
Role of Oxidative Stress in the Pathogenesis of Non-Alcoholic Fatty Liver Disease: Implications for Prevention and Therapy



Abstract
:1. Introduction
2. Antioxidant Balance in the Liver in Non-Alcoholic Fatty Liver Disease
2.1. Oxidative Stress Mechanisms in Non-Alcoholic Fatty Liver Disease
2.2. Oxidative Stress Biomarkers in NAFLD Patients
3. Lipotoxicity and Oxidative Stress
3.1. Lipid Metabolism and Oxidative Stress
3.2. Lipotoxicity in Non-Alcoholic Fatty Liver Disease
3.3. Cellular Dysfunction Triggered by Fatty Acids in the Liver
3.3.1. Mitochondrial Dysfunction
3.3.2. ER Stress
4. Antioxidants as a Therapy in NAFLD
4.1. Classical Antioxidants: Vitamin C and Vitamin E
4.2. Coffee Components: Caffeine and Coffee Polyphenols
4.3. Metformin
4.4. Hesperetin
5. Conclusions and Future Perspectives
Funding
Conflicts of Interest
Abbreviations
| OxS | Oxidative stress |
| ROS | Reactive oxygen species |
| NAFLD | Non-alcoholic liver disease |
| NASH | Non-alcoholic steatohepatitis |
| HFD | High fat diet |
| ER | Endoplasmic Reticulum |
| OxPhos | Oxidative phosphorylation |
| ARE | Antioxidant Response Element |
| TG | Triglycerides |
| FFA | Free fatty acids |
| SFAs | Saturated fatty acids |
| DAGs | Diacylglycerols |
| NEFAs | Non-esterified fatty acids |
| MUFAs | Monounsaturated fatty acids |
| NPCs | Non-parenchymal cells |
| LSECs | Liver sinusoidal endothelial cells |
| HSCs | Hepatic stellate cells |
| MRC | Mitochondrial respiratory chain |
References
- Li, S.; Tan, H.Y.; Wang, N.; Zhang, Z.J.; Lao, L.; Wong, C.W.; Feng, Y. The role of oxidative stress and antioxidants in liver diseases. Int. J. Mol. Sci. 2015, 16, 26087–26124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masarone, M.; Rosato, V.; Dallio, M.; Gravina, A.G.; Aglitti, A.; Loguercio, C.; Federico, A.; Persico, M. Role of Oxidative Stress in Pathophysiology of Nonalcoholic Fatty Liver Disease. Oxid Med. Cell Longev. 2018, 2018, 9547613. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Golabi, P.; de Avila, L.; Paik, J.M.; Srishord, M.; Fukui, N.; Qiu, Y.; Burns, L.; Afendy, A.; Nader, F. The global epidemiology of NAFLD and NASH in patients with type 2 diabetes: A systematic review and meta-analysis. J. Hepatol. 2019, 71, 793–801. [Google Scholar] [CrossRef] [PubMed]
- Madan, K.; Bhardwaj, P.; Thareja, S.; Gupta, S.D.; Saraya, A. Oxidant stress and antioxidant status among patients with nonalcoholic fatty liver disease (NAFLD). J. Clin. Gastroenterol. 2006, 40, 930–935. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, V.O.; Grattagliano, I.; Portincasa, P.; Palasciano, G. Systemic oxidative alterations are associated with visceral adiposity and liver steatosis in patients with metabolic syndrome. J. Nutr. 2006, 136, 3022–3026. [Google Scholar] [CrossRef]
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative stress and antioxidant defense. World Allergy Organ. J. 2012, 5, 9–19. [Google Scholar] [CrossRef] [Green Version]
- Medina, J.; Moreno-Otero, R. Pathophysiological basis for antioxidant therapy in chronic liver disease. Drugs 2005, 65, 2445–2461. [Google Scholar] [CrossRef] [PubMed]
- Cichoz-Lach, H.; Michalak, A. Oxidative stress as a crucial factor in liver diseases. World J. Gastroenterol. 2014, 20, 8082–8091. [Google Scholar] [CrossRef]
- Ore, A.; Akinloye, O.A. Oxidative stress and antioxidant biomarkers in clinical and experimental models of non-alcoholic fatty liver disease. Medicina 2019, 55, 26. [Google Scholar] [CrossRef] [Green Version]
- Abe, Y.; Hines, I.N.; Zibari, G.; Pavlick, K.; Gray, L.; Kitagawa, Y.; Grisham, M.B. Mouse model of liver ischemia and reperfusion injury: Method for studying reactive oxygen and nitrogen metabolites in vivo. Free Radic. Biol. Med. 2009, 46, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Al-Asmari, A.K.; Khan, A.Q.; Al-Masri, N. Mitigation of 5-fluorouracil-induced liver damage in rats by Vitamin C via targeting redox-sensitive transcription factors. Hum. Exp. Toxicol. 2016, 35, 1203–1213. [Google Scholar] [CrossRef] [PubMed]
- Smolková, K.; Mikó, E.; Kovács, T.; Leguina-Ruzzi, A.; Sipos, A.; Bai, P. Nuclear factor erythroid 2-related factor 2 in regulating cancer metabolism. Antioxid. Redox Signal. 2020, 33, 966–997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef] [PubMed]
- Kwak, M.K.; Itoh, K.; Yamamoto, M.; Kensler, T.W. Enhanced Expression of the Transcription Factor Nrf2 by Cancer Chemopreventive Agents: Role of Antioxidant Response Element-Like Sequences in the nrf2 Promoter. Mol. Cell. Biol. 2002, 22, 2883–2892. [Google Scholar] [CrossRef] [Green Version]
- Rushworth, S.A.; Zaitseva, L.; Murray, M.Y.; Shah, N.M.; Bowles, K.M.; MacEwan, D.J. The high Nrf2 expression in human acute myeloid leukemia is driven by NF-κB and underlies its chemo-resistance. Blood 2012, 120, 5188–5198. [Google Scholar] [CrossRef] [Green Version]
- Galicia-Moreno, M.; Lucano-Landeros, S.; Monroy-Ramirez, H.C.; Silva-Gomez, J.; Gutierrez-Cuevas, J.; Santos, A.; Armendariz-Borunda, J. Roles of NRF2 in liver diseases: Molecular, pharmacological, and epigenetic aspects. Antioxidants 2020, 9, 980. [Google Scholar] [CrossRef]
- Holmström, K.M.; Baird, L.; Zhang, Y.; Hargreaves, I.; Chalasani, A.; Land, J.M.; Stanyer, L.; Yamamoto, M.; Dinkova-Kostova, A.T.; Abramov, A.Y. Nrf2 impacts cellular bionergetics by controlling substrate availability for mitochondrial respiration. Biol. Open 2013, 2, 761–770. [Google Scholar] [CrossRef] [Green Version]
- Ludtmann, M.H.; Angelova, P.R.; Zhang, Y.; Abramov, A.Y.; Dinkova-Kostova, A.T. Nrf2 affects the efficiency of mitochondrial fatty acid oxidation. Biochem. J. 2014, 457, 415–424. [Google Scholar] [CrossRef] [Green Version]
- Yates, M.S.; Tran, Q.T.; Dolan, P.M.; Osburn, W.O.; Shin, S.; McCulloch, C.C.; Silkworth, J.B.; Taguchi, K.; Yamamoto, M.; Williams, C.R.; et al. Genetic versus chemoprotective activation of Nrf2 signaling: Overlapping yet distinct gene expression profiles between Keap1 knockout and triterpenoid-treated mice. Carcinogenesis 2009, 30, 1024–1031. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, Y.; Aleksunes, L.M.; Yeager, R.L.; Gyamfi, M.A.; Esterly, N.; Guo, G.L.; Klaassen, C.D. NF-E2-related factor 2 inhibits lipid accumulation and oxidative stress in mice fed a high-fat diet. J. Pharm. Exp. Ther. 2008, 325, 655–664. [Google Scholar] [CrossRef]
- Zhang, Y.-K.J.; Wu, K.C.; Liu, J.; Klaassen, C.D. Nrf2 deficiency improves glucose tolerance in mice fed a high-fat diet. Toxicol. Appl. Pharm. 2012, 264, 305–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Tabbi-Anneni, I.; Gunda, V.; Wang, L. Transcription factor Nrf2 regulates SHP and lipogenic gene expression in hepatic lipid metabolism. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 299, G1211–G1221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chambel, S.S.; Santos-Gonçalves, A.; Duarte, T.L. The dual role of Nrf2 in nonalcoholic fatty liver disease: Regulation of antioxidant defenses and hepatic lipid metabolism. Biomed Res. Int. 2015, 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chowdhry, S.; Nazmy, M.H.; Meakin, P.J.; Dinkova-Kostova, A.T.; Walsh, S.V.; Tsujita, T.; Dillon, J.F.; Ashford, M.L.; Hayes, J.D. Loss of Nrf2 markedly exacerbates nonalcoholic steatohepatitis. Free Radic. Biol. Med. 2010, 48, 357–371. [Google Scholar] [CrossRef]
