
Crude Saponin from Platycodon grandiflorum Attenuates Aβ-Induced Neurotoxicity via Antioxidant, Anti-Inflammatory and Anti-Apoptotic Signaling Pathways

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Preparation
2.2. Cell Culture
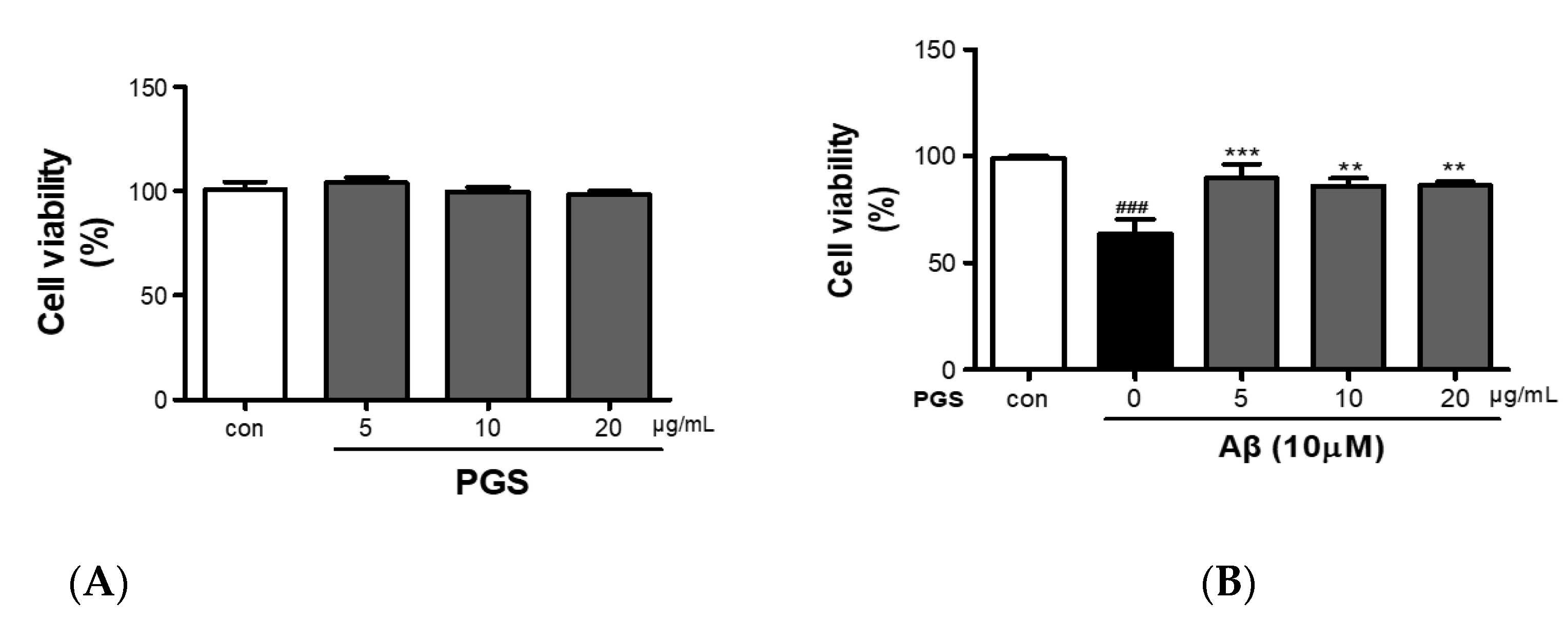
2.3. Cell Viability Assay
2.4. Measurement of Intracellular ROS Generation in HT22 Cells
2.5. Western Blot Analysis
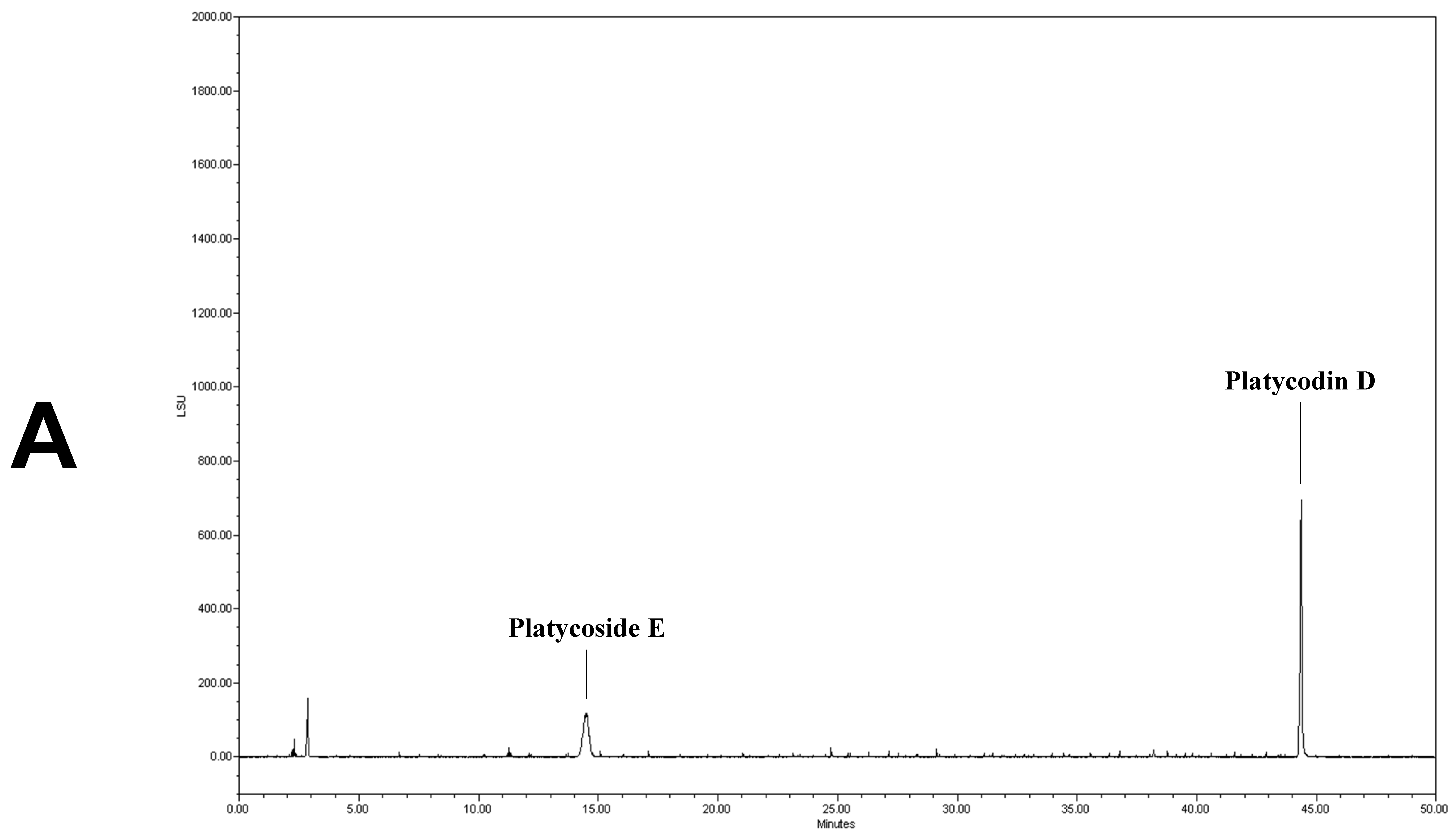
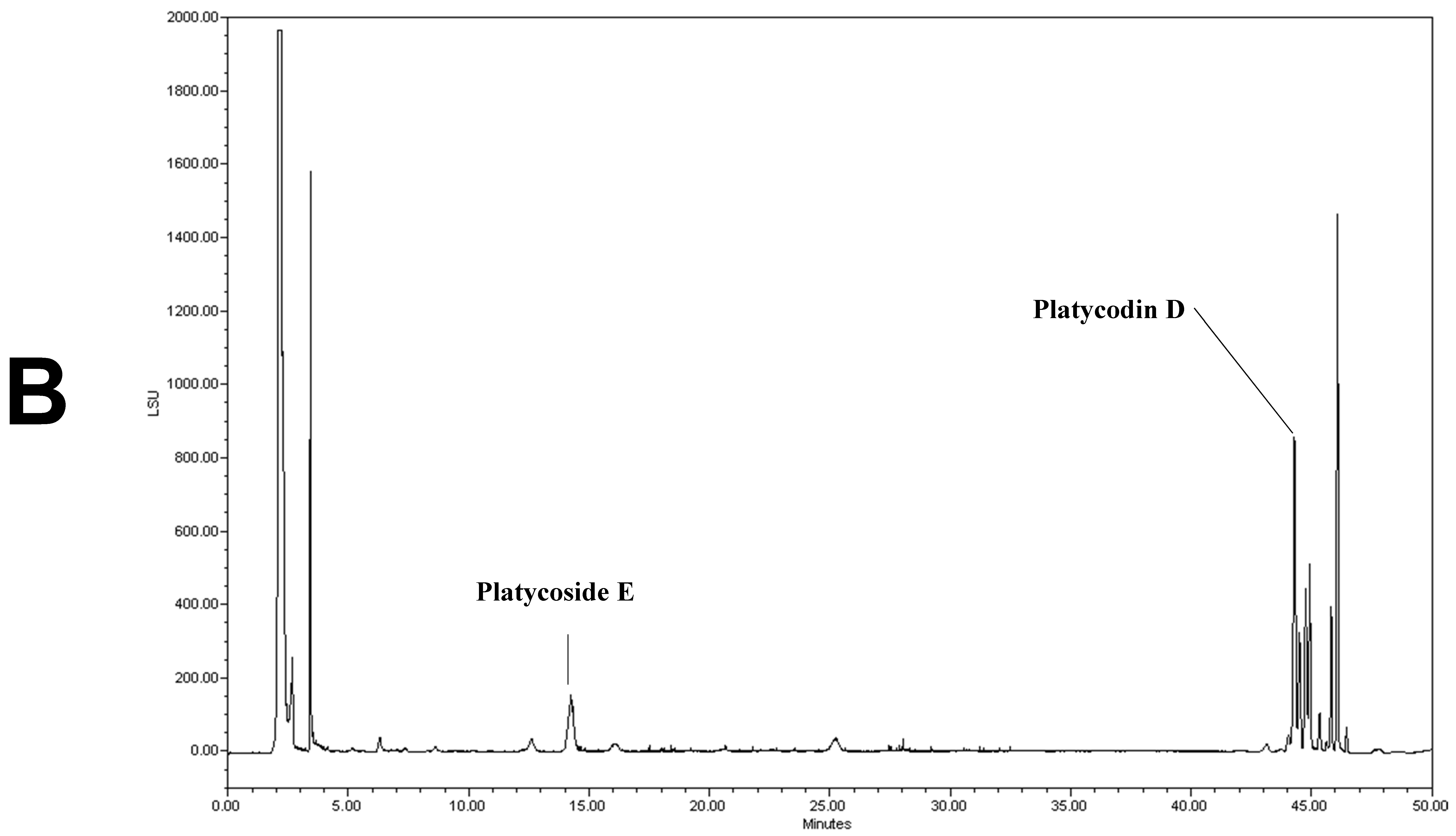
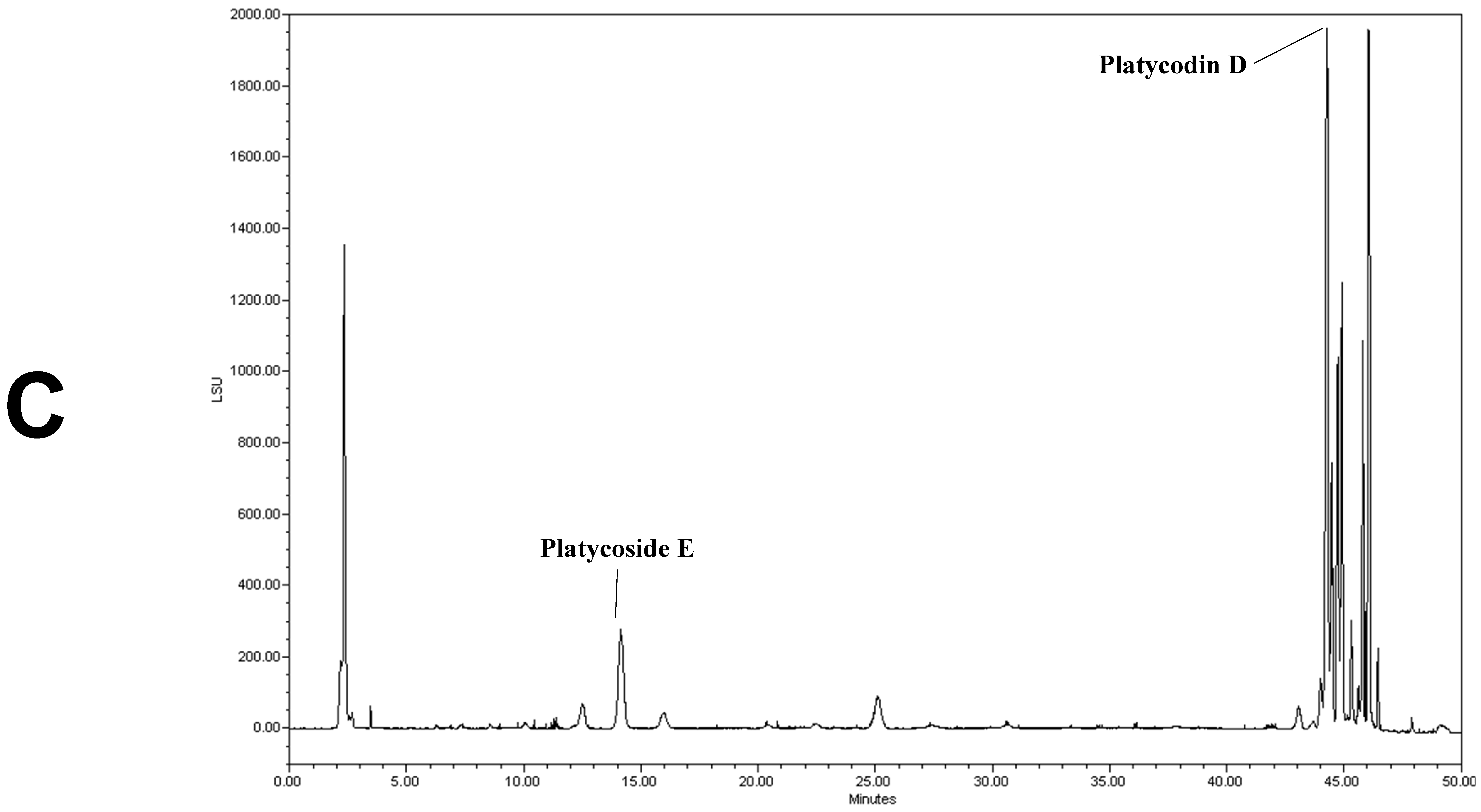
2.6. Analysis of Platycoside E and Platycodin D by HPLC-ELSD
2.7. Animals and PGS Administration
2.8. Preparation of Brain Tissue
2.9. Histological Analysis
2.10. Image Acquisition and Analysis
2.11. Statistical Analysis
3. Results
3.1. Analysis of Platycoside E and Platycodin D
3.2. Protective Effect of PGS against Aβ-Induced HT22 Cell Injury
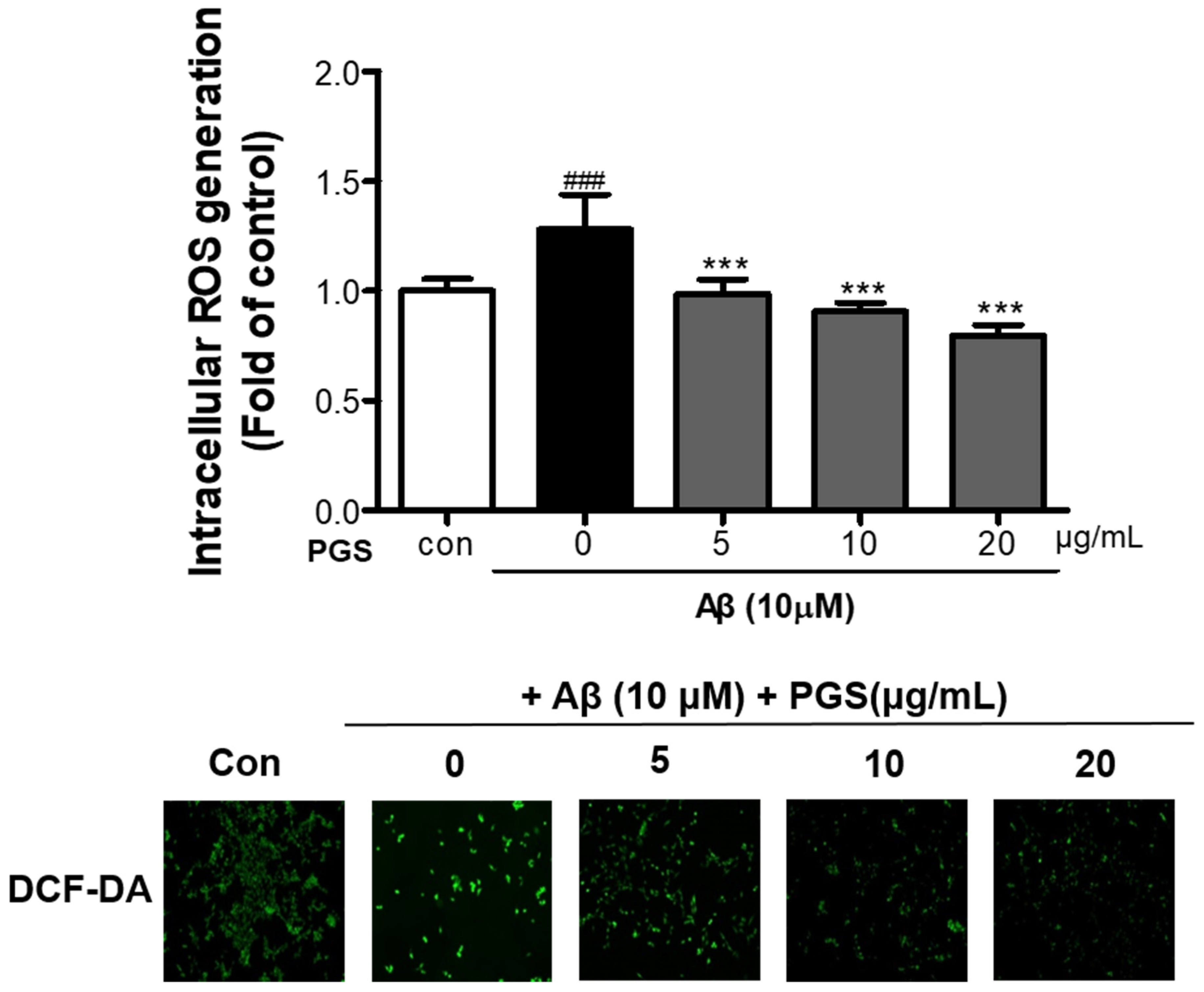
3.3. Effect of PGS on Aβ-Induced ROS Production in HT22 Cells
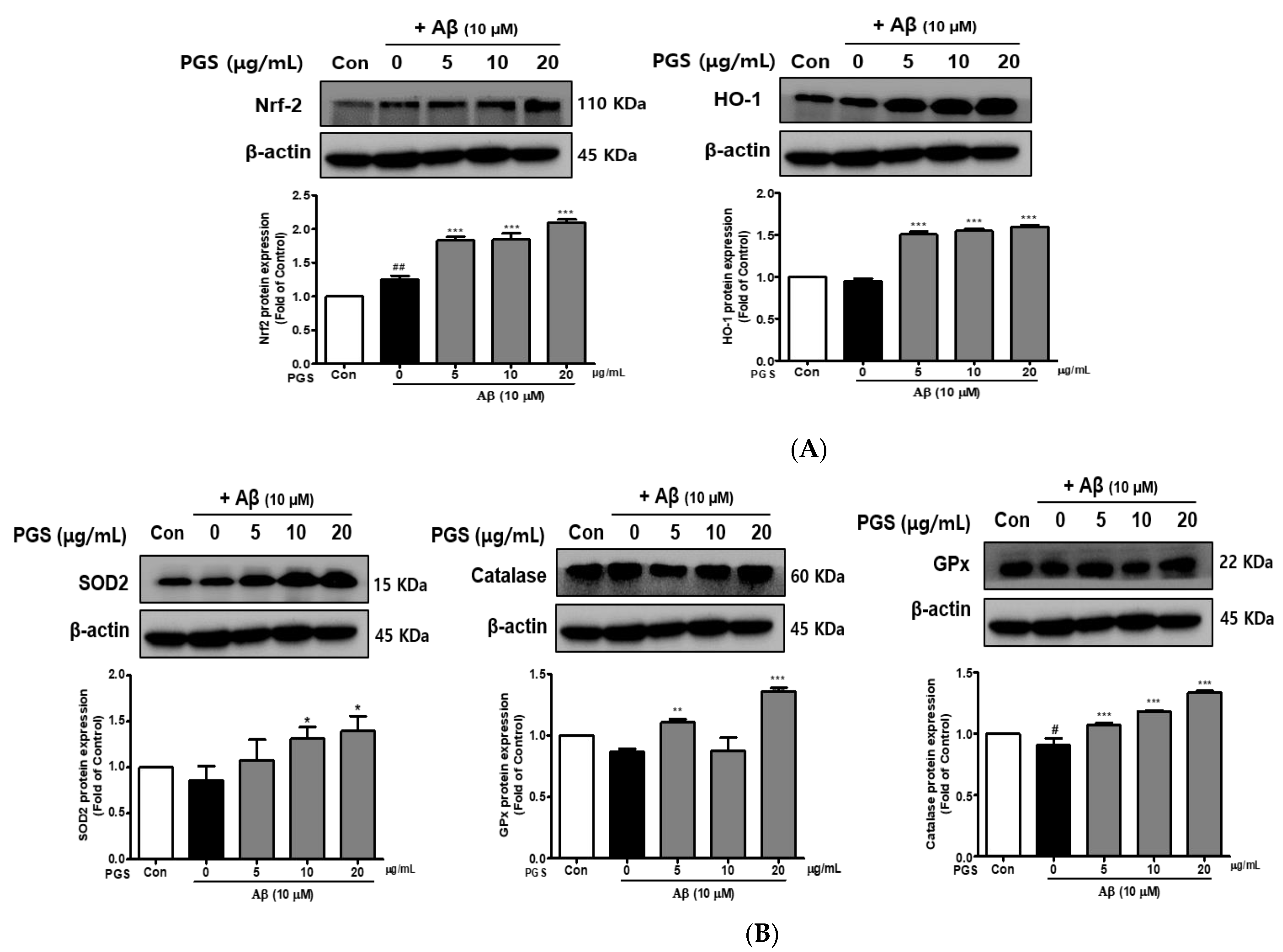
3.4. Effect of PGS on Antioxidant Enzymes in HT22 Cells
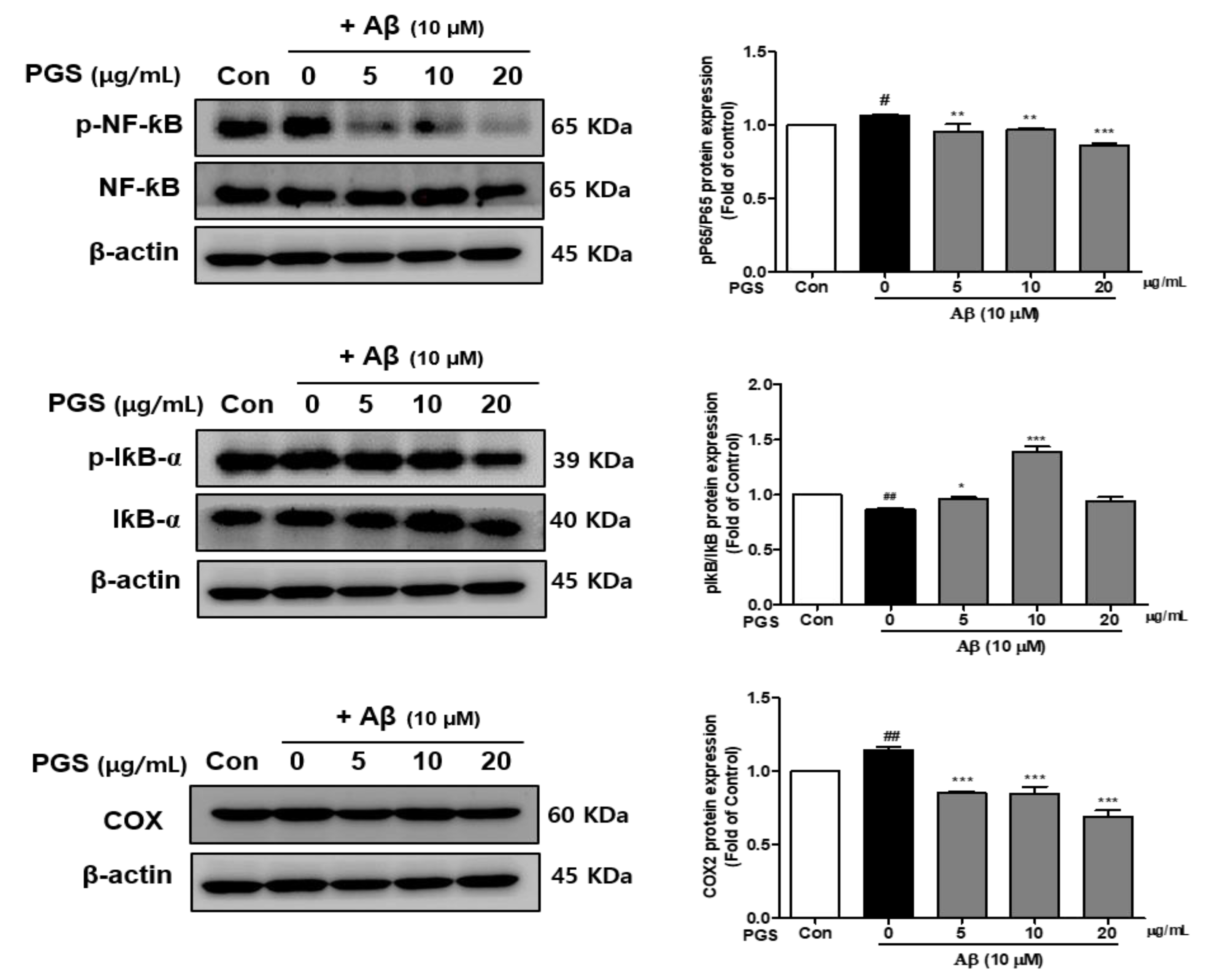
3.5. Effect of PGS on NF-κB Activation in Aβ-Induced HT22 Cells
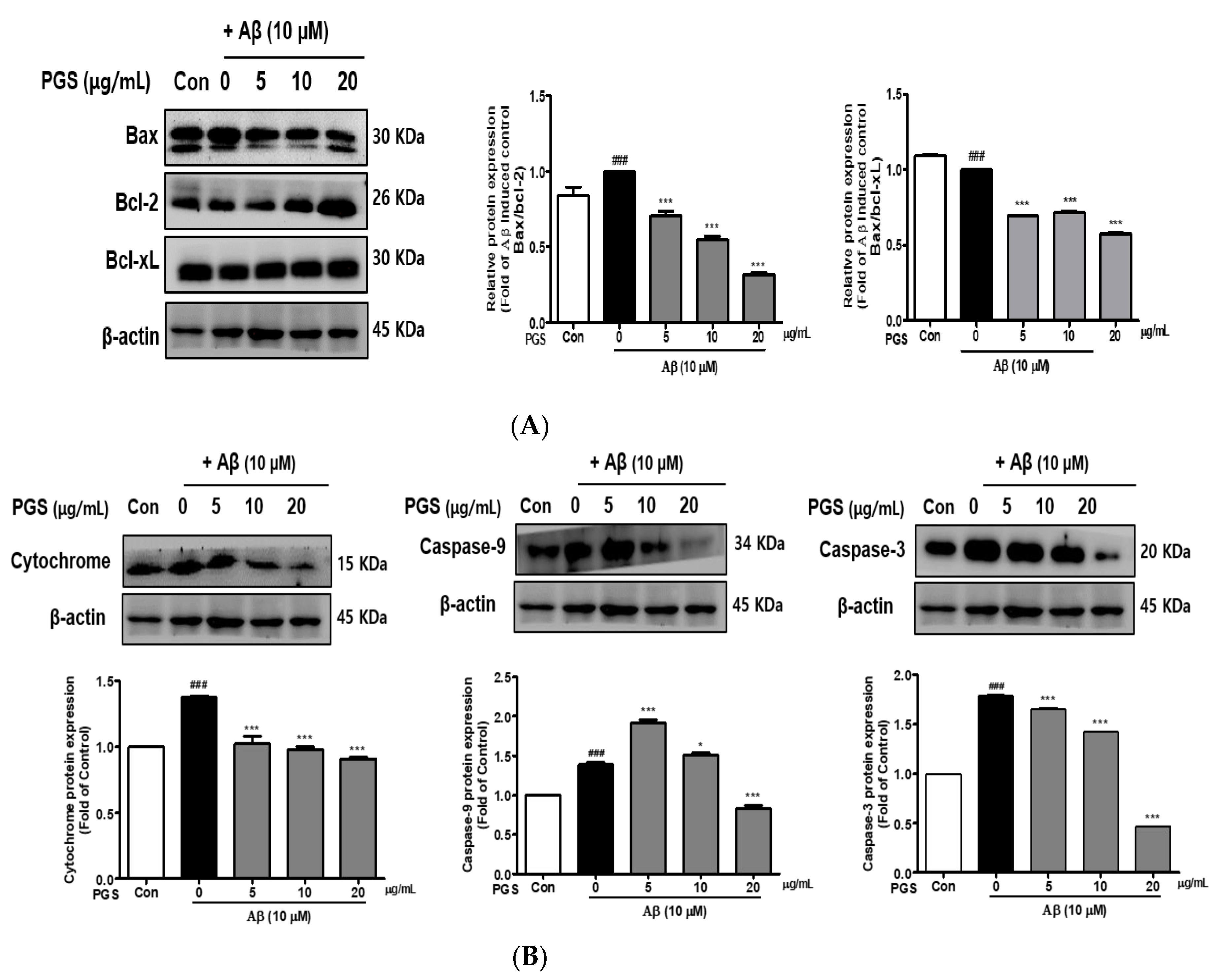
3.6. Effect of PGS on Apoptotic Protein Expression in Aβ-Induced HT22 Cells
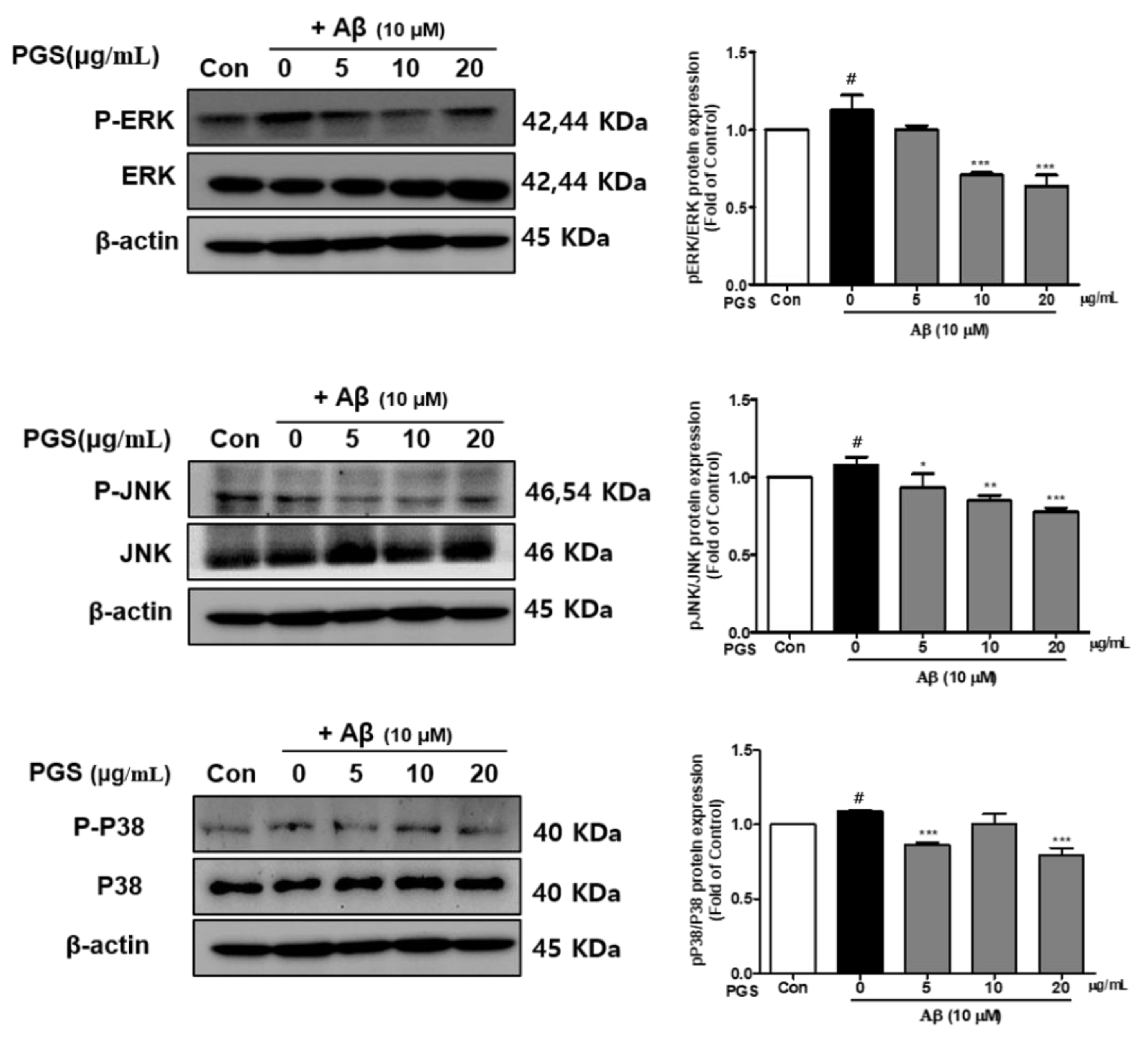
3.7. Effect of PGS on the MAPK Signaling Pathway in Aβ-Induced HT22 Cells
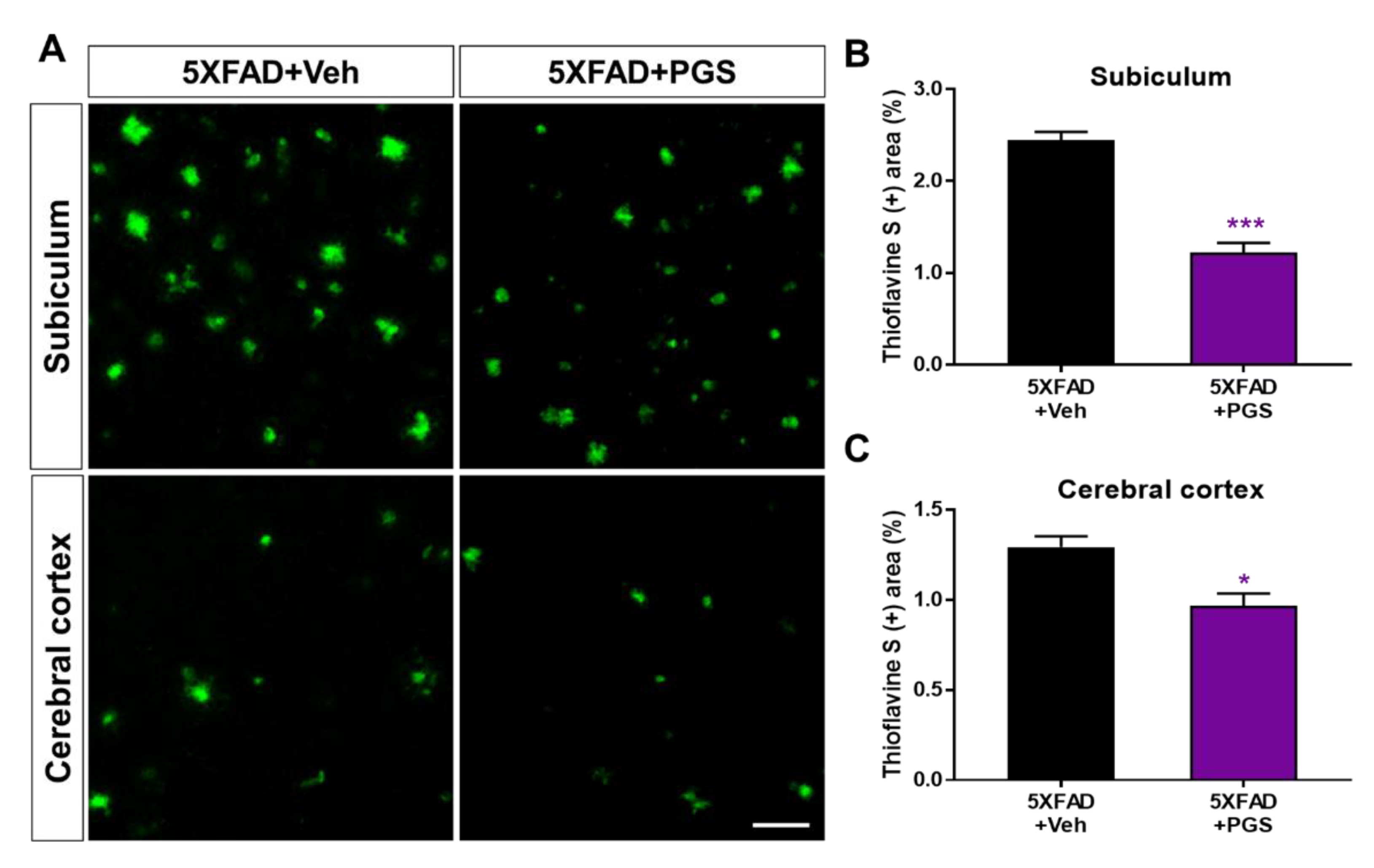
3.8. PGS Inhibits the Accumulation of Aβ in the Brain of 5XFAD Mice
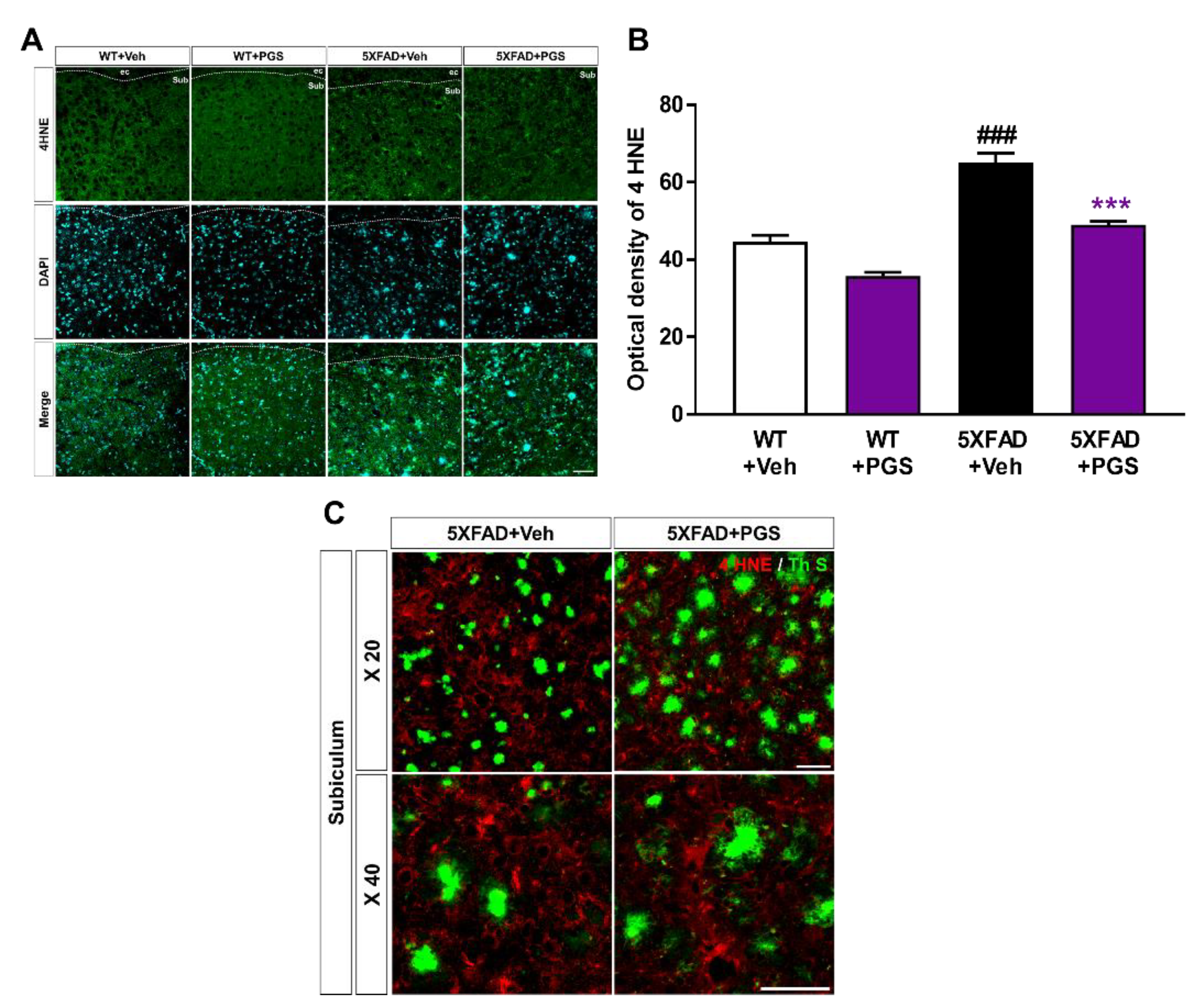
3.9. PGS Alleviates Oxidative Damage in the Brain of Aβ-Overexpressing Transgenic Mice
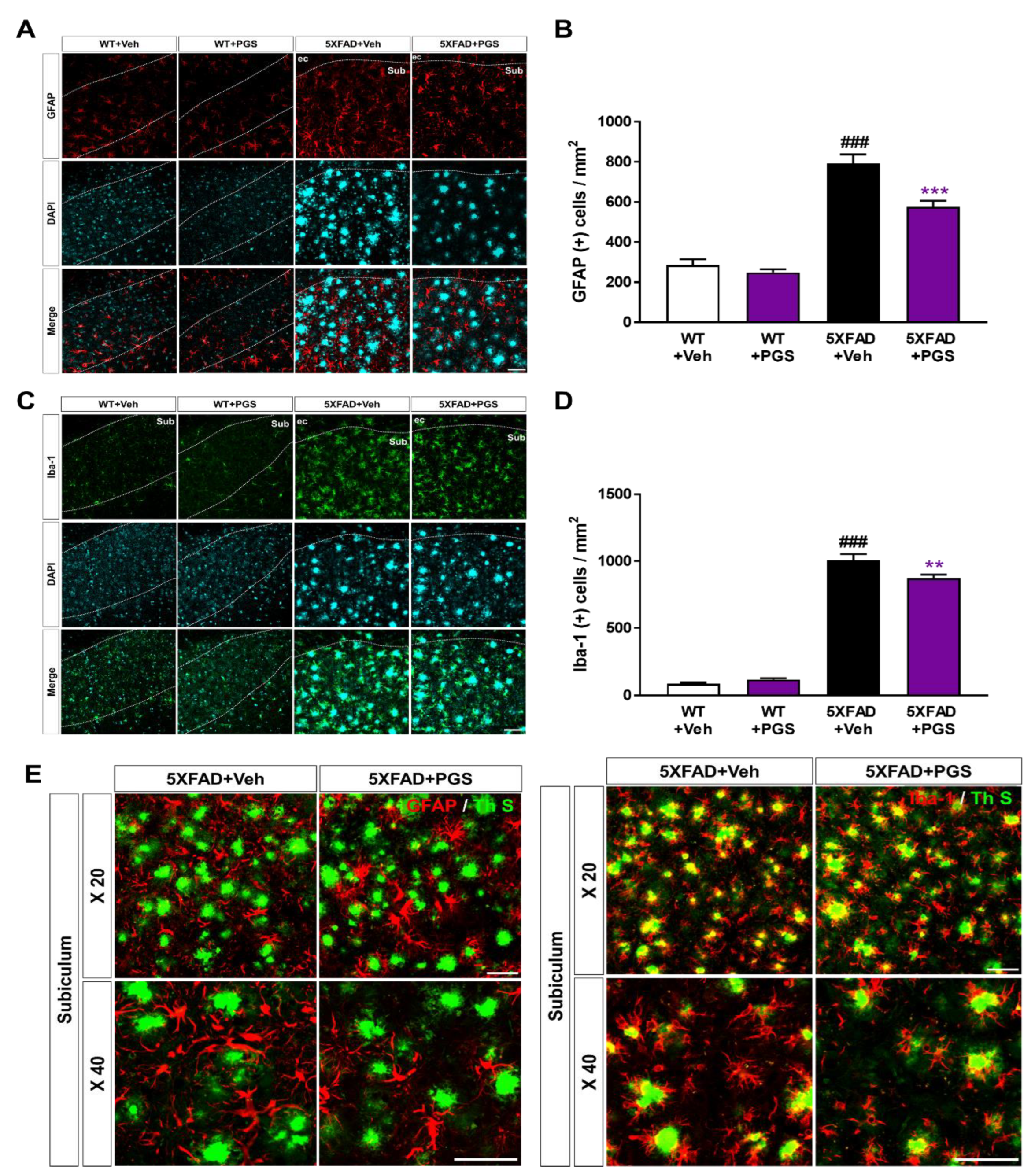
3.10. PGS Decreases Neuroinflammation in the Subiculum of 5XFAD Mice
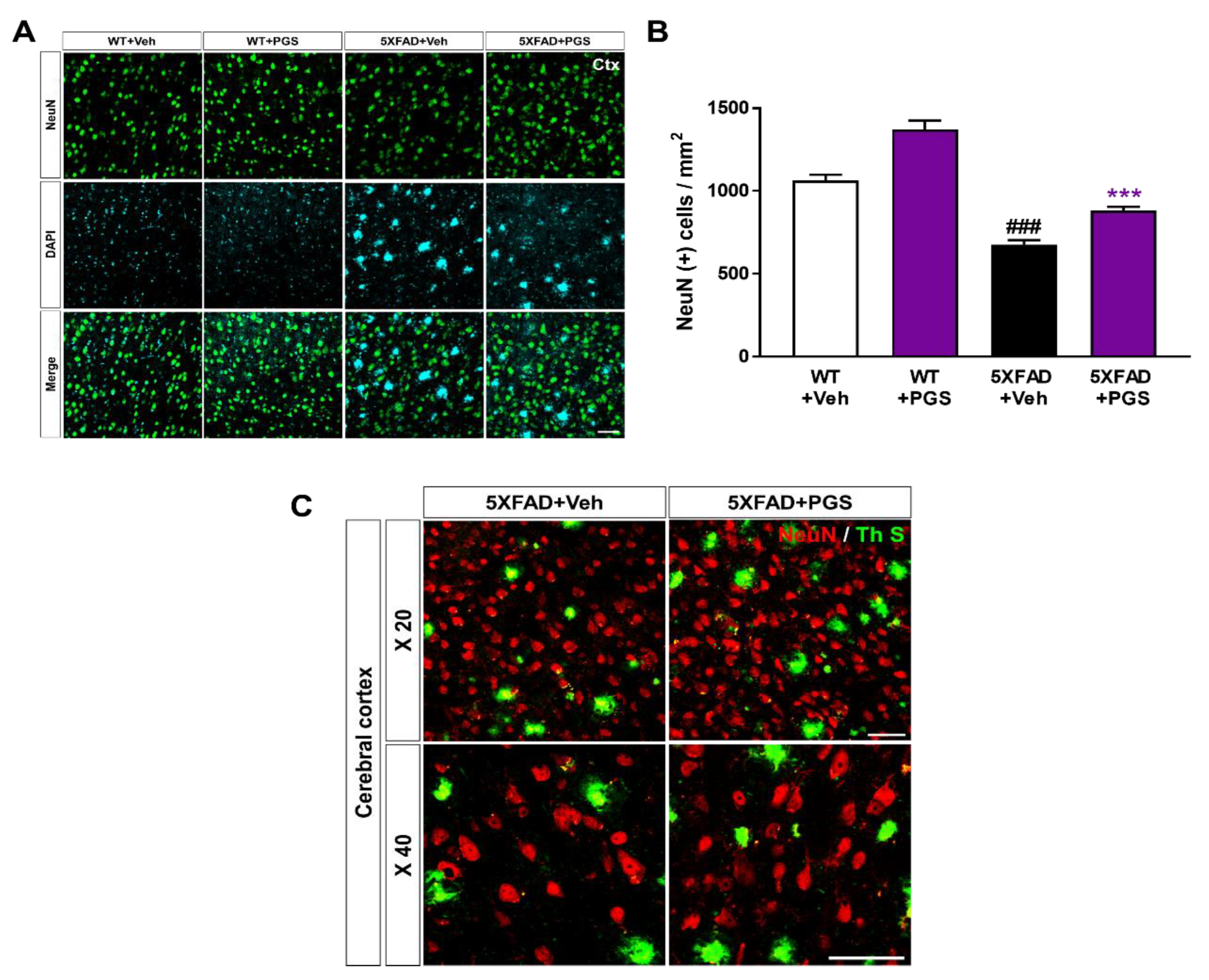
3.11. PGS Ameliorates Neurodegeneration in the Cerebral Cortex of an AD Animal Model
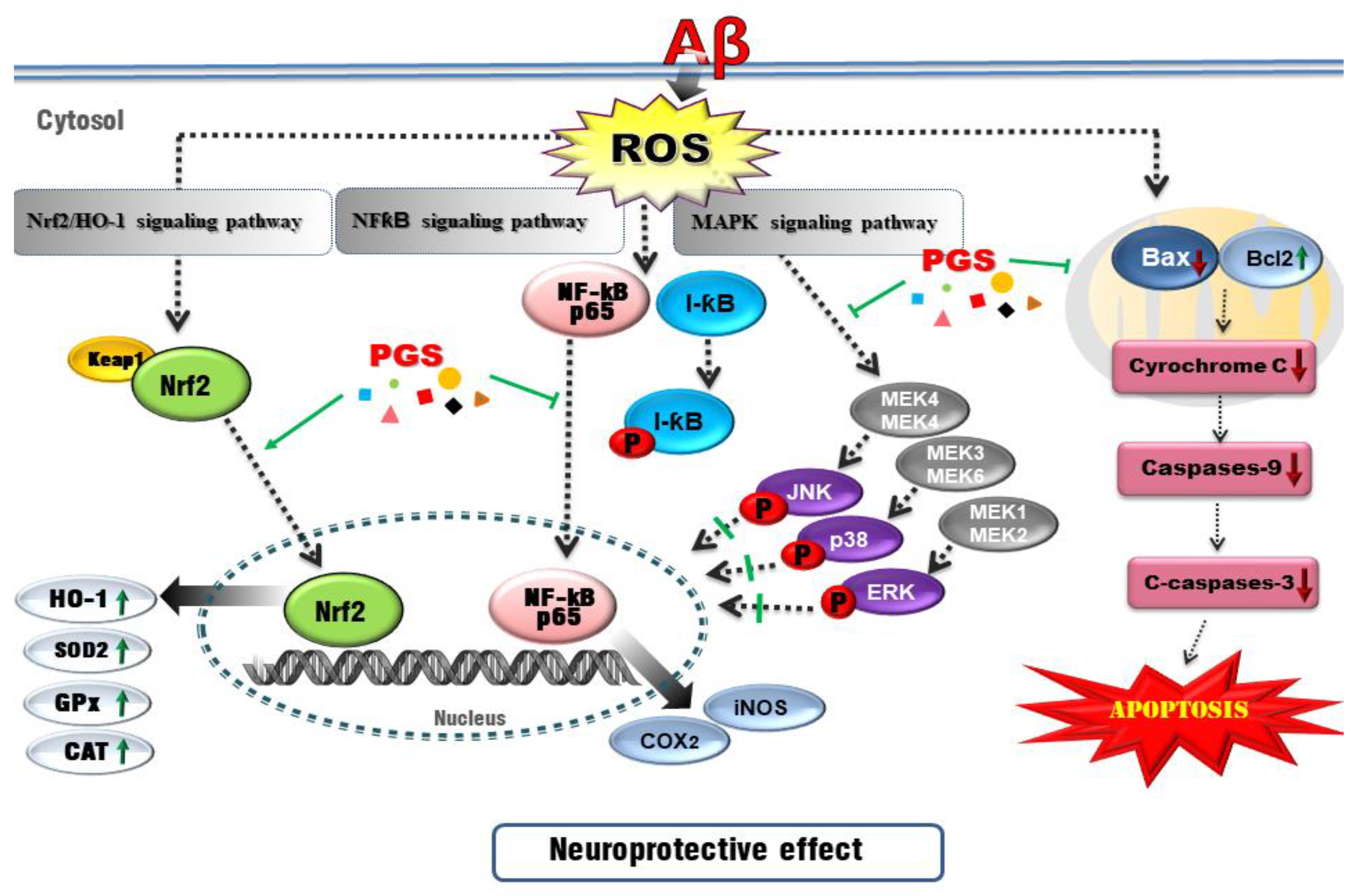
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Aβ | amyloid beta |
| AD | Alzheimer’s disease |
| CAT | catalase |
| COX-2 | cyclooxygenase 2 |
| GPx | glutathione peroxidase |
| HO-1 | heme oxygenase-1 |
| HT22 | hippocampal neuronal cells |
| IKBα | NF-κB inhibitor |
| Keap1 | Kelch-like ECH related protein 1 |
| MAPK | mitogen-activated protein kinase |
| NF- κB | nuclear factor kappa B |
| Nrf2 | nuclear factor erythroid 2 related factor 2 |
| ROS | reactive oxygen species |
| SOD | superoxide dismutase |
References
- Kriska, J.; Hermanova, Z.; Knotek, T.; Tureckova, J.; Anderova, M. On the Common Journey of Neural Cells through Ischemic Brain Injury and Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 9689. [Google Scholar] [CrossRef]
