
Changes in Glutathione Content in Liver Diseases: An Update

 ,
,  ,
,  and
and 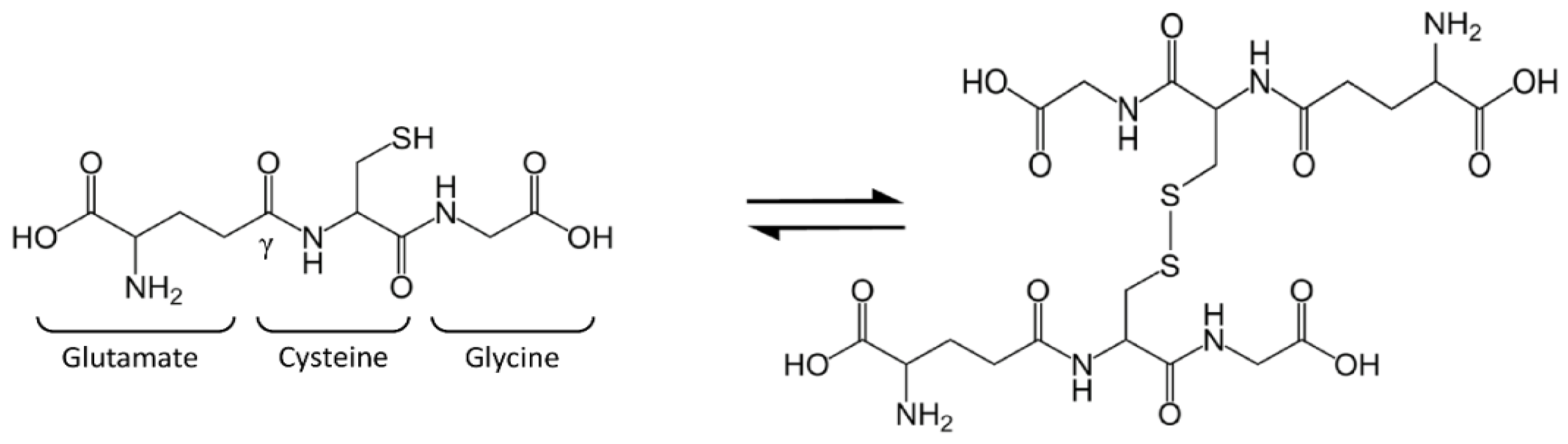{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Glutathione Synthesis and Metabolism
3. Glutathione Functions
3.1. Direct Antioxidant Activity
3.2. Detoxification of Endogenous Compounds and Xenobiotics
3.3. GSH in Protein Glutathionylation
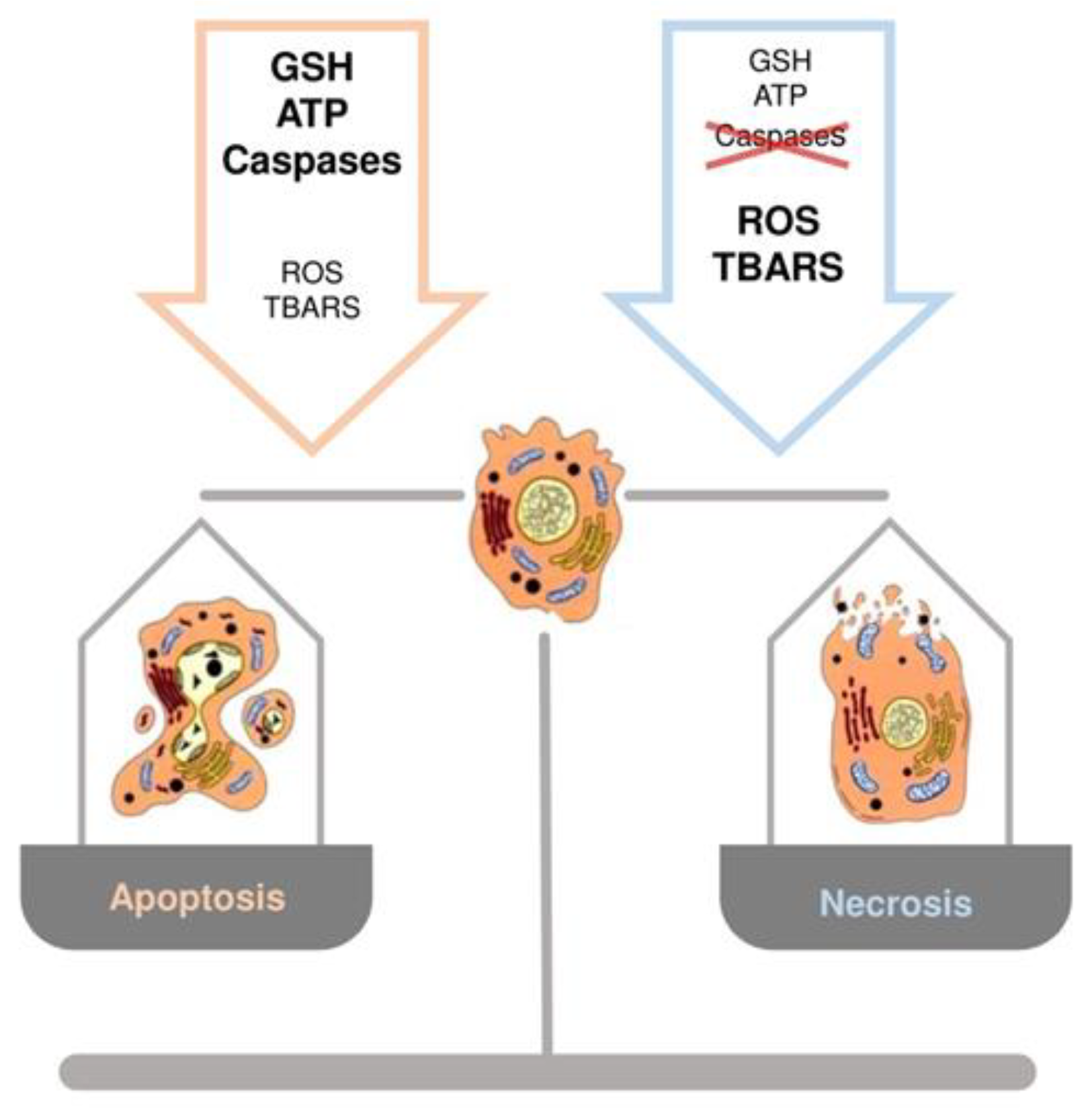
3.4. Hepatocyte Cell Death and GSH: Switch between Necrosis and Apoptosis
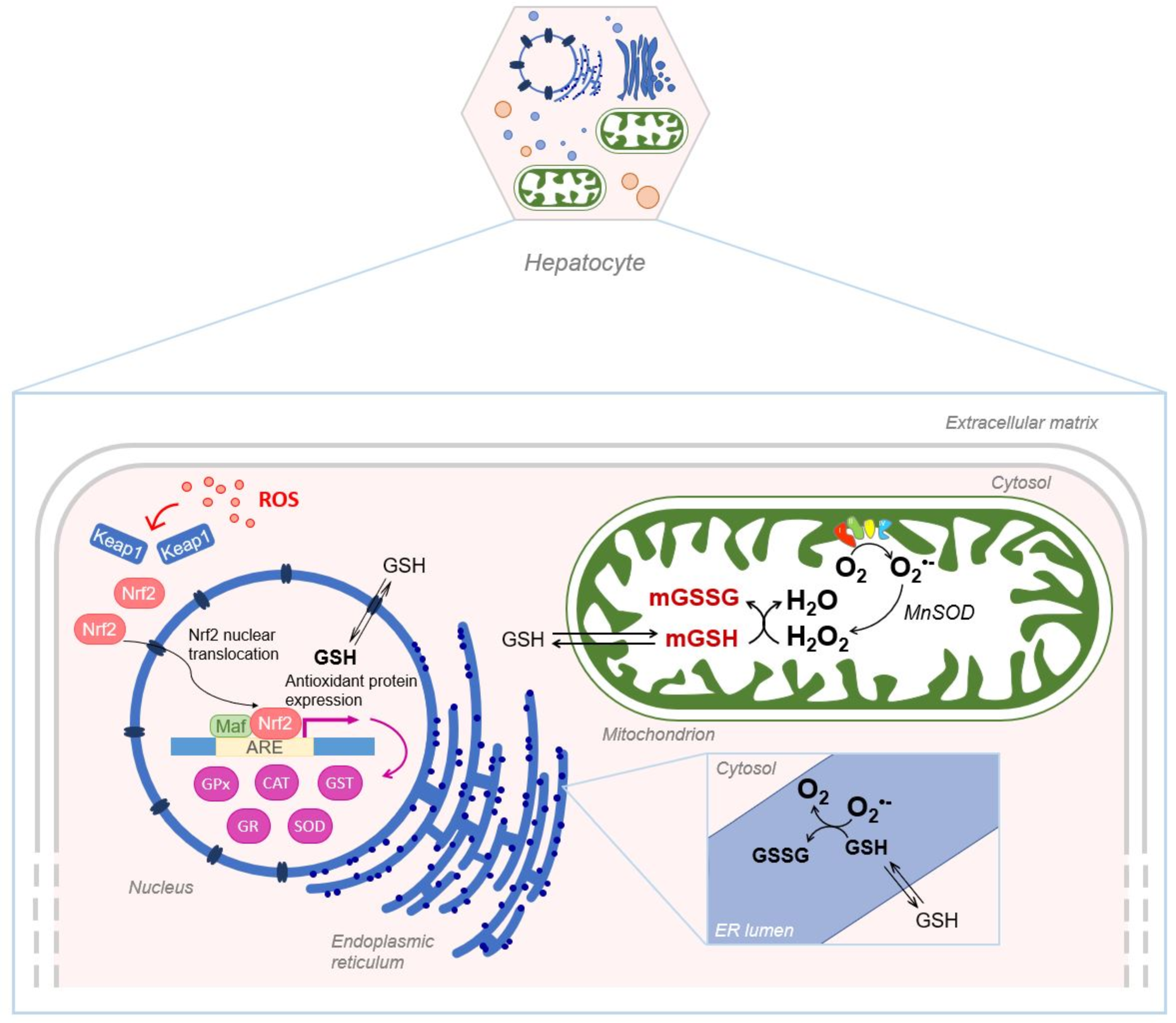
4. Liver and Intracellular Glutathione Compartmentalization
4.1. Nuclear Glutathione
4.2. Mitochondrial Glutathione
4.3. Endoplasmatic Reticulum Glutathione
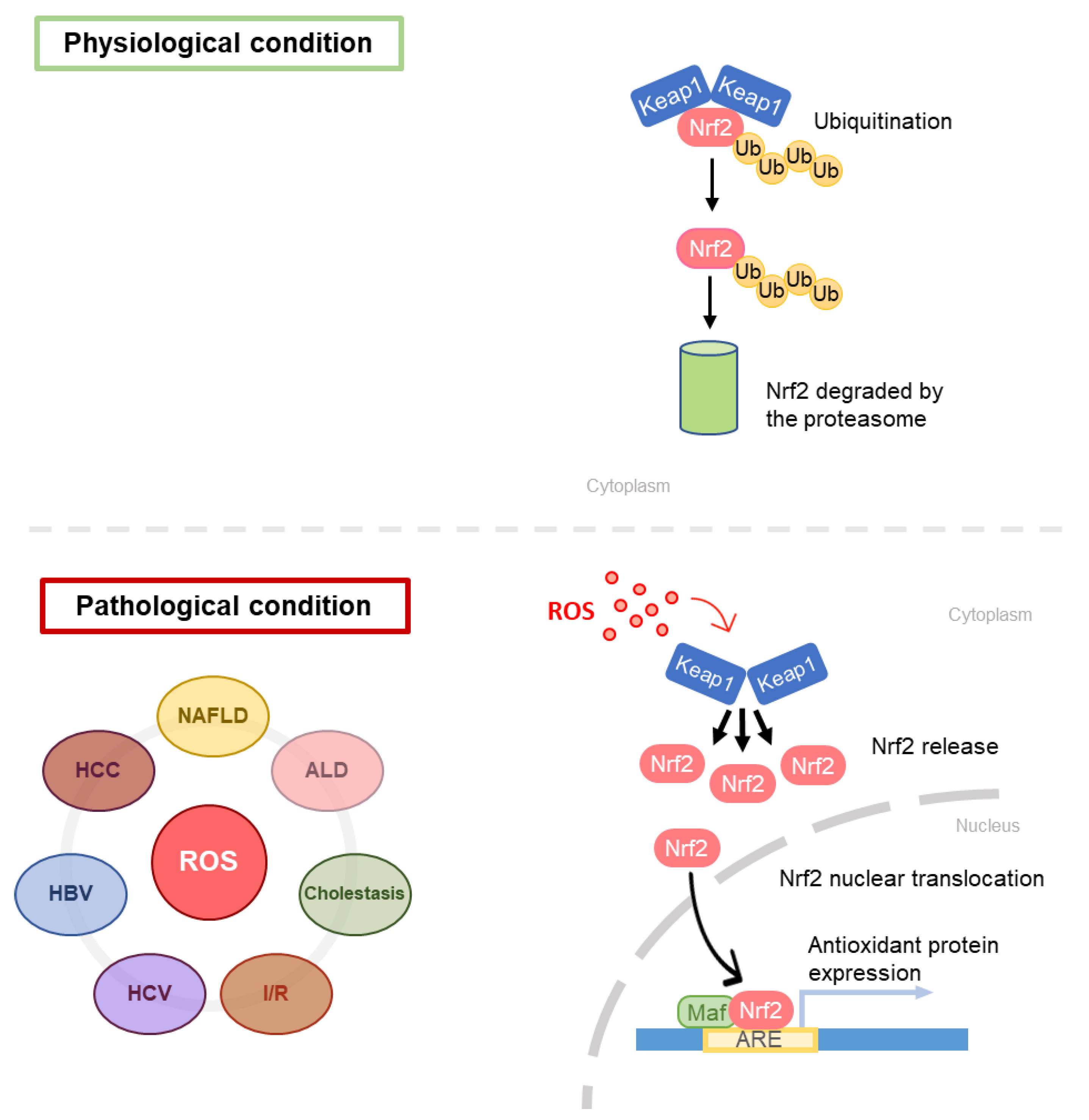
5. Glutathione and Liver Diseases
5.1. Non-Alcoholic Fatty Liver Disease
GSH in the Therapy of NAFLD
5.2. Alcoholic Liver Disease (ALD)
Antioxidant Approach to the Treatment of ALD
5.3. Chronic Cholestatic Liver Injury
Antioxidant Approach for the Treatment of Cholestasis
5.4. Hepatic Ischemia/Reperfusion Injury
Antioxidant Approach for the Treatment of Ischemia/Reperfusion Injury
5.5. Hepatitis C Virus (HCV) and Hepatitis B Virus (HBV)
Antioxidant Approach for the Treatment of Viral Hepatitis C and B
5.6. Hepatocellular Carcinoma (HCC)
Antioxidant Approach for the Treatment of Hepatocellular Carcinoma
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 2-AG | 2-arachidonoylglycerol |
| 4-HNE | 4-hydroxynonenal |
| ADH | Alcohol dehydrogenase |
| Akt | Protein kinase B |
| ALD | Alcoholic liver disease |
| ALDH | Aldehyde dehydrogenase |
| ALT | Alanine aminotransferase |
| AP-1 | Activator protein 1 |
| APP | Acute phase protein |
| ARE | Antioxidant response element |
| ASH | Alcoholic steatohepatitis |
| ATP | Adenosine triphosphate |
| BBS | Bombesin |
| BDL | Bile duct ligation |
| Bhe | Blue honeysuckle extract |
| CAT | Catalase |
| CB1R | Cannabinoid receptor-1 |
| CHB | Chronic hepatitis B |
| CYP2E1 | Cytochrome P450 2E1 |
| DIC | Dicarboxylate carrier |
| DILI | Drug-induced liver injury |
| DNMT | DNA methyltransferase |
| DTT | Dithiothreitol |
| ER | Endoplasmic reticulum |
| ETC | Electron transport chain |
| FA | Fatty acid |
| FCCP | carbonylcyanide p-(trifluoromethoxy)phenylhydrazone |
| GCL | l-glutamate l-cysteine γ-ligase |
| GCLC | GCL catalytic monomer |
| GCLM | GCL modifier monomer |
| GPx | Glutathione peroxide |
| GR | Glutathione reductase |
| Grx | Glutaredoxin |
| GS | GSH synthetase |
| GSH | Reduced glutathione |
| GSSH | Oxidized glutathione |
| GST | Glutathione S transferase |
| HBV | Hepatitis B virus |
| HCC | Hepatocellular carcinoma |
| HCV | Hepatitis C virus |
| HIF-1α | Hypoxia-inducible factor 1 alpha |
| HO-1 | Heme oxygenase-1 |
| HPE | Hypericum perforatum L. |
| HSP90 | Heat shock protein 90 |
| I/R | Ischemia/reperfusion |
| IL-6 | Interleukin-6 |
| IMM | Inner mitochondrial membrane |
| IR | Insulin resistance |
| JNK | c-Jun N-terminal kinases |
| Keap1 | Kelch-like ECH-associated protein 1 |
| LPS | Lipopolysaccharide |
| MAF | Musculoaponeurotic fibrosarcoma oncogene homolog (avian) |
| MAPK | Mitogen-activated protein kinase |
| MAT1A | Methionine adenosyltransferase-1 |
| Mcp1 | Monocyte chemoattractant protein 1 |
| MDA | Malondialdehyde |
| MEOS | Microsomal ethanol-oxidizing system |
| mGSH | Mitochondrial glutathione |
| MnSOD | Manganese-dependent superoxide dismutase |
| mTOR | Mammalian target of rapamycin |
| MTP | Microsomal triglyceride transfer protein |
| NAC | N-acetylcysteine |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| NAFLD | Non-alcoholic fatty liver disease |
| NAPQI | N-acetyl-p-bemzoquinone imine |
| NASH | Non-alcoholic steatohepatitis |
| NF-kB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NO | Nitric oxide |
| NOX | NADPH oxidase |
| NQO1 | NAD(P)H:quinone oxidoreductase 1 |
| Nrf2 | Nuclear factor erythroid 2-related factor 2 |
| NT | Neurotensin |
| OGC | 2-oxoglutarate carrier |
| OMM | Outer mitochondrial membrane |
| PAI1 | Plasminogen activator inhibitor 1 |
| PDI | Disulfide isomerase protein family |
| PI3K | Phosphoinositide 3-kinase |
| RNS | Reactive nitrogen species |
| ROS | Reactive oxygen species |
| SAM | S-adenosyl-l-methionine |
| SLG | S-d-lactoylglutathione |
| SOD | Superoxide dismutase |
| SREBP-1c | Sterol regulatory element-binding protein 1C |
| TBARS | Thiobarbituric acid reactive substances |
| TNF-α | Tumor necrosis factor alpha |
| UDCA | Ursodeoxycholic acid |
| WD | Western diet |
| xCT | Cystine/glutamate antiporter |
| γ-GT | Gamma-glutamyltransferase |
References
- Pizzorno, J. Glutathione! Integr. Med. 2014, 13, 8–12. [Google Scholar]
- Lu, S.C. Glutathione synthesis. Biochim. Biophys. Acta Gen. Subj. 2013, 1830, 3143–3153. [Google Scholar] [CrossRef] [Green Version]
- Lushchak, V.I. Glutathione Homeostasis and Functions: Potential Targets for Medical Interventions. J. Amino Acids 2012, 2012, 1–26. [Google Scholar] [CrossRef] [Green Version]
- Zitka, O.; Skalickova, S.; Gumulec, J.; Masarik, M.; Adam, V.; Hubalek, J.; Trnkova, L.; Kruseova, J.; Eckschlager, T.; Kizek, R. Redox status expressed as GSH:GSSG ratio as a marker for oxidative stress in paediatric tumour patients. Oncol. Lett. 2012, 4, 1247–1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chai, Y.C.; Ashraf, S.S.; Rokutan, K.; Johnston, R.B.; Thomas, J.A. S-thiolation of individual human neutrophil proteins including actin by stimulation of the respiratory burst: Evidence against a role for glutathione disulfide. Arch. Biochem. Biophys. 1994, 310, 273–281. [Google Scholar] [CrossRef]