- Zhang, Y.K.J.; Yeager, R.L.; Tanaka, Y.; Klaassen, C.D. Enhanced expression of Nrf2 in mice attenuates the fatty liver produced by a methionine- and choline-deficient diet. Toxicol. Appl. Pharm. 2010, 245, 326–334. [Google Scholar] [CrossRef] [Green Version]
- Sun, B.; Karin, M. NF-κB signaling, liver disease and hepatoprotective agents. Oncogene 2008, 27, 6228–6244. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-Mediated Cellular Signaling. Oxid. Med. Cell. Longev. 2016, 2016. [Google Scholar] [CrossRef] [Green Version]
- Morgan, M.J.; Liu, Z.G. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [Green Version]
- Lingappan, K. NF-κB in Oxidative Stress. Curr. Opin. Toxicol. 2018, 7, 81–86. [Google Scholar] [CrossRef]
- Oliveira-Marques, V.; Marinho, H.S.; Cyrne, L.; Antunes, F. Role of hydrogen peroxide in NF-κB activation: From inducer to modulator. Antioxid. Redox Signal. 2009, 11, 2223–2243. [Google Scholar] [CrossRef]
- Liu, G.-H.; Qu, J.; Shen, X. NF-κB:p65 antagonizes Nrf2-ARE pathway by depriving CBP from Nrf2 and facilitating recruitment of HDAC3 to MafK. Biochim. Biophys. Acta BBA 2008, 1783, 713–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, H.; Wang, H.; Wang, X.; Zhu, L.; Mao, L. The Absence of Nrf2 Enhances NF-kB-Dependent Inflammation following Scratch Injury in Mouse Primary Cultured Astrocytes. Mediat. Inflamm. 2012, 2021, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Zhang, W.; Wang, J.; Yang, H.; Zhao, X.; Zhou, Q.; Wang, H.; Li, L.; Du, G. Activation of Nrf2 signaling by salvianolic acid C attenuates NF-κB mediated inflammatory response both in vivo and in vitro. Int. Immunopharmacol. 2018, 63, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.M.; Guo, Z.X.; Zhao, Y.L.; Wang, Q.J.; Gao, Y.S.; Yu, T.; Chen, Y.K.; Chen, X.M.; Wang, G.Q. L-carnitine regulated Nrf2/Keap1 activation in vitro and in vivo and protected oxidized fish oil-induced inflammation response by inhibiting the NF-κB signaling pathway in Rhynchocypris lagowski Dybowski. Fish Shellfish Immunol. 2019, 93, 1100–1110. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Dou, W.; Ni, Z.; Wen, Q.; Zhang, R.; Qin, M.; Wang, X.; Tang, H.; Cao, Y.; Wang, J.; et al. Deletion of Nrf2 leads to hepatic insulin resistance via the activation of NF-κB in mice fed a high-fat diet. Mol. Med. Rep. 2016, 14, 1323–1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Q.; Xia, Z.; Liong, E.C.; Tipoe, G.L. Chronic aerobic exercise improves insulin sensitivity and modulates Nrf2 and NF-κB/IκBα pathways in the skeletal muscle of rats fed with a high fat diet. Mol. Med. Rep. 2019, 20, 4963–4972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ou, Q.; Weng, Y.; Wang, S.; Zhao, Y.; Zhang, F.; Zhou, J.; Wu, X. Silybin Alleviates Hepatic Steatosis and Fibrosis in NASH Mice by Inhibiting Oxidative Stress and Involvement with the Nf-κB Pathway. Dig. Dis. Sci. 2018, 63, 3398–3408. [Google Scholar] [CrossRef]
- Hodges, J.K.; Sasaki, G.Y.; Bruno, R.S. Anti-inflammatory activities of green tea catechins along the gut–liver axis in nonalcoholic fatty liver disease: Lessons learned from preclinical and human studies. J. Nutr. Biochem. 2020, 85, 108478. [Google Scholar] [CrossRef]
- Damba, T.; Bourgonje, A.R.; Abdulle, A.E.; Pasch, A.; Sydor, S.; van den Berg, E.H.; Gansevoort, R.T.; Bakker, S.J.; Blokzijl, H.; Dullaart, R.P.; et al. Oxidative stress is associated with suspected non-alcoholic fatty liver disease and all-cause mortality in the general population. Liver Int. 2020, 40, 2148–2159. [Google Scholar] [CrossRef]
- Koruk, M.; Taysi, S.; Savas, M.C.; Yilmaz, O.; Akcay, F.; Karakok, M. Oxidative Stress Enzymatic Antioxidant Status in Patients with Nonalcoholic Steatohepatitis. Ann. Clin. Lab. Sci. 2004, 34, 57–62. [Google Scholar]
- Świderska, M.; Maciejczyk, M.; Zalewska, A.; Pogorzelska, J.; Flisiak, R.; Chabowski, A. Oxidative stress biomarkers in the serum and plasma of patients with non-alcoholic fatty liver disease (NAFLD). Can plasma AGE be a marker of NAFLD? Oxidative stress biomarkers in NAFLD patients. Free Radic. Res. 2019, 53, 841–850. [Google Scholar] [CrossRef] [PubMed]
- Coelho, J.M.; Cansanção, K.; de Mello Perez, R.; Leite, N.C.; Padilha, P.; Ramalho, A.; Peres, W. Association between serum and dietary antioxidant micronutrients and advanced liver fibrosis in non-alcoholic fatty liver disease: An observational study. PeerJ 2020, 8, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, A.; Geng, Y.; Sepulveda, R.; Solis, N.; Torres, J.; Arab, J.P.; Barrera, F.; Cabrera, D.; Moshage, H.; Arrese, M. Chemical hypoxia induces pro-inflammatory signals in fat-laden hepatocytes and contributes to cellular crosstalk with Kupffer cells through extracellular vesicles. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165753. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.M.; Ni, X.X.; Xu, Q.Y.; Wang, Q.; Li, X.Y.; Hua, J. Regulation of lipid-induced macrophage polarization through modulating peroxisome proliferator-activated receptor-gamma activity affects hepatic lipid metabolism via a Toll-like receptor 4/NF-κB signaling pathway. J. Gastroenterol. Hepatol. 2020, 35, 1998–2008. [Google Scholar] [CrossRef] [PubMed]
- Kazankov, K.; Jørgensen, S.M.D.; Thomsen, K.L.; Møller, H.J.; Vilstrup, H.; George, J.; Schuppan, D.; Grønbæk, H. The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 145–159. [Google Scholar] [CrossRef]
- Bessone, F.; Razori, M.V.; Roma, M.G. Molecular pathways of nonalcoholic fatty liver disease development and progression. Cell. Mol. Life Sci. 2019, 76, 99–128. [Google Scholar] [CrossRef]
- Mendez-Sanchez, N.; Cruz-Ramon, V.C.; Ramirez-Perez, O.L.; Hwang, J.P.; Barranco-Fragoso, B.; Cordova-Gallardo, J. New aspects of lipotoxicity in nonalcoholic steatohepatitis. Int. J. Mol. Sci. 2018, 19, 2034. [Google Scholar] [CrossRef] [Green Version]
- Palomer, X.; Pizarro-Delgado, J.; Barroso, E.; Vázquez-Carrera, M. Palmitic and Oleic Acid: The Yin and Yang of Fatty Acids in Type 2 Diabetes Mellitus. Trends Endocrinol. Metab. 2017, 29, 178–190. [Google Scholar] [CrossRef]
- Fucho, R.; Casals, Ń.; Serra, D.; Herrero, L. Ceramides and mitochondrial fatty acid oxidation in obesity. FASEB J. 2017, 31, 1263–1272. [Google Scholar] [CrossRef] [Green Version]
- Engin, A.B. What Is Lipotoxicity? In Obesity and Lipotoxicity, Advances in Experimental Medicine and Biology; Springer: Berlin/Heidelberg, Germany, 2017; pp. 197–220. [Google Scholar]