- Dong, Y.-T.; Cao, K.; Tan, L.-C.; Wang, X.-L.; Qi, X.-L.; Xiao, Y.; Guan, Z.-Z. Stimulation of SIRT1 attenuates the level of oxidative stress in the brains of APP/PS1 double transgenic mice and in primary neurons exposed to oligomers of the amyloid-β peptide. J. Alzheimer Dis. 2018, 63, 283–301. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, C.M.; Solá, S.; Silva, R.; Brites, D. Bilirubin and amyloid-β peptide induce cytochrome c release through mitochondrial membrane permeabilization. Mol. Med. 2000, 6, 936–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varadarajan, S.; Kanski, J.; Aksenova, M.; Lauderback, C.; Butterfield, D.A. Different mechanisms of oxidative stress and neurotoxicity for Alzheimer‘s Aβ (1−42) and Aβ (25−35). J. Am. Chem. Soc. 2001, 123, 5625–5631. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.S.; Al Mamun, A.; Kabir, M.T.; Ashraf, G.M.; Bin-Jumah, M.N.; Abdel-Daim, M.M. Multi-target drug candidates for multifactorial Alzheimer’s disease: AChE and NMDAR as molecular targets. Mol. Neurobiol. 2021, 58, 281–303. [Google Scholar] [CrossRef]
- Boridy, S.; Takahashi, H.; Akiyoshi, K.; Maysinger, D. The binding of pullulan modified cholesteryl nanogels to Aβ oligomers and their suppression of cytotoxicity. Biomaterials 2009, 30, 5583–5591. [Google Scholar] [CrossRef] [PubMed]
- Blasko, I.; Stampfer-Kountchev, M.; Robatscher, P.; Veerhuis, R.; Eikelenboom, P.; Grubeck-Loebenstein, B. How chronic inflammation can affect the brain and support the development of Alzheimer’s disease in old age: The role of microglia and astrocytes. Aging Cell 2004, 3, 169–176. [Google Scholar] [CrossRef]
- Lee, A.Y.; Lee, M.H.; Lee, S.; Cho, E.J. Neuroprotective effect of alpha-linolenic acid against Aβ-mediated inflammatory responses in C6 glial cell. J. Agric. Food Chem. 2018, 66, 4853–4861. [Google Scholar] [CrossRef]
- Hwang, S.; Lim, J.W.; Kim, H. Inhibitory effect of lycopene on amyloid-β-induced apoptosis in neuronal cells. Nutrients 2017, 9, 883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carter, R.S.; Geyer, B.C.; Xie, M.; Acevedo-Suárez, C.A.; Ballard, D.W. Persistent activation of NF-κB by the tax transforming protein involves chronic phosphorylation of IκB kinase subunits IKKβ and IKKγ. J. Biol. Chem. 2001, 276, 24445–24448. [Google Scholar] [CrossRef] [Green Version]