- Samuele, A.; Mangiagalli, A.; Armentero, M.-T.; Fancellu, R.; Bazzini, E.; Vairetti, M.; Ferrigno, A.; Richelmi, P.; Nappi, G.; Blandini, F. Oxidative stress and pro-apoptotic conditions in a rodent model of Wilson’s disease. Biochim. Biophys. Acta 2005, 1741, 325–330. [Google Scholar] [CrossRef] [Green Version]
- Ferrigno, A.; Rizzo, V.; Bianchi, A.; Di Pasqua, L.G.; Berardo, C.; Richelmi, P.; Vairetti, M. Changes in ADMA/DDAH Pathway after Hepatic Ischemia/Reperfusion Injury in Rats: The Role of Bile. Biomed. Res. Int. 2014, 2014, 627434. [Google Scholar] [CrossRef] [Green Version]
- Vairetti, M.; Ferrigno, A.; Rizzo, V.; Richelmi, P.; Cillo, U.; Imberti, R. Liver damage during ischemia/reperfusion and glutathione: Implications for potential organ donors. Transplant. Proc. 2007, 39, 1768–1770. [Google Scholar] [CrossRef]
- Vairetti, M.; Ferrigno, A.; Bertone, R.; Richelmi, P.; Bertè, F.; Freitas, I. Apoptosis vs. necrosis: Glutathione-mediated cell death during rewarming of rat hepatocytes. Biochim. Biophys. Acta 2005, 1740, 367–374. [Google Scholar] [CrossRef] [Green Version]
- Aquilano, K.; Baldelli, S.; Ciriolo, M.R. Glutathione: New roles in redox signalling for an old antioxidant. Front. Pharmacol. 2014, 5, 196. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.P.; Sies, H. The Redox Code. Antioxid. Redox Signal. 2015, 23, 734–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silvagno, F.; Vernone, A.; Pescarmona, G.P. The role of glutathione in protecting against the severe inflammatory response triggered by covid-19. Antioxidants 2020, 9, 624. [Google Scholar] [CrossRef] [PubMed]
- Ookhtens, M.; Kaplowitz, N. Role of the Liver in Interorgan Homeostasis of Glutathione and Cyst(e)ine. Semin. Liver Dis. 1998, 18, 313–329. [Google Scholar] [CrossRef]
- Richie, J.P.; Komninou, D.; Leutzinger, Y.; Kleinman, W.; Orentreich, N.; Malloy, V.; Zimmerman, J.A. Tissue glutathione and cysteine levels in methionine-restricted rats. Nutrition 2004, 20, 800–805. [Google Scholar] [CrossRef]
- Maddineni, S.; Nichenametla, S.; Sinha, R.; Wilson, R.P.; Richie, J.P. Methionine restriction affects oxidative stress and glutathione-related redox pathways in the rat. Exp. Biol. Med. 2013, 238, 392–399. [Google Scholar] [CrossRef]
- Solis, W.A.; Dalton, T.P.; Dieter, M.Z.; Freshwater, S.; Harrer, J.M.; He, L.; Shertzer, H.G.; Nebert, D.W. Glutamate-cysteine ligase modifier subunit: Mouse Gclm gene structure and regulation by agents that cause oxidative stress. Biochem. Pharmacol. 2002, 63, 1739–1754. [Google Scholar] [CrossRef]
- Neumann, C.A.; Cao, J.; Manevich, Y. Peroxiredoxin 1 and its role in cell signaling. Cell Cycle 2009, 8, 4072–4078. [Google Scholar] [CrossRef] [Green Version]
- Soriano, F.X.; Baxter, P.; Murray, L.M.; Sporn, M.B.; Gillingwater, T.H.; Hardingham, G.E. Transcriptional regulation of the AP-1 and Nrf2 target gene sulfiredoxin. Mol. Cells 2009, 27, 279–282. [Google Scholar] [CrossRef] [Green Version]
- Hayes, J.D.; Chanas, S.A.; Henderson, C.J.; McMahon, M.; Sun, C.; Moffat, G.J.; Wolf, C.R.; Yamamoto, M. The Nrf2 transcription factor contributes both to the basal expression of glutathione S-transferases in mouse liver and to their induction by the chemopreventive synthetic antioxidants, butylated hydroxyanisole and ethoxyquin. In Proceedings of the Biochemical Society Transactions; Portland Press Ltd.: London, UK, 2000; Volume 28, pp. 33–41. [Google Scholar]
- Kobayashi, A.; Kang, M.-I.; Okawa, H.; Ohtsuji, M.; Zenke, Y.; Chiba, T.; Igarashi, K.; Yamamoto, M. Oxidative Stress Sensor Keap1 Functions as an Adaptor for Cul3-Based E3 Ligase To Regulate Proteasomal Degradation of Nrf2. Mol. Cell. Biol. 2004, 24, 7130–7139. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.D.; Lo, S.-C.; Cross, J.V.; Templeton, D.J.; Hannink, M. Keap1 Is a Redox-Regulated Substrate Adaptor Protein for a Cul3-Dependent Ubiquitin Ligase Complex. Mol. Cell. Biol. 2004, 24, 10941–10953. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Fu, J.; Sun, J.; Liu, D.; Chen, C.; Wang, H.; Hou, Y.; Xu, Y.; Pi, J. Is Nrf2-ARE a potential target in NAFLD mitigation? Curr. Opin. Toxicol. 2019, 13, 35–44. [Google Scholar] [CrossRef]
- Grant, C.M.; MacIver, F.H.; Dawes, I.W. Glutathione synthetase is dispensable for growth under both normal and oxidative stress conditions in the yeast Saccharomyces cerevisiae due to an accumulation of the dipeptide γ-glutamylcysteine. Mol. Biol. Cell 1997, 8, 1699–1707. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Forman, H.J.; Choi, J. γ-glutamyl transpeptidase in glutathione biosynthesis. Methods Enzymol. 2005, 401, 468–483. [Google Scholar] [PubMed]
- Pompella, A.; Corti, A.; Paolicchi, A.; Giommarelli, C.; Zunino, F. γ-Glutamyltransferase, redox regulation and cancer drug resistance. Curr. Opin. Pharmacol. 2007, 7, 360–366. [Google Scholar] [CrossRef] [PubMed]
- Qian, S.; Mumick, S.; Nizner, P.; Tota, M.R.; Menetski, J.; Reitman, M.L.; MacNeil, D.J. Deficiency in cytosolic malic enzyme does not increase acetaminophen- induced hepato-toxicity. Basic Clin. Pharmacol. Toxicol. 2008, 103, 36–42. [Google Scholar] [CrossRef]
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging, and diseases. Clin. Interv. Aging 2018, 13, 757–772. [Google Scholar] [CrossRef] [Green Version]
- Berardo, C.; Di Pasqua, L.G.; Cagna, M.; Richelmi, P.; Vairetti, M.; Ferrigno, A. Nonalcoholic Fatty Liver Disease and Non-Alcoholic Steatohepatitis: Current Issues and Future Perspectives in Preclinical and Clinical Research. Int. J. Mol. Sci. 2020, 21, 9646. [Google Scholar] [CrossRef]
- Sengupta, R.; Holmgren, A. Thioredoxin and thioredoxin reductase in relation to reversible S-nitrosylation. Antioxid. Redox Signal. 2013, 18, 259–269. [Google Scholar] [CrossRef]