- Korbecki, J.; Bajdak-Rusinek, K. The effect of palmitic acid on inflammatory response in macrophages: An overview of molecular mechanisms. Inflamm. Res. 2019, 68, 915–932. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, P.; Leray, V.; Diez, M.; Serisier, S.; Bloc’h, J.L.; Siliart, B.; Dumon, H. Liver lipid metabolism. J. Anim. Physiol. Anim. Nutr. 2008, 92, 272–283. [Google Scholar] [CrossRef] [PubMed]
- Satapati, S.; Kucejova, B.; Duarte, J.A.; Fletcher, J.A.; Reynolds, L.; Sunny, N.E.; He, T.; Nair, L.A.; Livingston, K.; Fu, X.; et al. Mitochondrial metabolism mediates oxidative stress and inflammation in fatty liver. J. Clin. Investig. 2015, 125, 4447–4462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savic, D.; Hodson, L.; Neubauer, S.; Pavlides, M. The importance of the fatty acid transporter l-carnitine in non-alcoholic fatty liver disease (Nafld). Nutrients 2020, 12, 2178. [Google Scholar] [CrossRef] [PubMed]
- Malaguarnera, M.; Gargante, M.P.; Russo, C.; Antic, T.; Vacante, M.; Malaguarnera, M.; Avitabile, T.; Volti, G.L.; Galvano, F. L-carnitine supplementation to diet: A new tool in treatment of nonalcoholic steatohepatitisa randomized and controlled clinical trial. Am. J. Gastroenterol. 2010, 105, 1338–1345. [Google Scholar] [CrossRef]
- Xia, Y.; Li, Q.; Zhong, W.; Dong, J.; Wang, Z.; Wang, C. L-carnitine ameliorated fatty liver in high-calorie diet/STZ-induced type 2 diabetic mice by improving mitochondrial function. Diabetol. Metab. Syndr. 2010, 3, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Videla, L.A.; Rodrigo, R.; Orellana, M.; Fernandez, V.; Tapia, G.; Quinones, L.; Varela, N.; Contreras, J.; Lazarte, R.; Csendes, A.; et al. Oxidative stress-related parameters in the liver of non-alcoholic fatty liver disease patients. Clin. Sci. 2004, 106, 261–268. [Google Scholar] [CrossRef] [Green Version]
- Musso, G.; Gambino, R.; Cassader, M. Recent insights into hepatic lipid metabolism in non-alcoholic fatty liver disease (NAFLD). Prog. Lipid Res. 2009, 48, 1–26. [Google Scholar] [CrossRef]
- Lambert, J.E.; Ramos-Roman, M.A.; Browning, J.D.; Parks, E.J. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology 2014, 146, 726–735. [Google Scholar] [CrossRef]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Investig. 2005, 115, 1343–1351. [Google Scholar] [CrossRef] [Green Version]
- Horst, K.W.T.; Serlie, M.J. Fructose Consumption, Lipogenesis, and Non-Alcoholic Fatty Liver Disease Enhanced Reader. Nutrients 2017, 9, 1–20. [Google Scholar]
- Cherkaoui-Malki, M.; Surapureddi, S.; El Hajj, H.I.; Vamecq, J.; Andreoletti, P. Hepatic Steatosis and Peroxisomal Fatty Acid Beta-oxidation. Curr. Drug Metab. 2012, 13, 1412–1421. [Google Scholar] [CrossRef] [PubMed]
- Carta, G.; Murru, E.; Banni, S.; Manca, C. Palmitic acid: Physiological role, metabolism and nutritional implications. Front. Physiol. 2017, 8, 902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alkhouri, N.; Dixon, L.J.; Feldstein, A.E. Lipotoxicity in nonalcoholic fatty liver disease: Not all lipids are created equal. Expert Rev. Gastroenterol. Hepatol. 2009, 3, 445–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Listenberger, L.L.; Han, X.; Lewis, S.E.; Cases, S.; Farese, R.V.; Ory, D.S.; Schaffer, J.E. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc. Natl. Acad. Sci. USA 2003, 100, 3077–3082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosma, M.; Dapito, D.H.; Drosatos-Tampakaki, Z.; Huiping-Son, N.; Huang, L.S.; Kersten, S.; Drosatos, K.; Goldberg, I.J. Sequestration of fatty acids in triglycerides prevents endoplasmic reticulum stress in an in vitro model of cardiomyocyte lipotoxicity. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2014, 1841, 1648–1655. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, K.; Yang, L.; McCall, S.; Huang, J.; Yu, X.X.; Pandey, S.K.; Bhanot, S.; Monia, B.P.; Li, Y.X.; Diehl, A.M. Inhibiting Triglyceride Synthesis Improves Hepatic Steatosis but Exacerbates Liver Damage and Fibrosis in Obese Mice with Nonalcoholic Steatohepatitis. Hepatology 2007, 45, 1366–1374. [Google Scholar] [CrossRef]
- Monetti, M.; Levin, M.C.; Watt, M.J.; Sajan, M.P.; Marmor, S.; Hubbard, B.K.; Stevens, R.D.; Bain, J.R.; Newgard, C.B.; Farese, R.V., Sr.; et al. Dissociation of Hepatic Steatosis and Insulin Resistance in Mice Overexpressing DGAT in the Liver. Cell Metab. 2007, 6, 69–78. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Hong, M.; Tan, H.Y.; Wang, N.; Feng, Y. Insights into the Role and Interdependence of Oxidative Stress and Inflammation in Liver Diseases. Oxid. Med. Cell. Longev. 2016, 2016. [Google Scholar] [CrossRef]
- Li, Z.Z.; Berk, M.; McIntyre, T.M.; Feldstein, A.E. Hepatic lipid partitioning and liver damage in nonalcoholic fatty liver disease: Role of stearoyl-Coa desaturase. J. Biol. Chem. 2009, 284, 5637–5644. [Google Scholar] [CrossRef] [Green Version]
- Magee, N.; Zou, A.; Zhang, Y. Pathogenesis of Nonalcoholic Steatohepatitis: Interactions between Liver Parenchymal and Nonparenchymal Cells. Biomed Res. Int. 2016, 2016. [Google Scholar] [CrossRef] [Green Version]
- Svegliati-Baroni, G.; Saccomanno, S.; van Goor, H.; Jansen, P.; Benedetti, A.; Moshage, H. Involvement of reactive oxygen species and nitric oxide radicals in activation and proliferation of rat hepatic stellate cells. Liver Int. 2001, 21, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernández, A.; Reyes, D.; Geng, Y.; Arab, J.P.; Cabrera, D.; Sepulveda, R.; Solis, N.; Buist-Homan, M.; Arrese, M.; Moshage, H. Extracellular vesicles derived from fat-laden hepatocytes undergoing chemical hypoxia promote a pro-fibrotic phenotype in hepatic stellate cells. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165857. [Google Scholar] [CrossRef] [PubMed]
- Pierantonelli, I.; Svegliati-Baroni, G. Nonalcoholic Fatty Liver Disease: Basic Pathogenetic Mechanisms in the Progression from NAFLD to NASH. Transplantation 2019, 103, E1–E13. [Google Scholar] [CrossRef] [PubMed]
- Schoeler, M.; Caesar, R. Dietary lipids, gut microbiota and lipid metabolism. Rev. Endocr. Metab. Disord. 2019, 20, 461–472. [Google Scholar] [CrossRef] [Green Version]
- Aron-Wisnewsky, J.; Vigliotti, C.; Witjes, J.; Le, P.; Holleboom, A.G.; Verheij, J.; Nieuwdorp, M.; Clément, K. Gut microbiota and human NAFLD: Disentangling microbial signatures from metabolic disorders. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 279–297. [Google Scholar] [CrossRef]
- Wang, S.; Huang, M.; You, X.; Zhao, J.; Chen, L.; Wang, L.; Luo, Y.; Chen, Y. Gut microbiota mediates the anti-obesity effect of calorie restriction in mice. Sci. Rep. 2018, 8, 2–15. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, J.; Igarashi, M.; Watanabe, K.; Karaki, S.I.; Mukouyama, H.; Kishino, S.; Li, X.; Ichimura, A.; Irie, J.; Sugimoto, Y.; et al. Gut microbiota confers host resistance to obesity by metabolizing dietary polyunsaturated fatty acids. Nat. Commun. 2019, 10, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Marchesi, J.R.; Adams, D.H.; Fava, F.; Hermes, G.D.; Hirschfield, G.M.; Hold, G.; Quraishi, M.N.; Kinross, J.; Smidt, H.; Tuohy, K.M.; et al. The gut microbiota and host health: A new clinical frontier. Gut 2016, 65, 330–339. [Google Scholar] [CrossRef] [Green Version]