- Haake, A.; Nguyen, K.; Friedman, L.; Chakkamparambil, B.; Grossberg, G.T. An update on the utility and safety of cholinesterase inhibitors for the treatment of Alzheimer’s disease. Expert Opin. Drug Saf. 2020, 19, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Foyet, H.S.; Abaïssou, H.H.N.; Wado, E.; Acha, E.A.; Alin, C. Emilia coccinae (SIMS) G extract improves memory impairment, cholinergic dysfunction, and oxidative stress damage in scopolamine-treated rats. BMC Complement. Altern. Med. 2015, 15, 333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, S.; Sangma, T.; Shukla, S.K.; Mediratta, P.K. Effect of Cinnamomum zeylanicum extract on scopolamine-induced cognitive impairment and oxidative stress in rats. Nutr. Neurosci. 2015, 18, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Maheshwari, K.; Singh, V. Protective effects of Punica granatum seeds extract against aging and scopolamine induced cognitive impairments in mice. Afr. J. Tradit. Complement. Altern. Med. 2009, 6, 49–56. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.-H.; Jung, B.-Y.; Jung, S.-K.; Lee, C.-H.; Lee, H.-S.; Kim, B.-H.; Kim, S.-K. Evaluation of antioxidant activity of Platycodon grandiflorum. Environ. Health Toxicol. 2010, 25, 85–94. [Google Scholar]
- Zheng, J.; He, J.; Ji, B.; Li, Y.; Zhang, X. Antihyperglycemic effects of Platycodon grandiflorum (Jacq.) A. DC. extract on streptozotocin-induced diabetic mice. Plant Foods Hum. Nutr. 2007, 62, 7–11. [Google Scholar] [CrossRef]
- Hwang, K.-A.; Hwang, Y.-J.; Im, P.R.; Hwang, H.-J.; Song, J.; Kim, Y.-J. Platycodon grandiflorum extract reduces high-fat diet-induced obesity through regulation of adipogenesis and lipogenesis pathways in mice. J. Med. Food 2019, 22, 993–999. [Google Scholar] [CrossRef]
- Kim, K.-S.; Ezaki, O.; Ikemoto, S.; Itakura, H. Effects of Platycodon grandiflorum feeding on serum and liver lipid concentrations in rats with diet-induced hyperlipidemia. J. Nutr. Sci. Vitaminol. 1995, 41, 485–491. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Harding, S.; Marinangeli, C.; Kim, Y.; Jones, P. Hypocholesterolemic and anti-obesity effects of saponins from Platycodon grandiflorum in hamsters fed atherogenic diets. J. Food Sci. 2008, 73, H195–H200. [Google Scholar] [CrossRef]
- Hwang, S.Y.; Choi, H.M.; Lim, S.-Y. Total phenolics of dried Platycodon grandiflorum and its effect on growth of human cancer cell lines. Korean J. Food Sci. Technol. 2013, 45, 84–89. [Google Scholar] [CrossRef]
- Lee, J.-Y.; Hwang, W.-I.; Lim, S.-T. Antioxidant and anticancer activities of organic extracts from Platycodon grandiflorum A. De Candolle roots. J. Ethnopharmacol. 2004, 93, 409–415. [Google Scholar] [CrossRef]
- Fu, X.-J.; Liu, H.-B.; Wang, P.; Guan, H.-S. A study on the antioxidant activity and tissues selective inhibition of lipid peroxidation by saponins from the roots of Platycodon grandiflorum. Am. J. Chin. Med. 2009, 37, 967–975. [Google Scholar] [CrossRef]