- Allocati, N.; Masulli, M.; Di Ilio, C.; Federici, L. Glutathione transferases: Substrates, inihibitors and pro-drugs in cancer and neurodegenerative diseases. Oncogenesis 2018, 7, 8. [Google Scholar] [CrossRef]
- Labrou, N.E.; Papageorgiou, A.C.; Pavli, O.; Flemetakis, E. Plant GSTome: Structure and functional role in xenome network and plant stress response. Curr. Opin. Biotechnol. 2015, 32, 186–194. [Google Scholar] [CrossRef]
- Kumar, S.; Trivedi, P.K. Glutathione S-transferases: Role in combating abiotic stresses including arsenic detoxification in plants. Front. Plant Sci. 2018, 9, 751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid. Med. Cell. Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef]
- Mattson, M.P. Roles of the lipid peroxidation product 4-hydroxynonenal in obesity, the metabolic syndrome, and associated vascular and neurodegenerative disorders. Exp. Gerontol. 2009, 44, 625–633. [Google Scholar] [CrossRef] [Green Version]
- Dalleau, S.; Baradat, M.; Guéraud, F.; Huc, L. Cell death and diseases related to oxidative stress:4-hydroxynonenal (HNE) in the balance. Cell Death Differ. 2013, 20, 1615–1630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dohnal, V.; Wu, Q.; Kuča, K. Metabolism of aflatoxins: Key enzymes and interindividual as well as interspecies differences. Arch. Toxicol. 2014, 88, 1635–1644. [Google Scholar] [CrossRef] [PubMed]
- Stevens, J.F.; Maier, C.S. Acrolein: Sources, metabolism, and biomolecular interactions relevant to human health and disease. Mol. Nutr. Food Res. 2008, 52, 7–25. [Google Scholar] [CrossRef] [Green Version]
- Carmella, S.G.; Chen, M.; Zhang, Y.; Zhang, S.; Hatsukami, D.K.; Hecht, S.S. Quantitation of acrolein-derived (3-hydroxypropyl)mercapturic acid in human urine by liquid chromatography-atmospheric pressure chemical ionization tandem mass spectrometry: Effects of cigarette smoking. Chem. Res. Toxicol. 2007, 20, 986–990. [Google Scholar] [CrossRef] [Green Version]
- Moyer, A.M.; Fridley, B.L.; Jenkins, G.D.; Batzler, A.J.; Pelleymounter, L.L.; Kalari, K.R.; Ji, Y.; Chai, Y.; Nordgren, K.K.S.; Weinshilboum, R.M. Acetaminophen-NAPQI hepatotoxicity: A cell line model system genome-wide association study. Toxicol. Sci. 2011, 120, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Marzullo, L. An update of N-acetylcysteine treatment for acute acetaminophen toxicity in children. Curr. Opin. Pediatr. 2005, 17, 239–245. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, H.F. Molecular and cellular aspects of thiol-disulfide exchange. Adv. Enzymol. Relat. Areas Mol. Biol. 1990, 63, 69–172. [Google Scholar]
- Mailloux, R.J.; Willmore, W.G. S-glutathionylation reactions in mitochondrial function and disease. Front. Cell Dev. Biol. 2014, 2, 68. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Ye, Z.W.; Singh, S.; Townsend, D.M.; Tew, K.D. An evolving understanding of the S-glutathionylation cycle in pathways of redox regulation. Free Radic. Biol. Med. 2018, 120, 204–216. [Google Scholar] [CrossRef]
- Han, D.; Hanawa, N.; Saberi, B.; Kaplowitz, N. Mechanisms of liver injury. III. Role of glutathione redox status in liver injury. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 291, G1–G7. [Google Scholar] [CrossRef] [Green Version]
- Yuan, L.; Kaplowitz, N. Glutathione in liver diseases and hepatotoxicity. Mol. Asp. Med. 2009, 30, 29–41. [Google Scholar] [CrossRef]
- Vairetti, M.; Griffini, P.; Pietrocola, G.; Richelmi, P.; Freitas, I. Cold-induced apoptosis in isolated rat hepatocytes: Protective role of glutathione. Free Radic. Biol. Med. 2001, 31, 954–961. [Google Scholar] [CrossRef]
- Yin, F.; Sancheti, H.; Cadenas, E. Mitochondrial thiols in the regulation of cell death pathways. Antioxid. Redox Signal. 2012, 17, 1714–1727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marí, M.; de Gregorio, E.; de Dios, C.; Roca-Agujetas, V.; Cucarull, B.; Tutusaus, A.; Morales, A.; Colell, A. Mitochondrial Glutathione: Recent Insights and Role in Disease. Antioxidants 2020, 9, 909. [Google Scholar] [CrossRef]
- Colell, A.; Garcia-Ruiz, C.; Miranda, M.; Ardite, E.; Mari, M.; Morales, A.; Corrales, F.; Kaplowitz, N.; Fernandez-Checa, J.C. Selective glutathione depletion of mitochondria by ethanol sensitizes hepatocytes to tumor necrosis factor. Gastroenterology 1998, 115, 1541–1551. [Google Scholar] [CrossRef]
- Abei, M.; Harada, S.; Tanaka, N.; McNeil, M.; Osuga, T. Immunohistochemical localization of human liver glutathione S-transferase (GST) isozymes with special reference to polymorphic GST1. Biochim. Biophys. Acta (BBA) Protein Struct. Mol. 1989, 995, 279–284. [Google Scholar] [CrossRef]
- Bellomo, G.; Vairetti, M.; Stivala, L.; Mirabelli, F.; Richelmi, P.; Orrenius, S. Demonstration of nuclear compartmentalization of glutathione in hepatocytes. Proc. Natl. Acad. Sci. USA 1992, 89, 4412–4416. [Google Scholar] [CrossRef] [Green Version]
- Scirè, A.; Cianfruglia, L.; Minnelli, C.; Bartolini, D.; Torquato, P.; Principato, G.; Galli, F.; Armeni, T. Glutathione compartmentalization and its role in glutathionylation and other regulatory processes of cellular pathways. BioFactors 2019, 45, 152–168. [Google Scholar] [CrossRef]
- Bellomo, G.; Palladini, G.; Vairetti, M. Intranuclear distribution, function and fate of glutathione and glutathione-S-conjugate in living rat hepatocytes studied by fluorescence microscopy. Microsc. Res. Tech. 1997, 36, 243–252. [Google Scholar] [CrossRef]