- Ji, Y.; Yin, Y.; Sun, L.; Zhang, W. The Molecular and Mechanistic Insights Based on Gut–Liver Axis: Nutritional Target for Non-Alcoholic Fatty Liver Disease (NAFLD) Improvement. Int. J. Mol. Sci. 2020, 21, 3066. [Google Scholar] [CrossRef]
- Ogawa, Y.; Imajo, K.; Honda, Y.; Kessoku, T.; Tomeno, W.; Kato, S.; Fujita, K.; Yoneda, M.; Saito, S.; Saigusa, Y.; et al. Palmitate-induced lipotoxicity is crucial for the pathogenesis of nonalcoholic fatty liver disease in cooperation with gut-derived endotoxin. Sci. Rep. 2018, 8, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Mansouri, A.; Gattolliat, C.H.; Asselah, T. Mitochondrial Dysfunction and Signaling in Chronic Liver Diseases. Gastroenterology 2018, 155, 629–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Ruiz, C.; Fernández-Checa, J.C. Mitochondrial Oxidative Stress and Antioxidants Balance in Fatty Liver Disease. Hepatol. Commun. 2018, 2, 1425–1439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mann, J.P.; Raponi, M.; Nobili, V. Clinical implications of understanding the association between oxidative stress and pediatric NAFLD. Expert Rev. Gastroenterol. Hepatol. 2017, 11, 371–382. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Carreras, M.; Del Hoyo, P.; Martín, M.A.; Rubio, J.C.; Martín, A.; Castellano, G.; Colina, F.; Arenas, J.; Solis-Herruzo, J.A. Defective hepatic mitochondrial respiratory chain in patients with nonalcoholic steatohepatitis. Hepatology 2003, 38, 999–1007. [Google Scholar] [CrossRef]
- Chen, Z.; Tian, R.; She, Z.; Cai, J.; Li, H. Role of oxidative stress in the pathogenesis of nonalcoholic fatty liver disease. Free Radic. Biol. Med. 2020, 152, 116–141. [Google Scholar] [CrossRef]
- Demine, S.; Renard, P.; Arnould, T. Mitochondrial Uncoupling: A Key Controller of Biological Processes in Physiology and Diseases. Cells 2019, 8, 795. [Google Scholar] [CrossRef] [Green Version]
- Hauck, A.K.; Bernlohr, D.A. Oxidative stress and lipotoxicity. J. Lipid Res. 2016, 57, 1976–1986. [Google Scholar] [CrossRef] [Green Version]
- Musso, G.; Cassader, M.; Paschetta, E.; Gambino, R. Bioactive Lipid Species and Metabolic Pathways in Progression and Resolution of Nonalcoholic Steatohepatitis. Gastroenterology 2018, 155, 282–302. [Google Scholar] [CrossRef]
- Ajith, T.A. Role of mitochondria and mitochondria-targeted agents in non-alcoholic fatty liver disease. Clin. Exp. Pharm. Physiol. 2018, 45, 413–421. [Google Scholar] [CrossRef]
- Sookoian, S.; Flichman, D.; Scian, R.; Rohr, C.; Dopazo, H.; Gianotti, T.F.; Martino, J.S.; Castaño, G.O.; Pirola, C.J. Mitochondrial genome architecture in non-alcoholic fatty liver disease. J. Pathol. 2016, 240, 437–449. [Google Scholar] [CrossRef]
- Garcia-Martinez, I.; Santoro, N.; Chen, Y.; Hoque, R.; Ouyang, X.; Caprio, S.; Shlomchik, M.J.; Coffman, R.L.; Candia, A.; Mehal, W.Z. Hepatocyte mitochondrial DNA drives nonalcoholic steatohepatitis by activation of TLR9. J. Clin. Investig. 2016, 126, 859–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apostolova, N.; Victor, V.M. Molecular strategies for targeting antioxidants to mitochondria: Therapeutic implications. Antioxid. Redox Signal. 2015, 22, 686–729. [Google Scholar] [CrossRef] [PubMed]
- Sheu, S.S.; Nauduri, D.; Anders, M.W. Targeting antioxidants to mitochondria: A new therapeutic direction. Biochim. Biophys. Acta Mol. Basis Dis. 2006, 1762, 256–265. [Google Scholar] [CrossRef] [Green Version]
- Broome, S.C.; Woodhead, J.S.T.; Merry, T.L. Mitochondria-targeted antioxidants and skeletal muscle function. Antioxidants 2018, 7, 107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjørndal, B.; Alterås, E.K.; Lindquist, C.; Svardal, A.; Skorve, J.; Berge, R.K. Associations between fatty acid oxidation, hepatic mitochondrial function, and plasma acylcarnitine levels in mice. Nutr. Metab. 2018, 15, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.; Zhang, Y.; Park, S.Y.; Joseph, A.M.; Han, C.; Park, H.J.; Kalavalapalli, S.; Chun, S.K.; Morgan, D.; Kim, J.S.; et al. Mitochondrial ATP transporter depletion protects mice against liver steatosis and insulin resistance. Nat. Commun. 2017, 8, 2017. [Google Scholar] [CrossRef]
- Pospisilik, J.A.; Knauf, C.; Joza, N.; Benit, P.; Orthofer, M.; Cani, P.D.; Ebersberger, I.; Nakashima, T.; Sarao, R.; Neely, G.; et al. Targeted deletion of AIF decreases mitochondrial oxidative phosphorylation and protects from obesity and diabetes. Cell 2007, 131, 476–491. [Google Scholar] [CrossRef]
- Ibdah, J.A.; Perlegas, P.; Zhao, Y.; Angdisen, J.; Borgerink, H.; Shadoan, M.K.; Wagner, J.D.; Matern, D.; Rinaldo, P.; Cline, J.M. Mice heterozygous for a defect in mitochondrial trifunctional protein develop hepatic steatosis and insulin resistance. Gastroenterology 2005, 128, 1381–1390. [Google Scholar] [CrossRef]
- Nassir, F.; Rector, R.S.; Hammoud, G.M.; Ibdah, J.A. Pathogenesis and Prevention of Hepatic Steatosis. Gastroenterol. Hepatol. 2015, 11, 167–175. [Google Scholar]
- Shimizu, Y.; Hendershot, L.M. Oxidative Folding- Cellular Strategies for Dealingwith the Resultant Equimolar Production of Reactive Oxygen Species. Antioxidants 2009, 11, 2317–2331. [Google Scholar]
- Ghemrawi, R.; Battaglia-Hsu, S.-F.; Arnold, C. Endoplasmic Reticulum Stress in Metabolic Disorders. Cells 2018, 7, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geng, M.H.; Faber, K.N.; de Meijer, V.E.; Blokzijl, H. How does hepatic lipid accumulation lead to lipotoxicity in non-alcoholic fatty liver disease? Hepatol. Int. 2020. [Google Scholar]
- Burban, A.; Sharanek, A.; Guguen-Guillouzo, C.; Guillouzo, A. Endoplasmic reticulum stress precedes oxidative stress in antibiotic-induced cholestasis and cytotoxicity in human hepatocytes. Free Radic. Biol. Med. 2018, 115, 166–178. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Kaufman, R.J. The role of ER stress in lipid metabolism and lipotoxicity. J. Lipid Res. 2016, 57, 1329–1338. [Google Scholar] [CrossRef] [Green Version]
- Zeeshan, H.M.A.; Lee, G.H.; Kim, H.R.; Chae, H.J. Endoplasmic reticulum stress and associated ROS. Int. J. Mol. Sci. 2016, 17, 327. [Google Scholar] [CrossRef] [Green Version]
- Santos, C.X.C.; Nabeebaccus, A.A.; Shah, A.M.; Camargo, L.L.; Filho, S.V.; Lopes, L.R. Endoplasmic reticulum stress and nox-mediated reactive oxygen species signaling in the peripheral vasculature: Potential role in hypertension. Antioxid. Redox Signal. 2014, 20, 121–134. [Google Scholar] [CrossRef]
- Ashraf, N.U.; Sheikh, T.A. Endoplasmic reticulum stress and Oxidative stress in the pathogenesis of Non-alcoholic fatty liver disease. Free Radic. Res. 2015, 49, 1405–1418. [Google Scholar] [CrossRef]