- Konish, Y.; Denda, A.; Inui, S.; Takahashi, S.; Ueda, N.; Namiki, M. Production of pancreatic acinar cell carcinoma by combined administration of 4-hydroxyaminoquinoline 1-oxide and azaserine in partial pancreatectomized rats. Cancer Lett. 1978, 4, 229–234. [Google Scholar] [CrossRef]
- Liu, Y.-Y.; Sun, W.-H.; Li, B.-Z.; Shang, N.; Wang, Y.; Lv, W.-Q.; Li, D.; Wang, L.-J. Value-added application of Platycodon grandiflorus (Jacq.) A. DC. roots (PGR) by ultrasound-assisted extraction (UAE) process to improve physicochemical quality, structural characteristics and functional properties. Food Chem. 2021, 363, 130354. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Xue, J.; Wu, J.; Yoo, D.; Lee, S.; Kim, Y.; Uddin, M.; Park, S. Variation of triterpenoid saponin content in Platycodon grandiflorum (Jacq.) ADC. Asian J. Chem. 2012, 24, 1268–1270. [Google Scholar]
- Li, W.; Zhang, W.; Xiang, L.; Wang, Z.; Zheng, Y.-N.; Wang, Y.-P.; Zhang, J.; Chen, L. Platycoside N: A new oleanane-type triterpenoid saponin from the roots of Platycodon grandiflorum. Molecules 2010, 15, 8702–8708. [Google Scholar] [CrossRef] [PubMed]
- Güçlü-Üstündağ, Ö.; Mazza, G. Saponins: Properties, applications and processing. Crit. Rev. Food Sci. Nutr. 2007, 47, 231–258. [Google Scholar] [CrossRef]
- Son, I.H.; Park, Y.H.; Lee, S.I.; Yang, H.D.; Moon, H.-I. Neuroprotective activity of triterpenoid saponins from Platycodi radix against glutamate-induced toxicity in primary cultured rat cortical cells. Molecules 2007, 12, 1147–1152. [Google Scholar] [CrossRef] [Green Version]
- Nam, Y.; Shin, S.J.; Park, Y.H.; Kim, M.-J.; Jeon, S.G.; Lee, H.; Choi, Y.; Kim, T.-J.; Shin, S.M.; Kim, J.-J. Platycodon grandiflorum Root Protects against Aβ-Induced Cognitive Dysfunction and Pathology in Female Models of Alzheimer’s Disease. Antioxidants 2021, 10, 207. [Google Scholar] [CrossRef]
- Kim, J.-I.; Jeon, S.G.; Kim, K.A.; Kim, J.-J.; Song, E.J.; Jeon, Y.; Kim, E.; Lee, K.B.; Kwak, J.H.; Moon, M. Platycodon grandiflorus root extract improves learning and memory by enhancing synaptogenesis in mice hippocampus. Nutrients 2017, 9, 794. [Google Scholar] [CrossRef] [Green Version]
- Sakthivel, K.; Guruvayoorappan, C. Amentoflavone inhibits iNOS, COX-2 expression and modulates cytokine profile, NF-κB signal transduction pathways in rats with ulcerative colitis. Int. Immunopharmacol. 2013, 17, 907–916. [Google Scholar] [CrossRef] [PubMed]
- Shao, J.; Li, Y.; Wang, Z.; Xiao, M.; Yin, P.; Lu, Y.; Qian, X.; Xu, Y.; Liu, J. 7b, a novel naphthalimide derivative, exhibited anti-inflammatory effects via targeted-inhibiting TAK1 following down-regulation of ERK1/2-and p38 MAPK-mediated activation of NF-κB in LPS-stimulated RAW264. 7 macrophages. Int. Immunopharmacol. 2013, 17, 216–228. [Google Scholar] [CrossRef] [PubMed]