- Yeh, H.I.; Hsieh, C.H.; Wang, L.Y.; Tsai, S.P.; Hsu, H.Y.; Tam, M.F. Mass spectrometric analysis of rat liver cytosolic glutathione S-transferases: Modifications are limited to N-terminal processing. Biochem. J. 1995, 308, 69–75. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, J.Z.; De Maria, F.; Turella, P.; Federici, G.; Mattei, M.; Fabrini, R.; Dawood, K.F.; Massimi, M.; Caccuri, A.M.; Ricci, G. Glutathione transferases sequester toxic dinitrosyl-iron complexes in cells: A protection mechanism against excess nitric oxide. J. Biol. Chem. 2007, 282, 6364–6371. [Google Scholar] [CrossRef] [Green Version]
- Stella, L.; Pallottini, V.; Moreno, S.; Leoni, S.; De Maria, F.; Turella, P.; Federici, G.; Fabrini, R.; Dawood, K.F.; Lo Bello, M.; et al. Electrostatic association of glutathione transferase to the nuclear membrane: Evidence of an enzyme defense barrier at the nuclear envelope. J. Biol. Chem. 2007, 282, 6372–6379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soboll, S.; Grundel, S.; Harris, J.; Kolb-Bachofen, V.; Ketterer, B.; Sies, H. The content of glutathione and glutathione S-transferases and the glutathione peroxidase activity in rat liver nuclei determined by a non-aqueous technique of cell fractionation. Biochem. J. 1995, 311, 889–894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres, L.; Sandoval, J.; Penella, E.; Zaragozá, R.; García, C.; Rodríguez, J.L.; Viña, J.R.; García-Trevijano, E.R. In vivo GSH depletion induces c-myc expression by modulation of chromatin protein complexes. Free Radic. Biol. Med. 2009, 46, 1534–1542. [Google Scholar] [CrossRef] [PubMed]
- García-Giménez, J.L.; Markovic, J.; Dasí, F.; Queval, G.; Schnaubelt, D.; Foyer, C.H.; Pallardó, F.V. Nuclear glutathione. Biochim. Biophys. Acta Gen. Subj. 2013, 1830, 3304–3316. [Google Scholar] [CrossRef]
- Brown, K.E.; Meleah Mathahs, M.; Broadhurst, K.A.; Coleman, M.C.; Ridnour, L.A.; Schmidt, W.N.; Spitz, D.R. Increased hepatic telomerase activity in a rat model of iron overload: A role for altered thiol redox state? Free Radic. Biol. Med. 2007, 42, 228–235. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Checa, J.C.; Kaplowitz, N. Hepatic mitochondrial glutathione: Transport and role in disease and toxicity. Toxicol. Appl. Pharmacol. 2005, 204, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Kojer, K.; Bien, M.; Gangel, H.; Morgan, B.; Dick, T.P.; Riemer, J. Glutathione redox potential in the mitochondrial intermembrane space is linked to the cytosol and impacts the Mia40 redox state. EMBO J. 2012, 31, 3169–3182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martensson, J.; Lai, J.C.K.; Meister, A. High-affinity Transport of Glutathione is Part of a Multicomponent System Essential for Mitochondrial Function. Proc. Natl. Acad. Sci. USA 1990, 87, 7185–7189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colell, A.; García-Ruiz, C.; Morales, A.; Ballesta, A.; Ookhtens, M.; Rodés, J.; Kaplowitz, N.; Fernández-Checa, J.C. Transport of reduced glutathione in hepatic mitochondria and mitoplasts from ethanol-treated rats: Effect of membrane physical properties and S-adenosyl-L-methionine. Hepatology 1997, 26, 699–708. [Google Scholar] [CrossRef]
- Zhong, Q.; Putt, D.A.; Xu, F.; Lash, L.H. Hepatic mitochondrial transport of glutathione: Studies in isolated rat liver mitochondria and H4IIE rat hepatoma cells. Arch. Biochem. Biophys. 2008, 474, 119–127. [Google Scholar] [CrossRef] [Green Version]
- Armeni, T.; Cianfruglia, L.; Piva, F.; Urbanelli, L.; Luisa Caniglia, M.; Pugnaloni, A.; Principato, G. S-D-Lactoylglutathione can be an alternative supply of mitochondrial glutathione. Free Radic. Biol. Med. 2014, 67, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Lluis, J.M.; Colell, A.; García-Ruiz, C.; Kaplowitz, N.; Fernández-Checa, J.C. Acetaldehyde impairs mitochondrial glutathione transport in HepG2 cells through endoplasmic reticulum stress. Gastroenterology 2003, 124, 708–724. [Google Scholar] [CrossRef]
- Coll, O.; Colell, A.; García-Ruiz, C.; Kaplowitz, N.; Fernández-Checa, J.C. Sensitivity of the 2-oxoglutarate carrier to alcohol intake contributes to mitochondrial glutathione depletion. Hepatology 2003, 38, 692–702. [Google Scholar] [CrossRef] [Green Version]
- Hayes, J.D.; Flanagan, J.U.; Jowsey, I.R. Glutathione transferases. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 51–88. [Google Scholar] [CrossRef] [PubMed]
- Gladyshev, V.N.; Liu, A.; Novoselov, S.V.; Krysan, K.; Sun, Q.A.; Kryukov, V.M.; Kryukov, G.V.; Lou, M.F. Identification and Characterization of a New Mammalian Glutaredoxin (Thioltransferase), Grx2. J. Biol. Chem. 2001, 276, 30374–30380. [Google Scholar] [CrossRef] [Green Version]
- Lluis, J.M.; Morales, A.; Blasco, C.; Colell, A.; Mari, M.; Garcia-Ruiz, C.; Fernandez-Checa, J.C. Critical role of mitochondrial glutathione in the survival of hepatocytes during hypoxia. J. Biol. Chem. 2005, 280, 3224–3232. [Google Scholar] [CrossRef] [Green Version]
- Görlach, A.; Klappa, P.; Kietzmann, T. The endoplasmic reticulum: Folding, calcium homeostasis, signaling, and redox control. Antioxid. Redox Signal. 2006, 8, 1391–1418. [Google Scholar] [CrossRef]
- Ellgaard, L. Catalysis of disulphide bond formation in the endoplasmic reticulum. Biochem. Soc. Trans. 2004, 32, 663–667. [Google Scholar] [CrossRef]
- Molteni, S.N.; Fassio, A.; Ciriolo, M.R.; Filomeni, G.; Pasqualetto, E.; Fagioli, C.; Sitia, R. Glutathione limits Ero1-dependent oxidation in the endoplasmic reticulum. J. Biol. Chem. 2004, 279, 32667–32673. [Google Scholar] [CrossRef] [Green Version]
- Kojer, K.; Riemer, J. Balancing oxidative protein folding: The influences of reducing pathways on disulfide bond formation. Biochim. Biophys. Acta Proteins Proteom. 2014, 1844, 1383–1390. [Google Scholar] [CrossRef]