- Fu, S.; Watkins, S.M.; Hotamisligil, G.S. The role of endoplasmic reticulum in hepatic lipid homeostasis and stress signaling. Cell Metab. 2012, 15, 623–634. [Google Scholar] [CrossRef] [Green Version]
- Pfaffenbach, K.T.; Lee, A.S. The critical role of GRP78 in physiologic and pathologic stress. Curr. Opin. Cell Biol. 2011, 23, 150–156. [Google Scholar] [CrossRef] [Green Version]
- Pfaffenbach, K.T.; Gentile, C.L.; Nivala, A.M.; Wang, D.; Wei, Y.; Pagliassotti, M.J. Linking endoplasmic reticulum stress to cell death in hepatocytes: Roles of C/EBP homologous protein and chemical chaperones in palmitate-mediated cell death. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E1027–E1035. [Google Scholar] [CrossRef] [Green Version]
- Colgan, S.M.; Tang, D.; Werstuck, G.H.; Austin, R.C. Endoplasmic reticulum stress causes the activation of sterol regulatory element binding protein-2. Int. J. Biochem. Cell Biol. 2007, 39, 1843–1851. [Google Scholar] [CrossRef] [PubMed]
- Basseri, S.; Austin, R.C. ER Stress and Lipogenesis: A Slippery Slope toward Hepatic Steatosis. Dev. Cell 2008, 15, 795–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferré, P.; Foufelle, F. Hepatic steatosis: A role for de novo lipogenesis and the transcription factor SREBP-1c. Diabetes, Obes. Metab. 2010, 12, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Liu, R. ER stress and hepatic lipid metabolism. Front. Genet. 2014, 5, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Viscarra, J.; Kim, S.J.; Sul, H.S. Transcriptional regulation of hepatic lipogenesis. Nat. Rev. Mol. Cell Biol. 2015, 16, 678–689. [Google Scholar] [CrossRef] [Green Version]
- Lauressergues, E.; Bert, E.; Duriez, P.; Hum, D.; Majd, Z.; Staels, B.; Cussac, D. Does endoplasmic reticulum stress participate in APD-induced hepatic metabolic dysregulation? Neuropharmacology 2012, 62, 784–796. [Google Scholar] [CrossRef]
- Kim, Y.R.; Lee, E.J.; Shin, K.O.; Kim, M.H.; Pewzner-Jung, Y.; Lee, Y.M.; Park, J.W.; Futerman, A.H.; Park, W.J. Hepatic triglyceride accumulation via endoplasmic reticulum stress-induced SREBP-1 activation is regulated by ceramide synthases. Exp. Mol. Med. 2019, 51, 1–16. [Google Scholar] [CrossRef]
- Rong, S.; Cortés, V.A.; Rashid, S.; Anderson, N.N.; McDonald, J.G.; Liang, G.; Moon, Y.A.; Hammer, R.E.; Horton, J.D. Expression of SREBP-1c requires SREBP-2-mediated generation of a sterol ligand for LXR in livers of mice. Elife 2017, 6, e25015. [Google Scholar] [CrossRef]
- Ye, J.; Rawson, R.B.; Komuro, R.; Chen, X.; Davé, U.P.; Prywes, R.; Brown, M.S.; Goldstein, J.L. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell 2000, 6, 1355–1364. [Google Scholar] [CrossRef]
- Xiao, G.; Zhang, T.; Yu, S.; Lee, S.; Calabuig-Navarro, V.; Yamauchi, J.; Ringquist, S.; Dong, H.H. ATF4 protein deficiency protects against high fructose-induced hypertriglyceridemia in mice. J. Biol. Chem. 2013, 288, 25350–25361. [Google Scholar] [CrossRef] [Green Version]
- Kammoun, H.L.; Chabanon, H.; Hainault, I.; Luquet, S.; Magnan, C.; Koike, T.; Ferré, P.; Foufelle, F. GRP78 expression inhibits insulin and ER stress-induced SREBP-1c activation and reduces hepatic steatosis in mice. J. Clin. Investig. 2009, 119, 1201–1215. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Zhang, C.; Zhang, K. Role of unfolded protein response in lipogenesis. World J. Hepatol. 2010, 2, 203–207. [Google Scholar] [CrossRef] [PubMed]
- Ren, L.P.; Chan, S.M.; Zeng, X.Y.; Laybutt, D.R.; Iseli, T.J.; Sun, R.Q.; Kraegen, E.W.; Cooney, G.J.; Turner, N.; Ye, J.M. Differing endoplasmic reticulum stress response to excess lipogenesis versus lipid oversupply in relation to hepatic steatosis and insulin resistance. PLoS ONE 2012, 7, e30816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Zhang, D.; Liu, X.; Li, X.; Liu, F.; Yu, Y.; Jia, S.; Zhou, Y.; Zhao, Y. Endoplasmic Reticulum Stress Affects Lipid Metabolism in Atherosclerosis Via CHOP Activation and Over-Expression of miR-33. Cell. Physiol. Biochem. 2018, 48, 1995–2010. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Chen, Z.; Lam, V.; Han, J.; Hassler, J.; Finck, B.N.; Davidson, N.O.; Kaufman, R.J. IRE1α-XBP1s induces PDI expression to increase MTP activity for hepatic VLDL assembly and lipid homeostasis. Cell Metab. 2012, 16, 473–486. [Google Scholar] [CrossRef] [Green Version]
- Puri, P.; Mirshahi, F.; Cheung, O.; Natarajan, R.; Maher, J.W.; Kellum, J.M.; Sanyal, A.J. Activation and Dysregulation of the Unfolded Protein Response in Nonalcoholic Fatty Liver Disease. Gastroenterology 2008, 134, 568–576. [Google Scholar] [CrossRef]
- Cunha, D.A.; Hekerman, P.; Ladrière, L.; Bazarra-Castro, A.; Ortis, F.; Wakeham, M.C.; Moore, F.; Rasschaert, J.; Cardozo, A.K.; Bellomo, E.; et al. Initiation and execution of lipotoxic ER stress in pancreatic β-cells. J. Cell Sci. 2008, 121, 2308–2318. [Google Scholar] [CrossRef] [Green Version]
- Cao, J.; Dai, D.L.; Yao, L.; Yu, H.H.; Ning, B.; Zhang, Q.; Chen, J.; Cheng, W.H.; Shen, W.; Yang, Z.X. Saturated fatty acid induction of endoplasmic reticulum stress and apoptosis in human liver cells via the PERK/ATF4/CHOP signaling pathway. Mol. Cell. Biochem. 2012, 364, 115–129. [Google Scholar] [CrossRef]
- Malhi, H.; Bronk, S.F.; Werneburg, N.W.; Gores, G.J. Free fatty acids induce JNK-dependent hepatocyte lipoapoptosis. J. Biol. Chem. 2006, 281, 12093–12101. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Zhang, H.; Zhong, Y. Hepatic GDF15 is regulated by CHOP of the unfolded protein response and alleviates NAFLD progression in obese mice. Biochem. Biophys. Res. Commun. 2018, 498, 388–394. [Google Scholar] [CrossRef]
- Fu, S.; Yang, L.; Li, P.; Hofmann, O.; Dicker, L.; Hide, W.; Lin, X.; Watkins, S.M.; Ivanov, A.R.; Hotamisligil, G.S. Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature 2011, 473, 528–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Della Pepa, G.; Vetrani, C.; Lombardi, G.; Bozzetto, L.; Annuzzi, G.; Rivellese, A.A. Isocaloric dietary changes and Non-Alcoholic fatty liver disease in high cardiometabolic risk individuals. Nutrients 2017, 9, 1065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cicero, A.F.G.; Colletti, A.; Bellentani, S. Nutraceutical approach to non-alcoholic fatty liver disease (NAFLD): The available clinical evidence. Nutrients 2018, 10, 1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Tan, H.Y.; Wang, N.; Cheung, F.; Hong, M.; Feng, Y. The Potential and Action Mechanism of Polyphenols in the Treatment of Liver Diseases. Oxid. Med. Cell. Longev. 2018, 2018. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Wang, T.; Li, J.; Wang, S.; Qiu, F.; Yu, H.; Zhang, Y.; Wang, T. Effects of natural products on fructose-induced nonalcoholic fatty liver disease (NAFLD). Nutrients 2017, 9, 96. [Google Scholar] [CrossRef] [Green Version]