- Bloom, G.S. Amyloid-β and tau: The trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 2014, 71, 505–508. [Google Scholar] [CrossRef] [Green Version]
- Christen, Y. Oxidative stress and Alzheimer disease. Am. J. Clin. Nutr. 2000, 71, 621S–629S. [Google Scholar] [CrossRef] [PubMed]
- Gella, A.; Durany, N. Oxidative stress in Alzheimer disease. Cell Adhes. Migr. 2009, 3, 88–93. [Google Scholar] [CrossRef] [Green Version]
- Bhatia, V.; Sharma, S. Role of mitochondrial dysfunction, oxidative stress and autophagy in progression of Alzheimer’s disease. J. Neurol. Sci. 2020, 421, 117253. [Google Scholar] [CrossRef]
- Cai, Z.; Hussain, M.D.; Yan, L.-J. Microglia, neuroinflammation, and beta-amyloid protein in Alzheimer’s disease. Int. J. Neurosci. 2014, 124, 307–321. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef]
- Verdile, G.; Keane, K.N.; Cruzat, V.F.; Medic, S.; Sabale, M.; Rowles, J.; Wijesekara, N.; Martins, R.N.; Fraser, P.E.; Newsholme, P. Inflammation and oxidative stress: The molecular connectivity between insulin resistance, obesity, and Alzheimer’s disease. Mediat. Inflamm. 2015, 2015, 105828. [Google Scholar] [CrossRef] [Green Version]
- Saeed, K.; Shah, S.A.; Ullah, R.; Alam, S.I.; Park, J.S.; Saleem, S.; Jo, M.H.; Kim, M.W.; Hahm, J.R.; Kim, M.O. Quinovic Acid Impedes Cholesterol Dyshomeostasis, Oxidative Stress, and Neurodegeneration in an Amyloid-β-Induced Mouse Model. Oxidative Med. Cell. Longev. 2020, 2020, 9523758. [Google Scholar] [CrossRef]
- Zhang, L.; Yu, H.; Zhao, X.; Lin, X.; Tan, C.; Cao, G.; Wang, Z. Neuroprotective effects of salidroside against beta-amyloid-induced oxidative stress in SH-SY5Y human neuroblastoma cells. Neurochem. Int. 2010, 57, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Yin, J.; Hong, J.; Li, G.; Zhang, B.; Liu, G.; Wan, Q.; Chen, L. Neuronal seipin knockout facilitates Aβ-induced neuroinflammation and neurotoxicity via reduction of PPARγ in hippocampus of mouse. J. Neuroinflamm. 2016, 13, 145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguirre-Rueda, D.; Guerra-Ojeda, S.; Aldasoro, M.; Iradi, A.; Obrador, E.; Ortega, A.; Mauricio, M.D.; Vila, J.M.; Valles, S.L. Astrocytes protect neurons from Aβ1-42 peptide-induced neurotoxicity increasing TFAM and PGC-1 and decreasing PPAR-γ and SIRT-1. Int. J. Med. Sci. 2015, 12, 48. [Google Scholar] [CrossRef] [Green Version]