- Bánhegyi, G.; Lusini, L.; Puskás, F.; Rossi, R.; Fulceri, R.; Braun, L.; Mile, V.; Di Simplicio, P.; Mandl, J.; Benedetti, A. Preferential transport of glutathione versus glutathione disulfide in rat liver microsomal vesicles. J. Biol. Chem. 1999, 274, 12213–12216. [Google Scholar] [CrossRef] [Green Version]
- Appenzeller-Herzog, C. Glutathione- and non-glutathione-based oxidant control in the endoplasmic reticulum. J. Cell Sci. 2011, 124, 847–855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef] [PubMed]
- Nassir, F.; Rector, R.S.; Hammoud, G.M.; Ibdah, J.A. Pathogenesis and prevention of hepatic steatosis. Gastroenterol. Hepatol. 2015, 11, 167–175. [Google Scholar]
- Peverill, W.; Powell, L.W.; Skoien, R. Evolving concepts in the pathogenesis of NASH: Beyond steatosis and inflammation. Int. J. Mol. Sci. 2014, 15, 8591–8638. [Google Scholar] [CrossRef] [PubMed]
- Pierantonelli, I.; Svegliati-Baroni, G. Nonalcoholic Fatty Liver Disease: Basic Pathogenetic Mechanisms in the Progression from NAFLD to NASH. Transplantation 2019, 103, e1–e13. [Google Scholar] [CrossRef] [PubMed]
- Jeznach-Steinhagen, A.; Ostrowska, J.; Czerwonogrodzka-Senczyna, A.; Boniecka, I.; Shahnazaryan, U.; Kuryłowicz, A. Dietary and pharmacological treatment of nonalcoholic fatty liver disease. Medicina 2019, 55, 166. [Google Scholar] [CrossRef] [Green Version]
- Moon, Y.-A. The SCAP/SREBP Pathway: A Mediator of Hepatic Steatosis. Endocrinol. Metab. 2017, 32, 6. [Google Scholar] [CrossRef] [PubMed]
- Seifert, E.L.; Estey, C.; Xuan, J.Y.; Harper, M.E. Electron transport chain-dependent and -independent mechanisms of mitochondrial H2O2 emission during long-chain fatty acid oxidation. J. Biol. Chem. 2010, 285, 5748–5758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Oxidative stress, cardiolipin and mitochondrial dysfunction in nonalcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 14205–14218. [Google Scholar] [CrossRef] [PubMed]
- Koruk, M.; Taysi, S.; Savas, M.C.; Yilmaz, O.; Akcay, F.; Karakok, M. Oxidative stress and enzymatic antioxidant status in patients with nonalcoholic steatohepatitis. Ann. Clin. Lab. Sci. 2004, 34, 57–62. [Google Scholar]
- Chambel, S.S.; Santos-Gonçalves, A.; Duarte, T.L. The dual role of Nrf2 in nonalcoholic fatty liver disease: Regulation of antioxidant defenses and hepatic lipid metabolism. Biomed. Res. Int. 2015, 2015, 597134. [Google Scholar] [CrossRef] [Green Version]
- Yates, M.S.; Tran, Q.T.; Dolan, P.M.; Osburn, W.O.; Shin, S.; McCulloch, C.C.; Silkworth, J.B.; Taguchi, K.; Yamamoto, M.; Williams, C.R.; et al. Genetic versus chemoprotective activation of Nrf2 signaling: Overlapping yet distinct gene expression profiles between Keap1 knockout and triterpenoid-treated mice. Carcinogenesis 2009, 30, 1024–1031. [Google Scholar] [CrossRef] [Green Version]
- Tsurusaki, S.; Tsuchiya, Y.; Koumura, T.; Nakasone, M.; Sakamoto, T.; Matsuoka, M.; Imai, H.; Yuet-Yin Kok, C.; Okochi, H.; Nakano, H.; et al. Hepatic ferroptosis plays an important role as the trigger for initiating inflammation in nonalcoholic steatohepatitis. Cell Death Dis. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Qi, J.; Kim, J.-W.; Zhou, Z.; Lim, C.-W.; Kim, B. Ferroptosis Affects the Progression of Nonalcoholic Steatohepatitis via the Modulation of Lipid Peroxidation–Mediated Cell Death in Mice. Am. J. Pathol. 2020, 190, 68–81. [Google Scholar] [CrossRef]
- Polyzos, S.A.; Kountouras, J.; Goulas, A.; Duntas, L. Selenium and selenoprotein P in nonalcoholic fatty liver disease. Hormones 2020, 19, 61–72. [Google Scholar] [CrossRef]
- Machado, M.V.; Ravasco, P.; Jesus, L.; Marques-Vidal, P.; Oliveira, C.R.; Proença, T.; Baldeiras, I.; Camilo, M.E.; Cortez-Pinto, H. Blood oxidative stress markers in non-alcoholic steatohepatitis and how it correlates with diet. Scand. J. Gastroenterol. 2008, 43, 95–102. [Google Scholar] [CrossRef]
- Schäfer, K.; Kyriakopoulos, A.; Gessner, H.; Grune, T.; Behne, D. Effects of selenium deficiency on fatty acid metabolism in rats fed fish oil-enriched diets. J. Trace Elem. Med. Biol. 2004, 18, 89–97. [Google Scholar] [CrossRef]
- Wu, J.; Zeng, C.; Yang, Z.; Li, X.; Lei, G.; Xie, D.; Wang, Y.; Wei, J.; Yang, T. Association Between Dietary Selenium Intake and the Prevalence of Nonalcoholic Fatty Liver Disease: A Cross-Sectional Study. J. Am. Coll. Nutr. 2020, 39, 103–111. [Google Scholar] [CrossRef]
- London, R.M.; George, J. Pathogenesis of NASH: Animal models. Clin. Liver Dis. 2007, 11, 55–74. [Google Scholar] [CrossRef] [PubMed]
- Montfort, C.V.; Matias, N.; Fernandez, A.; Fucho, R.; De La Rosa, L.C.; Martinez-Chantar, M.L.; Mato, J.M.; MacHida, K.; Tsukamoto, H.; Murphy, M.P.; et al. Mitochondrial GSH determines the toxic or therapeutic potential of superoxide scavenging in steatohepatitis. J. Hepatol. 2012, 57, 852–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ioannou, G.N. The Role of Cholesterol in the Pathogenesis of NASH. Trends Endocrinol. Metab. 2016, 27, 84–95. [Google Scholar] [CrossRef] [PubMed]
- Marí, M.; Caballero, F.; Colell, A.; Morales, A.; Caballeria, J.; Fernandez, A.; Enrich, C.; Fernandez-Checa, J.C.; García-Ruiz, C. Mitochondrial free cholesterol loading sensitizes to TNF- and Fas-mediated steatohepatitis. Cell Metab. 2006, 4, 185–198. [Google Scholar] [CrossRef]
- Grattagliano, I.; Caraceni, P.; Calamita, G.; Ferri, D.; Gargano, I.; Palasciano, G.; Portincasa, P. Severe liver steatosis correlates with nitrosative and oxidative stress in rats. Eur. J. Clin. Investig. 2008, 38, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Zhu, H.; Li, Y.; Lin, H.; Gabrielson, K.; Trush, M.A.; Diehl, A.M. Mitochondrial adaptations to obesity-related oxidant stress. Arch. Biochem. Biophys. 2000, 378, 259–268. [Google Scholar] [CrossRef]
