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, C.-C.; Kuro-o, M.; Rosenblatt, K.P.; Brobey, R.; Papaconstantinou, J. The ASK1-Signalosome Regulates P38 MAPK Activity in Response to Levels of Endogenous Oxidative Stress in the Klotho Mouse Models of Aging. Aging 2010, 2, 597–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papaconstantinou, J.; Hsieh, C.-C. Activation of Senescence and Aging Characteristics by Mitochondrially Generated ROS: How Are They Linked? Cell Cycle 2010, 9, 3831–3833. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, M.; Nishitoh, H.; Fujii, M.; Takeda, K.; Tobiume, K.; Sawada, Y.; Kawabata, M.; Miyazono, K.; Ichijo, H. Mammalian Thioredoxin Is a Direct Inhibitor of Apoptosis Signal-Regulating Kinase (ASK) 1. EMBO J. 1998, 17, 2596–2606. [Google Scholar] [CrossRef] [Green Version]
- Mammucari, C.; Rizzuto, R. Signaling Pathways in Mitochondrial Dysfunction and Aging. Mech. Ageing Dev. 2010, 131, 536–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasegawa, Y.; Toyama, K.; Uekawa, K.; Ichijo, H.; Kim-Mitsuyama, S. Role of ASK1/P38 Cascade in a Mouse Model of Alzheimer’s Disease and Brain Aging. J. Alzheimers Dis. 2018, 61, 259–263. [Google Scholar] [CrossRef]
- Furihata, T.; Takada, S.; Kakutani, N.; Maekawa, S.; Tsuda, M.; Matsumoto, J.; Mizushima, W.; Fukushima, A.; Yokota, T.; Enzan, N.; et al. Cardiac-Specific Loss of MitoNEET Expression Is Linked with Age-Related Heart Failure. Commun. Biol. 2021, 4, 138. [Google Scholar] [CrossRef]
- Kusminski, C.M.; Holland, W.L.; Sun, K.; Park, J.; Spurgin, S.B.; Lin, Y.; Askew, G.R.; Simcox, J.A.; McClain, D.A.; Li, C.; et al. MitoNEET-Driven Alterations in Adipocyte Mitochondrial Activity Reveal a Crucial Adaptive Process That Preserves Insulin Sensitivity in Obesity. Nat. Med. 2012, 18, 1539–1549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipper, C.H.; Stofleth, J.T.; Bai, F.; Sohn, Y.-S.; Roy, S.; Mittler, R.; Nechushtai, R.; Onuchic, J.N.; Jennings, P.A. Redox-Dependent Gating of VDAC by MitoNEET. Proc. Natl. Acad. Sci. USA 2019, 116, 19924–19929. [Google Scholar] [CrossRef] [Green Version]
- Ferecatu, I.; Gonçalves, S.; Golinelli-Cohen, M.-P.; Clémancey, M.; Martelli, A.; Riquier, S.; Guittet, E.; Latour, J.-M.; Puccio, H.; Drapier, J.-C.; et al. The Diabetes Drug Target MitoNEET Governs a Novel Trafficking Pathway to Rebuild an Fe-S Cluster into Cytosolic Aconitase/Iron Regulatory Protein 1. J. Biol. Chem. 2014, 289, 28070–28086. [Google Scholar] [CrossRef] [Green Version]
- Geldenhuys, W.J.; Benkovic, S.A.; Lin, L.; Yonutas, H.M.; Crish, S.D.; Sullivan, P.G.; Darvesh, A.S.; Brown, C.M.; Richardson, J.R. MitoNEET (CISD1) Knockout Mice Show Signs of Striatal Mitochondrial Dysfunction and a Parkinson’s Disease Phenotype. ACS Chem. Neurosci. 2017, 8, 2759–2765. [Google Scholar] [CrossRef]
- Amanakis, G.; Murphy, E. Cyclophilin D: An Integrator of Mitochondrial Function. Front. Physiol. 2020, 11, 595. [Google Scholar] [CrossRef] [PubMed]
- Carraro, M.; Checchetto, V.; Szabó, I.; Bernardi, P. F-ATP Synthase and the Permeability Transition Pore: Fewer Doubts, More Certainties. FEBS Lett. 2019, 593, 1542–1553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linard, D.; Kandlbinder, A.; Degand, H.; Morsomme, P.; Dietz, K.-J.; Knoops, B. Redox Characterization of Human Cyclophilin D: Identification of a New Mammalian Mitochondrial Redox Sensor? Arch. Biochem. Biophys. 2009, 491, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Vereczki, V.; Mansour, J.; Pour-Ghaz, I.; Bodnar, I.; Pinter, O.; Zelena, D.; Oszwald, E.; Adam-Vizi, V.; Chinopoulos, C. Cyclophilin D Regulates Lifespan and Protein Expression of Aging Markers in the Brain of Mice. Mitochondrion 2017, 34, 115–126. [Google Scholar] [CrossRef] [Green Version]
- Marí, M.; Colell, A. Mitochondrial Oxidative and Nitrosative Stress as a Therapeutic Target in Diseases. Antioxidants 2021, 10, 314. [Google Scholar] [CrossRef]