- Kong, Y.; Kong, L.-P.; Luo, T.; Li, G.-W.; Jiang, W.; Li, S.; Zhou, Y.; Wang, H.-Q. The protective effects of crocetin on aβ1-42-induced toxicity in Ht22 cells. CNS Neurol. Disord. Drug Targets 2014, 13, 1627–1632. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-J.; Shin, S.-R.; Yoon, K.-Y. Physicochemical properties of black doraji (Platycodon grandiflorum). Korean J. Food Sci. Technol. 2013, 45, 422–427. [Google Scholar] [CrossRef]
- Lee, K.S.; Seong, B.J.; Kim, S.I.; Jee, M.G.; Park, S.B.; Park, M.H.; Park, S.Y.; Kim, H.H. Changes in platycoside components and antimicrobial activities of bronchus disease-inducing bacteria of fermented Platycodon grandiflorum root by lactic acid bacteria. J. Korean Soc. Food Sci. Nutr. 2016, 45, 1017–1025. [Google Scholar] [CrossRef]
- Noh, J.-R.; Kim, Y.-H.; Gang, G.-T.; Hwang, J.-H.; Kim, S.-K.; Ryu, S.-Y.; Kim, Y.-S.; Lee, H.-S.; Lee, C.-H. Hepatoprotective effect of Platycodon grandiflorum against chronic ethanol-induced oxidative stress in C57BL/6 mice. Ann. Nutr. Metab. 2011, 58, 224–231. [Google Scholar] [CrossRef]
- Chung, J.W.; Noh, E.J.; Zhao, H.L.; Sim, J.-S.; Ha, Y.W.; Shin, E.M.; Lee, E.B.; Cheong, C.S.; Kim, Y.S. Anti-inflammatory activity of prosapogenin methyl ester of platycodin D via nuclear factor-kappaB pathway inhibition. Biol. Pharm. Bull. 2008, 31, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Guo, R.; Meng, Q.; Wang, B.; Li, F. Anti-inflammatory effects of Platycodin D on dextran sulfate sodium (DSS) induced colitis and E. coli Lipopolysaccharide (LPS) induced inflammation. Int. Immunopharmacol. 2021, 94, 107474. [Google Scholar] [CrossRef]
- Wang, G.; Guo, H.; Wang, X. Platycodin D protects cortical neurons against oxygen-glucose deprivation/reperfusion in neonatal hypoxic-ischemic encephalopathy. J. Cell. Biochem. 2019, 120, 14028–14034. [Google Scholar] [CrossRef]
- Choi, J.H.; Yoo, K.-Y.; Park, O.K.; Lee, C.H.; Won, M.-H.; Hwang, I.K.; Ryu, S.Y.; Kim, Y.S.; Yi, J.-S.; Bae, Y.-S. Platycodin D and 2″-o-acetyl-polygalacin D2 isolated from Platycodon grandiflorum protect ischemia/reperfusion injury in the gerbil hippocampus. Brain Res. 2009, 1279, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Xin, Z.; Liu, B.; Wang, J.; Wang, J.; Zhang, X.; Wang, Y.; Li, F. Platycodin D inhibits inflammatory response in LPS-stimulated primary rat microglia cells through activating LXRα–ABCA1 signaling pathway. Front. Immunol. 2018, 8, 1929. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, M.; Li, L.; Iwamoto, N.; Nakajima-Takagi, Y.; Kaneko, H.; Nakayama, Y.; Eguchi, M.; Wada, Y.; Kumagai, Y.; Yamamoto, M. The antioxidant defense system Keap1-Nrf2 comprises a multiple sensing mechanism for responding to a wide range of chemical compounds. Mol. Cell. Biol. 2009, 29, 493–502. [Google Scholar] [CrossRef] [Green Version]