- Lee, J.I.; Dominy, J.E.; Sikalidis, A.K.; Hirschberger, L.L.; Wang, W.; Stipanuk, M.H. HepG2/C3A cells respond to cysteine deprivation by induction of the amino acid deprivation/integrated stress response pathway. Physiol. Genom. 2008, 33, 218–229. [Google Scholar] [CrossRef] [Green Version]
- De Gottardi, A.; Vinciguerra, M.; Sgroi, A.; Moukil, M.; Ravier-Dall’Antonia, F.; Pazienza, V.; Pugnale, P.; Foti, M.; Hadengue, A. Microarray analyses and molecular profiling of steatosis induction in immortalized human hepatocytes. Lab. Investig. 2007, 87, 792–806. [Google Scholar] [CrossRef]
- Videla, L.A.; Rodrigo, R.; Orellana, M.; Fernandez, V.; Tapia, G.; Quiñones, L.; Varela, N.; Contreras, J.; Lazarte, R.; Csendes, A.; et al. Oxidative stress-related parameters in the liver of non-alcoholic fatty liver disease patients. Clin. Sci. 2004, 106, 261–268. [Google Scholar] [CrossRef] [Green Version]
- Cobbina, E.; Akhlaghi, F. Non-Alcoholic Fatty Liver Disease (NAFLD)-Pathogenesis, Classification, and Effect on Drug Metabolizing Enzymes and Transporters HHS Public Access. Drug Metab. Rev. 2017, 49, 197–211. [Google Scholar] [CrossRef]
- Caro, A.A.; Cederbaum, A.I. Oxidative Stress, Toxicology, and Pharmacology of CYP2E1. Annu. Rev. Pharmacol. Toxicol. 2004, 44, 27–42. [Google Scholar] [CrossRef]
- Singh, R.; Wang, Y.; Schattenberg, J.M.; Xiang, Y.; Czaja, M.J. Chronic oxidative stress sensitizes hepatocytes to death from 4-hydroxynonenal by JNK/c-Jun overactivation. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 297. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.; Cederbaum, A.I. Oxidative stress mediated toxicity exerted by ethanol-inducible CYP2E1. Toxicol. Appl. Pharmacol. 2005, 207 (Suppl. S2), 70–76. [Google Scholar] [CrossRef] [PubMed]
- Honda, Y.; Kessoku, T.; Sumida, Y.; Kobayashi, T.; Kato, T.; Ogawa, Y.; Tomeno, W.; Imajo, K.; Fujita, K.; Yoneda, M.; et al. Efficacy of glutathione for the treatment of nonalcoholic fatty liver disease: An open-label, single-arm, multicenter, pilot study. BMC Gastroenterol. 2017, 17, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Mardinoglu, A.; Bjornson, E.; Zhang, C.; Klevstig, M.; Söderlund, S.; Ståhlman, M.; Adiels, M.; Hakkarainen, A.; Lundbom, N.; Kilicarslan, M.; et al. Personal model-assisted identification of NAD+ and glutathione metabolism as intervention target in NAFLD. Mol. Syst. Biol. 2017, 13, 916. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Han, D.; Xu, R.; Wu, H.; Qu, C.; Wang, F.; Wang, X.; Zhao, Y. Glycine protects against high sucrose and high fat-induced non-alcoholic steatohepatitis in rats. Oncotarget 2016, 7, 80223–80237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.C.; Jiang, Z.M.; Li, D.M. Glutamine: A precursor of glutathione and its effect on liver. World J. Gastroenterol. 1999, 5, 143–146. [Google Scholar] [CrossRef] [PubMed]
- Sellmann, C.; Jin, C.J.; Degen, C.; De Bandt, J.P.; Bergheim, I. Oral Glutamine Supplementation Protects Female Mice from Nonalcoholic Steatohepatitis1-3. J. Nutr. 2015, 145, 2280–2286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sellmann, C.; Baumann, A.; Brandt, A.; Jin, C.J.; Nier, A.; Bergheim, I. Oral supplementation of glutamine attenuates the progression of nonalcoholic steatohepatitis in C57BL/6J mice. J. Nutr. 2017, 147, 2041–2049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Z.; Cai, F.; Lin, N.; Ye, J.; Zheng, Q.; Ding, G. Effects of glutamine on oxidative stress and nuclear factor-κB expression in the livers of rats with nonalcoholic fatty liver disease. Exp. Ther. Med. 2014, 7, 365–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Vries, N.; De Flora, S. N-acetyl-l-cysteine. J. Cell. Biochem. 1993, 53, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Mardinoglu, A.; Ural, D.; Zeybel, M.; Yuksel, H.H.; Uhlén, M.; Borén, J. The potential use of metabolic cofactors in treatment of NAFLD. Nutrients 2019, 11, 1578. [Google Scholar] [CrossRef] [Green Version]
- Thong-Ngam, D.; Samuhasaneeto, S.; Kulaputana, O.; Klaikeaw, N. N-acetylcysteine attenuates oxidative stress and liver pathology in rats with non-alcoholic steatohepatitis. World J. Gastroenterol. 2007, 13, 5127–5132. [Google Scholar] [CrossRef] [Green Version]
- Wellington, K.; Adis, B.J. Silymarin: A review of its clinical properties in the management of hepatic disorders. BioDrugs 2001, 15, 465–489. [Google Scholar] [CrossRef]
- Dixit, N.; Baboota, S.; Kohli, K.; Ahmad, S.; Ali, J. Silymarin: A review of pharmacological aspects and bioavailability enhancement approaches. Indian J. Pharmacol. 2007, 39, 172–179. [Google Scholar] [CrossRef] [Green Version]
- Frazier, T.H.; Mcclain, C.J.; Kershner, N.A.; Marsano, L.S. Treatment of alcoholic liver disease. Therap. Adv. Gastroenterol. 2011, 4, 63–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feher, J.; Lengyel, G. Silymarin in the Prevention and Treatment of Liver Diseases and Primary Liver Cancer. Curr. Pharm. Biotechnol. 2011, 13, 210–217. [Google Scholar] [CrossRef]