- Rafiei, H.; Omidian, K.; Bandy, B. Dietary polyphenols protect against oleic acid-induced steatosis in an in vitro model of NAFLD by modulating lipid metabolism and improving mitochondrial function. Nutrients 2019, 11, 541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musso, G.; Cassader, M.; Gambino, R. Non-alcoholic steatohepatitis: Emerging molecular targets and therapeutic strategies. Nat. Rev. Drug Discov. 2016, 15, 249–274. [Google Scholar] [CrossRef]
- Hosseini, H.; Teimouri, M.; Shabani, M.; Koushki, M.; Khorzoughi, R.B.; Namvarjah, F.; Izadi, P.; Meshkani, R. Resveratrol alleviates non-alcoholic fatty liver disease through epigenetic modification of the Nrf2 signaling pathway. Int. J. Biochem. Cell Biol. 2020, 119, 105667. [Google Scholar] [CrossRef]
- Xia, H.M.; Wang, J.; Xie, X.J.; Xu, L.J.; Tang, S.Q. Green tea polyphenols attenuate hepatic steatosis, and reduce insulin resistance and inflammation in high-fat diet-induced rats. Int. J. Mol. Med. 2019, 44, 1523–1530. [Google Scholar] [CrossRef]
- Yang, J.P.; Shin, J.H.; Seo, S.H.; Kim, S.G.; Lee, S.H.; Shin, E.H. Effects of antioxidants in reducing accumulation of fat in hepatocyte. Int. J. Mol. Sci. 2018, 19, 2563. [Google Scholar] [CrossRef] [Green Version]
- Singal, A.K.; Jampana, S.C.; Weinman, S.A. Antioxidants as Therapeutic Agents for Liver Disease Ashwani. Liver Int. 2011, 31, 1432–1448. [Google Scholar] [CrossRef] [Green Version]
- Casas-Grajales, S. Antioxidants in liver health. World J. Gastrointest. Pharm. Ther. 2015, 6, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivancovsky-Wajcman, D.; Fliss-Isakov, N.; Salomone, F.; Webb, M.; Shibolet, O.; Kariv, R.; Zelber-Sagi, S. Dietary vitamin E and C intake is inversely associated with the severity of nonalcoholic fatty liver disease. Dig. Liver Dis. 2019, 51, 1698–1705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hadzi-Petrushev, N.; Dimovska, K.; Jankulovski, N.; Mitrov, D.; Mladenov, M. Supplementation with Alpha-Tocopherol and Ascorbic Acid to Nonalcoholic Fatty Liver Disease’s Statin Therapy in Men. Adv. Pharm. Sci. 2018, 2018. [Google Scholar] [CrossRef] [PubMed]
- Traber, M.G.; Atkinson, J. Vitamin E, antioxidant and nothing more. Free Radic. Biol. Med. 2007, 43, 4–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perumpail, B.J.; Li, A.A.; John, N.; Sallam, S.; Shah, N.D.; Kwong, W.; Cholankeril, G.; Kim, D.; Ahmed, A. The Role of Vitamin E in the Treatment of NAFLD. Diseases 2018, 6, 86. [Google Scholar] [CrossRef] [Green Version]
- Sanyal, A.J.; Chalasani, N.; Kowdley, K.V.; McCullough, A.; Diehl, A.M.; Bass, N.M.; Neuschwander-Tetri, B.A.; Lavine, J.E.; Tonascia, J.; Unalp, A.; et al. Pioglitazone, Vitamin E, or Placebo for Nonalcoholic Steatohepatitis. N. Engl. J. Med. 2010, 362, 1675–1685. [Google Scholar] [CrossRef] [Green Version]
- Vilar-Gomez, E.; Vuppalanchi, R.; Gawrieh, S.; Ghabril, M.; Saxena, R.; Cummings, O.W.; Chalasani, N. Vitamin E Improves Transplant-Free Survival and Hepatic Decompensation among Patients with Nonalcoholic Steatohepatitis and Advanced Fibrosis. Hepatology 2020, 71, 495–509. [Google Scholar] [CrossRef] [Green Version]
- Raso, G.M.; Esposito, E.; Iacono, A.; Pacilio, M.; Cuzzocrea, S.; Canani, R.B.; Calignano, A.; Meli, R. Comparative therapeutic effects of metformin and vitamin E in a model of non-alcoholic steatohepatitis in the young rat. Eur. J. Pharm. 2009, 604, 125–131. [Google Scholar] [CrossRef]
- Oliveira, C.P.; da Costa Gayotto, L.C.; Tatai, C.; Della Nina, B.I.; Lima, E.S.; Abdalla, D.S.; Lopasso, F.P.; Laurindo, F.R.; Carrilho, F.J. Vitamin C and vitamin E in prevention of Nonalcoholic Fatty Liver Disease (NAFLD) in choline deficient diet fed rats. Nutr. J. 2003, 2, 1–5. [Google Scholar] [CrossRef]
- Machado, M.V.; Ravasco, P.; Jesus, L.; Marques-Vidal, P.; Oliveira, C.R.; Proença, T.; Baldeiras, I.; Camilo, M.E.; Cortez-Pinto, H. Blood oxidative stress markers in non-alcoholic steatohepatitis and how it correlates with diet. Scand. J. Gastroenterol. 2008, 43, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Pastori, D.; Baratta, F.; Carnevale, R.; Cangemi, R.; Del Ben, M.; Bucci, T.; Polimeni, L.; Labbadia, G.; Nocella, C.; Scardella, L.; et al. Similar Reduction of Cholesterol-Adjusted Vitamin E Serum Levels in Simple Steatosis and Non-Alcoholic Steatohepatitis. Clin. Transl. Gastroenterol. 2015, 6, e113–e116. [Google Scholar] [CrossRef] [PubMed]
- Bahcecioglu, I.H.; Yalniz, M.; Ilhan, N.; Ataseven, H.; Ozercan, I.H. Levels of serum vitamin A, alpha-tocopherol and malondialdehyde in patients with non-alcoholic steatohepatitis: Relationship with histopathologic severity. Int. J. Clin. Pract. 2005, 59, 318–323. [Google Scholar] [CrossRef] [PubMed]
- Tsou, P.; Wu, C.-J. Serum Vitamin E Levels of Adults with Nonalcoholic Fatty Liver Disease: An Inverse Relationship with All-Cause Mortality in Non-Diabetic but Not in Pre-Diabetic or Diabetic Subjects. J. Clin. Med. 2019, 8, 1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bril, F.; Biernacki, D.M.; Kalavalapalli, S.; Lomonaco, R.; Subbarayan, S.K.; Lai, J.; Tio, F.; Suman, A.; Orsak, B.K.; Hecht, J.; et al. Role of Vitamin E for nonalcoholic steatohepatitis in patients with type 2 diabetes: A randomized controlled trial. Diabetes Care 2019, 42, 1481–1488. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef]
- Podszun, M.C.; Alawad, A.S.; Lingala, S.; Morris, N.; Huang, W.C.A.; Yang, S.; Schoenfeld, M.; Rolt, A.; Ouwerkerk, R.; Valdez, K.; et al. Vitamin E treatment in NAFLD patients demonstrates that oxidative stress drives steatosis through upregulation of de-novo lipogenesis. Redox Biol. 2020, 37, 101710. [Google Scholar] [CrossRef]
- Huang, X.; Zhen, J.; Dong, S.; Zhang, H.; van Halm-Lutterodt, N.; Yuan, L. DHA and vitamin e antagonized the Aβ 25–35 -mediated neuron oxidative damage through activation of Nrf2 signaling pathways and regulation of CD36, SRB1 and FABP5 expression in PC12 cells. Food Funct. 2019, 10, 1049–1061. [Google Scholar] [CrossRef]
- Bozaykut, P.; Karademir, B.; Yazgan, B.; Sozen, E.; Siow, R.C.; Mann, G.E.; Ozer, N.K. Effects of vitamin e on peroxisome proliferator-activated receptor γ and nuclear factor-erythroid 2-related factor 2 in hypercholesterolemia-induced atherosclerosis. Free Radic. Biol. Med. 2014, 70, 174–181. [Google Scholar] [CrossRef]
- Mishra, P.; Paital, B.; Jena, S.; Swain, S.S.; Kumar, S.; Yadav, M.K.; Chainy, G.B.; Samanta, L. Possible activation of NRF2 by Vitamin E/Curcumin against altered thyroid hormone induced oxidative stress via NFĸB/AKT/mTOR/KEAP1 signalling in rat heart. Sci. Rep. 2019, 9, 1–16. [Google Scholar] [CrossRef]
- Fang, J.; Yin, H.; Yang, Z.; Tan, M.; Wang, F.; Chen, K.; Zuo, Z.; Shu, G.; Cui, H.; Ouyang, P.; et al. Vitamin E protects against cadmium-induced sub-chronic liver injury associated with the inhibition of oxidative stress and activation of Nrf2 pathway. Ecotoxicol. Environ. Saf. 2021, 208, 111610. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Xu, Y.; Ren, X.; Xiang, D.; Lei, K.; Zhang, C.; Liu, D. Vitamin E Ameliorates Lipid Metabolism in Mice with Nonalcoholic Fatty Liver Disease via Nrf2/CES1 Signaling Pathway. Dig. Dis. Sci. 2019, 64, 3182–3191. [Google Scholar] [CrossRef]