- Bresciani, A.; Missineo, A.; Gallo, M.; Cerretani, M.; Fezzardi, P.; Tomei, L.; Cicero, D.O.; Altamura, S.; Santoprete, A.; Ingenito, R. Nuclear factor (erythroid-derived 2)-like 2 (NRF2) drug discovery: Biochemical toolbox to develop NRF2 activators by reversible binding of Kelch-like ECH-associated protein 1 (KEAP1). Arch. Biochem. Biophys. 2017, 631, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Song, J.H.; Lee, H.-J.; Kang, K.S. Procyanidin C1 activates the Nrf2/HO-1 signaling pathway to prevent glutamate-induced apoptotic HT22 cell death. Int. J. Mol. Sci. 2019, 20, 142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouyang, Y.; Chen, Z.; Tan, M.; Liu, A.; Chen, M.; Liu, J.; Pi, R.; Fang, J. Carvedilol, a third-generation β-blocker prevents oxidative stress-induced neuronal death and activates Nrf2/ARE pathway in HT22 cells. Biochem. Biophys. Res. Commun. 2013, 441, 917–922. [Google Scholar] [CrossRef]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Chao, D.T.; Korsmeyer, S.J. BCL-2 family: Regulators of cell death. Annu. Rev. Immunol. 1998, 16, 395–419. [Google Scholar] [CrossRef]
- Manon, S.; Chaudhuri, B.; Guérin, M. Release of cytochrome c and decrease of cytochrome c oxidase in Bax-expressing yeast cells, and prevention of these effects by coexpression of Bcl-xL. FEBS Lett. 1997, 415, 29–32. [Google Scholar] [CrossRef] [Green Version]
- Seo, E.-J.; Fischer, N.; Efferth, T. Phytochemicals as inhibitors of NF-κB for treatment of Alzheimer’s disease. Pharmacol. Res. 2018, 129, 262–273. [Google Scholar] [CrossRef]
- Cachofeiro, V.; Goicochea, M.; De Vinuesa, S.G.; Oubiña, P.; Lahera, V.; Luño, J. Oxidative stress and inflammation, a link between chronic kidney disease and cardiovascular disease: New strategies to prevent cardiovascular risk in chronic kidney disease. Kidney Int. 2008, 74, S4–S9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ju Hwang, C.; Choi, D.-Y.; Park, M.H.; Hong, J.T. NF-κB as a key mediator of brain inflammation in Alzheimer’s disease. CNS Neurol. Disord. Drug Targets 2019, 18, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Steer, S.A.; Corbett, J.A. The role and regulation of COX-2 during viral infection. Viral Immunol. 2003, 16, 447–460. [Google Scholar] [CrossRef]
- Lee, S.; Shin, S.; Kim, H.; Han, S.; Kim, K.; Kwon, J.; Kwak, J.-H.; Lee, C.-K.; Ha, N.-J.; Yim, D. Anti-inflammatory function of arctiin by inhibiting COX-2 expression via NF-κB pathways. J. Inflamm. 2011, 8, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Hou, J.; Yan, X.; Leng, J.; Li, R.; Zhang, J.; Xing, J.; Chen, C.; Wang, Z.; Li, W. Platycodon grandiflorum saponins ameliorate cisplatin-induced acute nephrotoxicity through the NF-κB-mediated inflammation and PI3K/Akt/apoptosis signaling pathways. Nutrients 2018, 10, 1328. [Google Scholar] [CrossRef] [Green Version]