- De Freitas Carvalho, M.M.; Lage, N.N.; de Souza Paulino, A.H.; Pereira, R.R.; de Almeida, L.T.; da Silva, T.F.; de Brito Magalhães, C.L.; de Lima, W.G.; Silva, M.E.; Pedrosa, M.L.; et al. Effects of açai on oxidative stress, ER stress, and inflammation-related parameters in mice with high fat diet-fed induced NAFLD. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.W.; Lee, Y.S.; Seol, D.J.; Cho, I.J.; Kwang Ku, S.; Choi, J.S.; Lee, H.J. Anti-obesity and fatty liver-preventing activities of Lonicera caerulea in high-fat diet-fed mice. Int. J. Mol. Med. 2018, 42, 3047–3064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Wang, H.; Zhang, L.; Yuan, Y.; Yu, D. Codonopsis lanceolata polysaccharide CLPS alleviates high fat/high sucrose diet-induced insulin resistance via anti-oxidative stress. Int. J. Biol. Macromol. 2020, 145, 944–949. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, R.P.; Moore, M.P.; Moore, A.N.; Healy, J.C.; Roberts, M.D.; Rector, R.S.; Martin, J.S. Curcumin supplementation mitigates NASH development and progression in female Wistar rats. Physiol. Rep. 2018, 6. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Li, Q.; Chai, W.; Sun, C.; Zhang, T.; Zhao, C.; Yuan, Y.; Wang, X.; Liu, H.; Ye, H. Lactobacillus paracasei Jlus66 extenuate oxidative stress and inflammation via regulation of intestinal flora in rats with non alcoholic fatty liver disease. Food Sci. Nutr. 2019, 7, 2636–2646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, P.; Arora, A. Clinical presentation of alcoholic liver disease and non-alcoholic fatty liver disease: Spectrum and diagnosis. Transl. Gastroenterol. Hepatol. 2020, 5, 19. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Bataller, R. Alcoholic liver disease: Pathogenesis and new therapeutic targets. Gastroenterology 2011, 141, 1572–1585. [Google Scholar] [CrossRef] [Green Version]
- Teli, M.R.; Day, C.P.; James, O.F.W.; Burt, A.D.; Bennett, M.K. Determinants of progression to cirrhosis or fibrosis in pure alcoholic fatty liver. Lancet 1995, 346, 987–990. [Google Scholar] [CrossRef]
- Poynard, T.; Bedossa, P.; Opolon, P. Natural history of liver fibrosis progression in patients with chronic hepatitis C. Lancet 1997, 349, 825–832. [Google Scholar] [CrossRef]
- Singal, A.K.; Bataller, R.; Ahn, J.; Kamath, P.S.; Shah, V.H. ACG clinical guideline: Alcoholic liver disease. Am. J. Gastroenterol. 2018, 113, 175–194. [Google Scholar] [CrossRef]
- Begriche, K.; Igoudjil, A.; Pessayre, D.; Fromenty, B. Mitochondrial dysfunction in NASH: Causes, consequences and possible means to prevent it. Mitochondrion 2006, 6, 1–28. [Google Scholar] [CrossRef]
- Leung, T.M.; Nieto, N. CYP2E1 and oxidant stress in alcoholic and non-alcoholic fatty liver disease. J. Hepatol. 2013, 58, 395–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farrell, G.C.; Larter, C.Z. Nonalcoholic fatty liver disease: From steatosis to cirrhosis. Hepatology 2006, 43, S99–S112. [Google Scholar] [CrossRef] [PubMed]
- Landar, A.; Zmijewski, J.W.; Dickinson, D.A.; Le Goffe, C.; Johnson, M.S.; Milne, G.L.; Zanoni, G.; Vidari, G.; Morrow, J.D.; Darley-Usmar, V.M. Interaction of electrophilic lipid oxidation products with mitochondria in endothelial cells and formation of reactive oxygen species. Am. J. Physiol. Hear. Circ. Physiol. 2006, 290, H1777–H1787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, S.M. A review of the role of reactive oxygen and nitrogen species in alcohol-induced mitochondrial dysfunction. Free Radic. Res. 2003, 37, 585–596. [Google Scholar] [CrossRef] [PubMed]
- McKillop, I.H.; Schrum, L.W. Alcohol and liver cancer. Alcohol 2005, 35, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, C.C.; Bailey, S.M. Ethanol Consumption and Liver Mitochondria Function. Neurosignals 2001, 10, 271–282. [Google Scholar] [CrossRef]
- 1Lieber, C.S. Role of Oxidative Stress and Antioxidant Therapy in Alcoholic and Nonalcoholic Liver Diseases. Adv. Pharmacol. 1996, 38, 601–628. [Google Scholar] [CrossRef]
- Adachi, M.; Ishii, H. Role of mitochondria in alcoholic liver injury. Free Radic. Biol. Med. 2002, 32, 487–491. [Google Scholar] [CrossRef]
- Dupont, I.; Lucas, D.; Clot, P.; Ménez, C.; Albano, E. Cytochrome P4502E1 inducibility and hydroxyethyl radical formation among alcoholics. J. Hepatol. 1998, 28, 564–571. [Google Scholar] [CrossRef]
- Crabb, D.W.; Bosron, W.F.; Li, T.K. Ethanol metabolism. Pharmacol. Ther. 1987, 34, 59–73. [Google Scholar] [CrossRef]
- Seronello, S.; Sheikh, M.Y.; Choi, J. Redox regulation of hepatitis C in nonalcoholic and alcoholic liver. Free Radic. Biol. Med. 2007, 43, 869–882. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Ruiz, C.; Fernandez-Checa, J.C. Mitochondrial glutathione: Hepatocellular survival–death switch. J. Gastroenterol. Hepatol. 2006, 21, S3–S6. [Google Scholar] [CrossRef]
- García-Ruiz, C.; Morales, A.; Ballesta, A.; Rodés, J.; Kaplowitz, N.; Fernández-Checa, J.C. Effect of chronic ethanol feeding on glutathione and functional integrity of mitochondria in periportal and perivenous rat hepatocytes. J. Clin. Investig. 1994, 94, 193–201. [Google Scholar] [CrossRef] [Green Version]
- García-Ruiz, C.; Morales, A.; Colell, A.; Ballesta, A.; Rodés, J.; Kaplowitz, N.; Fernandez-Checa, J.C. Feeding S-adenosyl-l-methionine attenuates both ethanol-induced depletion of mitochondrial glutathione and mitochondrial dysfunction in periportal and perivenous rat hepatocytes. Hepatology 1995, 21, 207–214. [Google Scholar] [CrossRef]
- Colell, A.; Coll, O.; García-Ruiz, C.; París, R.; Tiribelli, C.; Kaplowitz, N.; Fernández-Checa, J.C. Tauroursodeoxycholic acid protects hepatocytes from ethanol-fed rats against tumor necrosis factor-induced cell death by replenishing mitochondrial glutathione. Hepatology 2001, 34, 964–971. [Google Scholar] [CrossRef]
- Neve, E.P.A.; Ingelman-Sundberg, M. Molecular basis for the transport of cytochrome P450 2E1 to the plasma membrane. J. Biol. Chem. 2000, 275, 17130–17135. [Google Scholar] [CrossRef] [Green Version]
- Bai, J.; Cederbaum, A.I. Overexpression of CYP2E1 in mitochondria sensitizes HepG2 cells to the toxicity caused by depletion of glutathione. J. Biol. Chem. 2006, 281, 5128–5136. [Google Scholar] [CrossRef] [Green Version]
- Bardag-Gorce, F.; Yuan, Q.X.; Li, J.; French, B.A.; Fang, C.; Ingelman-Sundberg, M.; French, S.W. The effect of ethanol-induced cytochrome p4502E1 on the inhibition of proteasome activity by alcohol. Biochem. Biophys. Res. Commun. 2000, 279, 23–29. [Google Scholar] [CrossRef]