- Wei, J.; Lei, G.H.; Fu, L.; Zeng, C.; Yang, T.; Peng, S.F. Association between dietary Vitamin C intake and non-alcoholic fatty liver disease: A cross-sectional study among middle-aged and older adults. PLoS ONE 2016, 11, e0147985. [Google Scholar] [CrossRef] [PubMed]
- Ipsen, D.H.; Tveden-Nyborg, P.; Lykkesfeldt, J. Does vitamin C deficiency promote fatty liver disease development? Nutrients 2014, 6, 5473–5499. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Ahn, J.; Shin, S.S.; Yoon, M. Ascorbic acid inhibits visceral obesity and nonalcoholic fatty liver disease by activating peroxisome proliferator-activated receptor α in high-fat-diet-fed C57BL/6J mice. Int. J. Obes. 2019, 43, 1620–1630. [Google Scholar] [CrossRef]
- Amanullah, I.; Khan, Y.H.; Anwar, I.; Gulzar, A.; Mallhi, T.H.; Raja, A.A. Effect of Vitamin E in non-alcoholic fatty liver disease: A systematic review and meta-analysis of randomised controlled trials. Postgrad. Med. J. 2019, 95, 601–611. [Google Scholar] [CrossRef]
- Poole, R.; Kennedy, O.J.; Roderick, P.; Fallowfield, J.A.; Hayes, P.C.; Parkes, J. Coffee consumption and health: Umbrella review of meta-analyses of multiple health outcomes. BMJ 2017, 359, j5024. [Google Scholar] [CrossRef] [Green Version]
- Kolb, H.; Kempf, K.; Martin, S. Health Effects of Coffee: Mechanism Unraveled? Nutrients 2020, 12, 1842. [Google Scholar] [CrossRef]
- Contaldo, F.; Santarpia, L.; Pasanisi, F. Chronic inflammatory liver diseases and coffee intake. Curr. Opin. Clin. Nutr. Metab. Care 2019, 22, 389–392. [Google Scholar] [CrossRef]
- Wijarnpreecha, K.; Thongprayoon, C.; Ungprasert, P. Coffee consumption and risk of nonalcoholic fatty liver disease: A systematic review and meta-analysis. Eur. J. Gastroenterol. Hepatol. 2017, 29, e8–e12. [Google Scholar] [CrossRef]
- Xiao, Q.; Sinha, R.; Graubard, B.I.; Freedman, N.D. Inverse associations of total and decaffeinated coffee with liver enzyme levels in National Health and Nutrition Examination Survey 1999–2010. Hepatology 2014, 60, 2091–2098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boettler, U.; Volz, N.; Teller, N.; Haupt, L.M.; Bakuradze, T.; Eisenbrand, G.; Bytof, G.; Lantz, I.; Griffiths, L.R.; Marko, D. Induction of antioxidative Nrf2 gene transcription by coffee in humans: Depending on genotype? Mol. Biol. Rep. 2012, 39, 7155–7162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boettler, U.; Sommerfeld, K.; Volz, N.; Pahlke, G.; Teller, N.; Somoza, V.; Lang, R.; Hofmann, T.; Marko, D. Coffee constituents as modulators of Nrf2 nuclear translocation and ARE (EpRE)-dependent gene expression. J. Nutr. Biochem. 2011, 22, 426–440. [Google Scholar] [CrossRef] [PubMed]
- Priftis, A.; Angeli-Terzidou, A.E.; Veskoukis, A.S.; Spandidos, D.A.; Kouretas, D. Cell-specific and roasting-dependent regulation of the Keap1/Nrf2 pathway by coffee extracts. Mol. Med. Rep. 2018, 17, 8325–8331. [Google Scholar] [CrossRef]
- Zheng, X.; Dai, W.; Chen, X.; Wang, K.; Zhang, W.; Liu, L.; Hou, J. Caffeine reduces hepatic lipid accumulation through regulation of lipogenesis and ER stress in zebrafish larvae. J. Biomed. Sci. 2015, 22, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Sinha, R.A.; Farah, B.L.; Singh, B.K.; Siddique, M.M.; Li, Y.; Wu, Y.; Ilkayeva, O.R.; Gooding, J.; Ching, J.; Zhou, J.; et al. Caffeine stimulates hepatic lipid metabolism by the autophagy-lysosomal pathway in mice. Hepatology 2014, 59, 1366–1380. [Google Scholar] [CrossRef]
- Salomone, F.; Galvano, F.; Volti, G.L. Molecular bases underlying the hepatoprotective effects of coffee. Nutrients 2017, 9, 85. [Google Scholar] [CrossRef] [Green Version]
- Du, X.; Huang, Q.; Guan, Y.; Lv, M.; He, X.; Fang, C.; Wang, X.; Sheng, J. Caffeine promotes conversion of palmitic acid to palmitoleic acid by inducing expression of fat-5 in Caenorhabditis elegans and scd1 in mice. Front. Pharm. 2018, 9, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Dai, X.; Yang, W.; Wang, H.; Zhao, H.; Yang, F.; Yang, Y.; Li, J.; Lv, X. Caffeine protects against alcohol-induced liver fibrosis by dampening the cAMP/PKA/CREB pathway in rat hepatic stellate cells. Int. Immunopharmacol. 2015, 25, 340–352. [Google Scholar] [CrossRef]
- Foretz, M.; Even, P.C.; Viollet, B. AMPK activation reduces hepatic lipid content by increasing fat oxidation in vivo. Int. J. Mol. Sci. 2018, 19, 2826. [Google Scholar] [CrossRef] [Green Version]
- Egawa, T.; Hamada, T.; Ma, X.; Karaike, K.; Kameda, N.; Masuda, S.; Iwanaka, N.; Hayashi, T. Caffeine activates preferentially α1-isoform of 5′AMP-activated protein kinase in rat skeletal muscle. Acta Physiol. 2011, 201, 227–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, S.; Liu, K.; Luo, H.; Xu, D.; Chen, L.; Zhang, L.; Wang, H. Caffeine programs hepatic SIRT1-related cholesterol synthesis and hypercholesterolemia via A2AR/cAMP/PKA pathway in adult male offspring rats. Toxicology 2019, 418, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Ceballos, M.; Arroyave, J.C.; Cortes-Mancera, F.M.; Röthlisberger, S. Chemopreventive effect of coffee against colorectal cancer and hepatocellular carcinoma. Int. J. Food Prop. 2019, 22, 536–555. [Google Scholar] [CrossRef]
- Zhang, Y.; Miao, L.; Zhang, H.; Wu, G.; Zhang, Z.; Lv, J. Chlorogenic acid against palmitic acid in endoplasmic reticulum stress-mediated apoptosis resulting in protective effect of primary rat hepatocytes. Lipids Health Dis. 2018, 17, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bazool, H.F.; Balas, M.; Gradinaru, D.; Margină, D.; Dinischiotu, A. Hepatoprotective effects of chlorogenic acid under hyperglycemic conditions. Rom. Biotechnol. Lett. 2017, X, 1–10. [Google Scholar]
- Bhandarkar, N.S.; Brown, L.; Panchal, S.K. Chlorogenic acid attenuates high-carbohydrate, high-fat diet–induced cardiovascular, liver, and metabolic changes in rats. Nutr. Res. 2019, 62, 78–88. [Google Scholar] [CrossRef]
- Wang, J.M.; Chen, R.X.; Zhang, L.L.; Ding, N.N.; Liu, C.; Cui, Y.; Cheng, Y.X. In vivo protective effects of chlorogenic acid against triptolide-induced hepatotoxicity and its mechanism. Pharm. Biol. 2018, 56, 626–631. [Google Scholar] [CrossRef] [Green Version]
- Shi, A.; Shi, H.; Wang, Y.; Liu, X.; Cheng, Y.; Li, H.; Zhao, H.; Wang, S.; Dong, L. Activation of Nrf2 pathway and inhibition of NLRP3 inflammasome activation contribute to the protective effect of chlorogenic acid on acute liver injury. Int. Immunopharmacol. 2018, 54, 125–130. [Google Scholar] [CrossRef]
- Mazza, A.; Fruci, B.; Garinis, G.A.; Giuliano, S.; Malaguarnera, R.; Belfiore, A. The role of metformin in the management of NAFLD. Exp. Diabetes Res. 2012, 2012. [Google Scholar] [CrossRef] [Green Version]