- Yang, R.; Liu, S.; Zhou, J.; Bu, S.; Zhang, J. Andrographolide attenuates microglia-mediated Aβ neurotoxicity partially through inhibiting NF-κB and JNK MAPK signaling pathway. Immunopharmacol. Immunotoxicol. 2017, 39, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.D.; Chen, X.C.; Zhu, Y.G.; Chen, L.M.; Zhang, J.; Huang, T.W.; Ye, Q.Y.; Huang, H.P. Tripchlorolide protects neuronal cells from microglia-mediated β-amyloid neurotoxicity through inhibiting NF-κB and JNK signaling. Glia 2009, 57, 1227–1238. [Google Scholar] [CrossRef] [PubMed]
- Shoji, M.; Iwakami, N.; Takeuchi, S.; Waragai, M.; Suzuki, M.; Kanazawa, I.; Lippa, C.F.; Ono, S.; Okazawa, H. JNK activation is associated with intracellular β-amyloid accumulation. Mol. Brain Res. 2000, 85, 221–233. [Google Scholar] [CrossRef]
- Jeong, S. Molecular and cellular basis of neurodegeneration in Alzheimer’s disease. Mol. Cells 2017, 40, 613. [Google Scholar] [PubMed] [Green Version]
- Nordengen, K.; Kirsebom, B.-E.; Henjum, K.; Selnes, P.; Gísladóttir, B.; Wettergreen, M.; Torsetnes, S.B.; Grøntvedt, G.R.; Aarsland, D.; Nilsson, L.N. Glial activation and inflammation along the Alzheimer’s disease continuum. J. Neuroinflamm. 2019, 16, 46. [Google Scholar] [CrossRef]














Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ji, Y.-J.; Kim, S.; Kim, J.-J.; Jang, G.Y.; Moon, M.; Kim, H.D. Crude Saponin from Platycodon grandiflorum Attenuates Aβ-Induced Neurotoxicity via Antioxidant, Anti-Inflammatory and Anti-Apoptotic Signaling Pathways. Antioxidants 2021, 10, 1968. https://doi.org/10.3390/antiox10121968
Ji Y-J, Kim S, Kim J-J, Jang GY, Moon M, Kim HD. Crude Saponin from Platycodon grandiflorum Attenuates Aβ-Induced Neurotoxicity via Antioxidant, Anti-Inflammatory and Anti-Apoptotic Signaling Pathways. Antioxidants. 2021; 10(12):1968. https://doi.org/10.3390/antiox10121968
Chicago/Turabian StyleJi, Yun-Jeong, Sujin Kim, Jwa-Jin Kim, Gwi Yeong Jang, Minho Moon, and Hyung Don Kim. 2021. "Crude Saponin from Platycodon grandiflorum Attenuates Aβ-Induced Neurotoxicity via Antioxidant, Anti-Inflammatory and Anti-Apoptotic Signaling Pathways" Antioxidants 10, no. 12: 1968. https://doi.org/10.3390/antiox10121968
APA StyleJi, Y. -J., Kim, S., Kim, J. -J., Jang, G. Y., Moon, M., & Kim, H. D. (2021). Crude Saponin from Platycodon grandiflorum Attenuates Aβ-Induced Neurotoxicity via Antioxidant, Anti-Inflammatory and Anti-Apoptotic Signaling Pathways. Antioxidants, 10(12), 1968. https://doi.org/10.3390/antiox10121968