- Navarro, A.; Bandez, M.J.; Lopez-Cepero, J.M.; Gómez, C.; Boveris, A. High Doses of Vitamin E Improve Mitochondrial Dysfunction in Rat Hippocampus and Frontal Cortex upon Aging. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 300, R827–R834. [Google Scholar] [CrossRef] [Green Version]
- Shertzer, H.G.; Krishan, M.; Genter, M.B. Dietary Whey Protein Stimulates Mitochondrial Activity and Decreases Oxidative Stress in Mouse Female Brain. Neurosci. Lett. 2013, 548, 159–164. [Google Scholar] [CrossRef] [Green Version]
- Apostolova, N.; Victor, V.M. Molecular Strategies for Targeting Antioxidants to Mitochondria: Therapeutic Implications. Antioxid. Redox Signal. 2015, 22, 686–729. [Google Scholar] [CrossRef]
- Franceschi, C.; Bonafè, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-Aging. An Evolutionary Perspective on Immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef]
- Franceschi, C.; Garagnani, P.; Vitale, G.; Capri, M.; Salvioli, S. Inflammaging and “Garb-Aging”. Trends Endocrinol. Metab. 2017, 28, 199–212. [Google Scholar] [CrossRef] [Green Version]
- Santello, M.; Volterra, A. TNFα in Synaptic Function: Switching Gears. Trends Neurosci. 2012, 35, 638–647. [Google Scholar] [CrossRef]
- Maier, F.C.; Wehrl, H.F.; Schmid, A.M.; Mannheim, J.G.; Wiehr, S.; Lerdkrai, C.; Calaminus, C.; Stahlschmidt, A.; Ye, L.; Burnet, M.; et al. Longitudinal PET-MRI Reveals β-Amyloid Deposition and RCBF Dynamics and Connects Vascular Amyloidosis to Quantitative Loss of Perfusion. Nat. Med. 2014, 20, 1485–1492. [Google Scholar] [CrossRef]
- Lerdkrai, C.; Asavapanumas, N.; Brawek, B.; Kovalchuk, Y.; Mojtahedi, N.; Olmedillas Del Moral, M.; Garaschuk, O. Intracellular Ca2+ Stores Control In Vivo Neuronal Hyperactivity in a Mouse Model of Alzheimer’s Disease. Proc. Natl. Acad. Sci. USA 2018, 115, E1279–E1288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brawek, B.; Schwendele, B.; Riester, K.; Kohsaka, S.; Lerdkrai, C.; Liang, Y.; Garaschuk, O. Impairment of In Vivo Calcium Signaling in Amyloid Plaque-Associated Microglia. Acta Neuropathol. 2014, 127, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Busche, M.A.; Eichhoff, G.; Adelsberger, H.; Abramowski, D.; Wiederhold, K.-H.; Haass, C.; Staufenbiel, M.; Konnerth, A.; Garaschuk, O. Clusters of Hyperactive Neurons near Amyloid Plaques in a Mouse Model of Alzheimer’s Disease. Science 2008, 321, 1686–1689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Odoj, K.; Brawek, B.; Asavapanumas, N.; Mojtahedi, N.; Heneka, M.T.; Garaschuk, O. In Vivo Mechanisms of Cortical Network Dysfunction Induced by Systemic Inflammation. Brain. Behav. Immun. 2021, 96, 113–126. [Google Scholar] [CrossRef]
- Riester, K.; Brawek, B.; Savitska, D.; Fröhlich, N.; Zirdum, E.; Mojtahedi, N.; Heneka, M.T.; Garaschuk, O. In Vivo Characterization of Functional States of Cortical Microglia during Peripheral Inflammation. Brain. Behav. Immun. 2020, 87, 243–255. [Google Scholar] [CrossRef]
- Brawek, B.; Skok, M.; Garaschuk, O. Changing Functional Signatures of Microglia along the Axis of Brain Aging. Int. J. Mol. Sci. 2021, 22, 1091. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lushchak, V.I.; Duszenko, M.; Gospodaryov, D.V.; Garaschuk, O. Oxidative Stress and Energy Metabolism in the Brain: Midlife as a Turning Point. Antioxidants 2021, 10, 1715. https://doi.org/10.3390/antiox10111715
Lushchak VI, Duszenko M, Gospodaryov DV, Garaschuk O. Oxidative Stress and Energy Metabolism in the Brain: Midlife as a Turning Point. Antioxidants. 2021; 10(11):1715. https://doi.org/10.3390/antiox10111715
Chicago/Turabian StyleLushchak, Volodymyr I., Michael Duszenko, Dmytro V. Gospodaryov, and Olga Garaschuk. 2021. "Oxidative Stress and Energy Metabolism in the Brain: Midlife as a Turning Point" Antioxidants 10, no. 11: 1715. https://doi.org/10.3390/antiox10111715
APA StyleLushchak, V. I., Duszenko, M., Gospodaryov, D. V., & Garaschuk, O. (2021). Oxidative Stress and Energy Metabolism in the Brain: Midlife as a Turning Point. Antioxidants, 10(11), 1715. https://doi.org/10.3390/antiox10111715