- Feng, W.H.; Bi, Y.; Li, P.; Yin, T.T.; Gao, C.X.; Shen, S.M.; Gao, L.J.; Yang, D.H.; Zhu, D.L. Effects of liraglutide, metformin and gliclazide on body composition in patients with both type 2 diabetes and non-alcoholic fatty liver disease: A randomized trial. J. Diabetes Investig. 2019, 10, 399–407. [Google Scholar] [CrossRef]
- Marchesini, G.; Roden, M.; Vettor, R. EASL-EASD-EASO Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease. J. Hepatol. 2016, 64, 1388–1402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandt, A.; Hernández-Arriaga, A.; Kehm, R.; Sánchez, V.; Jin, C.J.; Nier, A.; Baumann, A.; Camarinha-Silva, A.; Bergheim, I. Metformin attenuates the onset of non-alcoholic fatty liver disease and affects intestinal microbiota and barrier in small intestine. Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Y.M.; Lee, Y.H.; Kim, J.W.; Ham, D.S.; Kang, E.S.; Cha, B.S.; Lee, H.C.; Lee, B.W. Metformin alleviates hepatosteatosis by restoring SIRT1-mediated autophagy induction via an AMP-activated protein kinase-independent pathway. Autophagy 2015, 11, 46–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez-Martın, P.; Komatsu, M. p62/SQSTM1—Steering the cell through health and disease. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef] [Green Version]
- De la Rosa, L.C.; Vrenken, T.E.; Buist-Homan, M.; Faber, K.N.; Moshage, H. Metformin protects primary rat hepatocytes against oxidative stress-induced apoptosis. Pharm. Res. Perspect. 2015, 3, 1–12. [Google Scholar] [CrossRef]
- Geng, Y.; Villanueva, A.H.; Oun, A.; Buist-Homan, M.; Blokzijl, H.; Faber, K.N.; Dolga, A.; Moshage, H. Protective effect of metformin against palmitate-induced hepatic cell death. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165621. [Google Scholar] [CrossRef]
- Rouabhia, S.; Milic, N.; Abenavoli, L. Metformin in the treatment of non-alcoholic fatty liver disease: Safety, efficacy and mechanism. Expert Rev. Gastroenterol. Hepatol. 2014, 8, 343–349. [Google Scholar] [CrossRef]
- Suraweera, T.L.; Rupasinghe, H.P.V.; Dellaire, G.; Xu, Z. Regulation of Nrf2/are pathway by dietary flavonoids: A friend or foe for cancer management? Antioxidants 2020, 9, 973. [Google Scholar] [CrossRef]
- Parhiz, H.; Roohbakhsh, A.; Soltani, F.; Rezaee, R.; Iranshahi, M. Antioxidant and anti-inflammatory properties of the citrus flavonoids hesperidin and hesperetin: An updated review of their molecular mechanisms and experimental models. Phyther. Res. 2015, 29, 323–331. [Google Scholar] [CrossRef]
- Bai, X.; Yang, P.; Zhou, Q.; Cai, B.; Buist-Homan, M.; Cheng, H.; Jiang, J.; Shen, D.; Li, L.; Luo, X.; et al. The protective effect of the natural compound hesperetin against fulminant hepatitis in vivo and in vitro. Br. J. Pharm. 2017, 174, 41–56. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Li, X.F.; Chen, Y.; Zhu, S.; Li, H.D.; Chen, S.Y.; Wang, J.N.; Pan, X.Y.; Bu, F.T.; Huang, C.; et al. Hesperetin derivative attenuates CCl4-induced hepatic fibrosis and inflammation by Gli-1-dependent mechanisms. Int. Immunopharmacol. 2019, 76, 105838. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Ding, H.W.; Li, H.D.; Huang, H.M.; Li, X.F.; Yang, Y.; Zhang, Y.L.; Pan, X.Y.; Huang, C.; Meng, X.M.; et al. Hesperetin derivative-14 alleviates inflammation by activating PPAR-γ in mice with CCl4-induced acute liver injury and LPS-treated RAW264.7 cells. Toxicol. Lett. 2017, 274, 51–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheraghpour, M.; Imani, H.; Ommi, S.; Alavian, S.M.; Karimi-Shahrbabak, E.; Hedayati, M.; Yari, Z.; Hekmatdoost, A. Hesperidin improves hepatic steatosis, hepatic enzymes, and metabolic and inflammatory parameters in patients with nonalcoholic fatty liver disease: A randomized, placebo-controlled, double-blind clinical trial. Phyther. Res. 2019, 33, 2118–2125. [Google Scholar] [CrossRef] [PubMed]
- Ikram, M.; Muhammad, T.; Rehman, S.U.; Khan, A.; Jo, M.G.; Ali, T.; Kim, M.O. Hesperetin Confers Neuroprotection by Regulating Nrf2/TLR4/NF-κB Signaling in an Aβ Mouse Model. Mol. Neurobiol. 2019, 56, 6293–6309. [Google Scholar] [CrossRef] [PubMed]
- Geng, Y.; Wu, Z.; Buist-Homan, M.; Blokzijl, H.; Moshage, H. Hesperetin protects against palmitate-induced cellular toxicity via induction of GRP78 in hepatocytes. Toxicol. Appl. Pharm. 2020, 404, 115183. [Google Scholar] [CrossRef]



{kind=link}
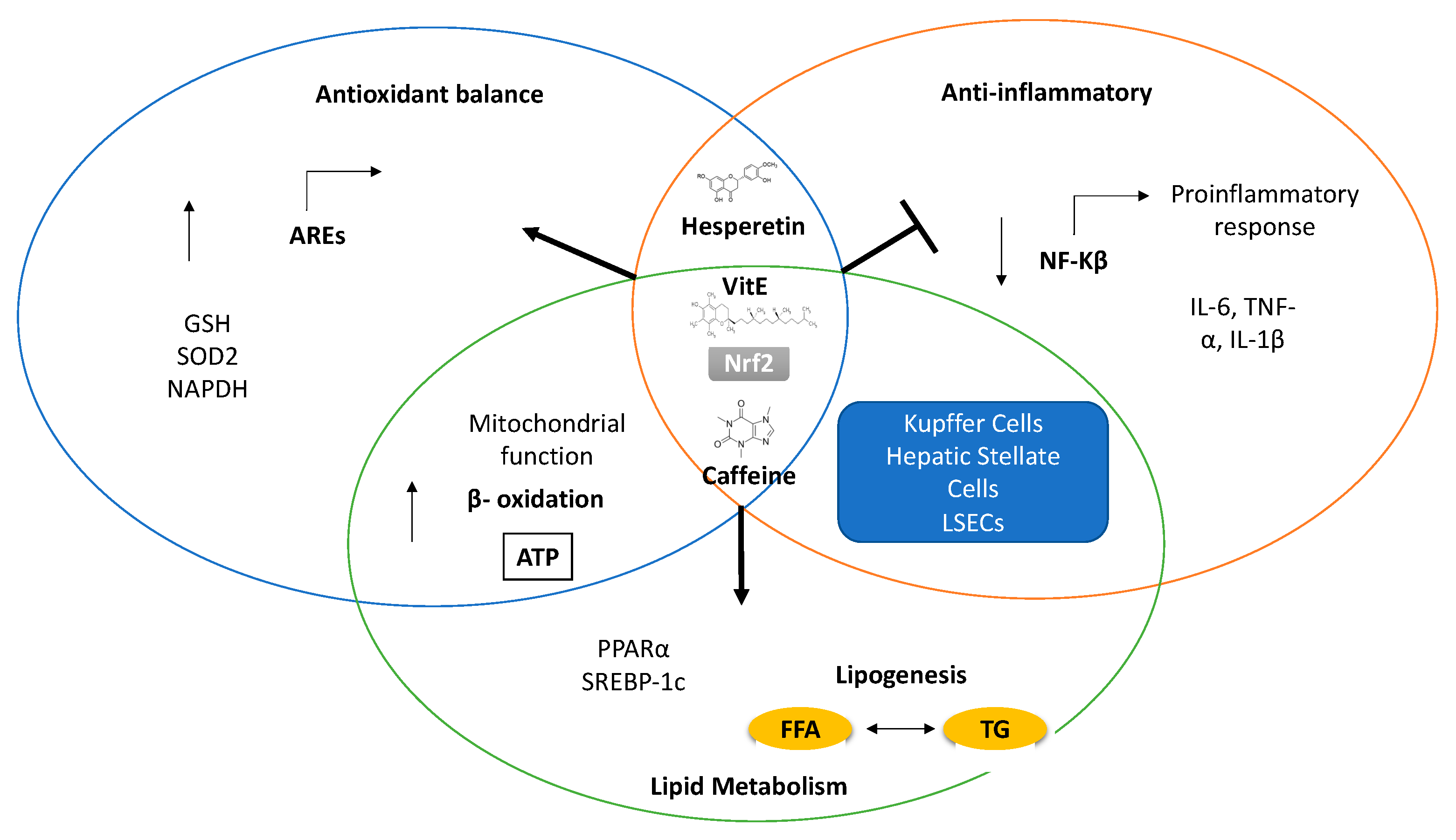{kind=link}
| Antioxidant Compound | Therapeutic Effect | Model | Mechanism | References |
|---|---|---|---|---|
| Vitamin E | Lipid metabolism improvement; Decrease HSC activation | NAFLD patients | Stabilizing free radicals and prevention against lipid peroxidation. -Nrf2 pathway | [148,149,150,151,152,153,154,155,156,157,158,159,160,161,162,163] |
| Vitamin C | Suppress HFD-induced visceral obesity | HFD 1 C57BL/6J mice NAFLD patients | Increased expression PPARα-dependent fatty acid β-oxidation genes. | [164,165,166] |
| Caffeine | Reduces hepatic lipid accumulation | Zebrafish HFD 1 model | -Nrf2 pathway-ER stress prevention -AMPK activation -Increasing fatty oxidation | [176,177,178,179,180,181,182] |
| Coffee polyphenols | Reduce hepatic lipid accumulation, proinflammatory cytokine expression. Attenuated fibrosis | Mice models HFD 1, MCD 2 and CDAA 3 | Nrf2-ARE pathway activation -Lipogenesis regulation -NLRP3 inflammasome -ER stress and apoptosis protection | [184,185,186,187] |
| Metformin | protection against palmitate cell death. | Primary Rat hepatocytes HepG2 | -Decrease ROS production -Increase SOD expression -mitochondrial restoration -Autophagy induction through AMPK | [188,189,190,191,192,193,194,195,196,197] |
| Hesperetin | protection against palmitate cell death | Primary Rat Hepatocytes HepG2 | -ERK/Nrf2 Induction-ER stress protection -NF-kB modulation | [198,199,200,201,202,203,204,205,206] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arroyave-Ospina, J.C.; Wu, Z.; Geng, Y.; Moshage, H. Role of Oxidative Stress in the Pathogenesis of Non-Alcoholic Fatty Liver Disease: Implications for Prevention and Therapy. Antioxidants 2021, 10, 174. https://doi.org/10.3390/antiox10020174
Arroyave-Ospina JC, Wu Z, Geng Y, Moshage H. Role of Oxidative Stress in the Pathogenesis of Non-Alcoholic Fatty Liver Disease: Implications for Prevention and Therapy. Antioxidants. 2021; 10(2):174. https://doi.org/10.3390/antiox10020174
Chicago/Turabian StyleArroyave-Ospina, Johanna C., Zongmei Wu, Yana Geng, and Han Moshage. 2021. "Role of Oxidative Stress in the Pathogenesis of Non-Alcoholic Fatty Liver Disease: Implications for Prevention and Therapy" Antioxidants 10, no. 2: 174. https://doi.org/10.3390/antiox10020174
APA StyleArroyave-Ospina, J. C., Wu, Z., Geng, Y., & Moshage, H. (2021). Role of Oxidative Stress in the Pathogenesis of Non-Alcoholic Fatty Liver Disease: Implications for Prevention and Therapy. Antioxidants, 10(2), 174. https://doi.org/10.3390/antiox10020174