- Butura, A.; Nilsson, K.; Morgan, K.; Morgan, T.R.; French, S.W.; Johansson, I.; Schuppe-Koistinen, I.; Ingelman-Sundberg, M. The impact of CYP2E1 on the development of alcoholic liver disease as studied in a transgenic mouse model. J. Hepatol. 2009, 50, 572–583. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Cederbaum, A.I. Adenovirus-Mediated Expression of CYP2E1 Produces Liver Toxicity in Mice. Toxicol. Sci. 2006, 91, 365–371. [Google Scholar] [CrossRef] [Green Version]
- Cederbaum, A.I. Nrf2 and antioxidant defense against CYP2E1 toxicity. Subcell. Biochem. 2013, 67, 105–130. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, H.; Sato, H.; Kuriyama-Matsumura, K.; Sato, K.; Maebara, K.; Wang, H.; Tamba, M.; Itoh, K.; Yamamoto, M.; Bannai, S. Electrophile response element-mediated induction of the cystine/glutamate exchange transporter gene expression. J. Biol. Chem. 2002, 277, 44765–44771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, W.I.; Osei-Hyiaman, D.; Park, O.; Liu, J.; Bátkai, S.; Mukhopadhyay, P.; Horiguchi, N.; Harvey-White, J.; Marsicano, G.; Lutz, B.; et al. Paracrine Activation of Hepatic CB1 Receptors by Stellate Cell-Derived Endocannabinoids Mediates Alcoholic Fatty Liver. Cell Metab. 2008, 7, 227–235. [Google Scholar] [CrossRef] [Green Version]
- Choi, W.M.; Kim, H.H.; Kim, M.H.; Cinar, R.; Yi, H.S.; Eun, H.S.; Kim, S.H.; Choi, Y.J.; Lee, Y.S.; Kim, S.Y.; et al. Glutamate Signaling in Hepatic Stellate Cells Drives Alcoholic Steatosis. Cell Metab. 2019, 30, 877–889.e7. [Google Scholar] [CrossRef] [PubMed]
- Ferrigno, A.; Berardo, C.; Di Pasqua, L.G.; Cagna, M.; Siciliano, V.; Richelmi, P.; Vairetti, M. The selective blockade of metabotropic glutamate receptor-5 attenuates fat accumulation in an in vitro model of benign steatosis. Eur. J. Histochem. 2020, 64. [Google Scholar] [CrossRef] [PubMed]
- Lucena, M.I.; Andrade, R.J.; de la Cruz, J.P.; Rodriguez-Mendizabal, M.; Blanco, E.; Sánchez de la Cuesta, F. Effects of silymarin MZ-80 on oxidative stress in patients with alcoholic cirrhosis. Int. J. Clin. Pharmacol. Ther. 2002, 40, 2–8. [Google Scholar] [CrossRef]
- Ronis, M.J.J.; Butura, A.; Sampey, B.P.; Shankar, K.; Prior, R.L.; Korourian, S.; Albano, E.; Ingelman-Sundberg, M.; Petersen, D.R.; Badger, T.M. Effects of N-acetylcysteine on ethanol-induced hepatotoxicity in rats fed via total enteral nutrition. Free Radic. Biol. Med. 2005, 39, 619–630. [Google Scholar] [CrossRef] [Green Version]
- Ozaras, R.; Tahan, V.; Aydin, S.; Uzun, H.; Kaya, S.; Senturk, H. N-acetylcysteine attenuates alcohol-induced oxidative stress in the rat. World J. Gastroenterol. 2003, 9, 125–128. [Google Scholar] [CrossRef]
- Nguyen-Khac, E.; Thevenot, T.; Piquet, M.-A.; Benferhat, S.; Goria, O.; Chatelain, D.; Tramier, B.; Dewaele, F.; Ghrib, S.; Rudler, M.; et al. Glucocorticoids plus N -Acetylcysteine in Severe Alcoholic Hepatitis. N. Engl. J. Med. 2011, 365, 1781–1789. [Google Scholar] [CrossRef] [Green Version]
- Stewart, S.; Prince, M.; Bassendine, M.; Hudson, M.; James, O.; Jones, D.; Record, C.; Day, C.P. A randomized trial of antioxidant therapy alone or with corticosteroids in acute alcoholic hepatitis. J. Hepatol. 2007, 47, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Purohit, V.; Abdelmalek, M.F.; Barve, S.; Benevenga, N.J.; Halsted, C.H.; Kaplowitz, N.; Kharbanda, K.K.; Liu, Q.Y.; Lu, S.C.; McClain, C.J.; et al. Role of S-adenosylmethionine, folate, and betaine in the treatment of alcoholic liver disease: Summary of a symposium. Am. J. Clin. Nutr. 2007, 86, 14–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caballería, J.; Parés, A.; Brú, C.; Mercader, J.; Plaza, A.G.; Caballería, L.; Clemente, G.; Rodrigo, L.; Rodés, J. Metadoxine accelerates fatty liver recovery in alcoholic patients: Results of a randomized double-blind, placebo-control trial. J. Hepatol. 1998, 28, 54–60. [Google Scholar] [CrossRef]
- Han, K.H.; Hashimoto, N.; Fukushima, M. Relationships among alcoholic liver disease, antioxidants, and antioxidant enzymes. World J. Gastroenterol. 2016, 22, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Gao, C.; Xing, M.; Li, Y.; Zhu, L.; Wang, D.; Yang, X.; Liu, L.; Yao, P. Quercetin prevents ethanol-induced dyslipidemia and mitochondrial oxidative damage. Food Chem. Toxicol. 2012, 50, 1194–1200. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Li, Y.; Yu, H.; Gao, C.; Liu, L.; Xing, M.; Yao, P. Quercetin attenuates chronic ethanol hepatotoxicity: Implication of “free” iron uptake and release. Food Chem. Toxicol. 2014, 67, 131–138. [Google Scholar] [CrossRef]
- Yin, H.Q.; Kim, Y.C.; Chung, Y.S.; Kim, Y.C.; Shin, Y.K.; Lee, B.H. Honokiol reverses alcoholic fatty liver by inhibiting the maturation of sterol regulatory element binding protein-1c and the expression of its downstream lipogenesis genes. Toxicol. Appl. Pharmacol. 2009, 236, 124–130. [Google Scholar] [CrossRef]
- Zhang, P.; Ma, D.; Wang, Y.; Zhang, M.; Qiang, X.; Liao, M.; Liu, X.; Wu, H.; Zhang, Y. Berberine protects liver from ethanol-induced oxidative stress and steatosis in mice. Food Chem. Toxicol. 2014, 74, 225–232. [Google Scholar] [CrossRef]
- Bao, W.; Li, K.; Rong, S.; Yao, P.; Hao, L.; Ying, C.; Zhang, X.; Nussler, A.; Liu, L. Curcumin alleviates ethanol-induced hepatocytes oxidative damage involving heme oxygenase-1 induction. J. Ethnopharmacol. 2010, 128, 549–553. [Google Scholar] [CrossRef]
- Forsyth, C.B.; Farhadi, A.; Jakate, S.M.; Tang, Y.; Shaikh, M.; Keshavarzian, A. Lactobacillus GG treatment ameliorates alcohol-induced intestinal oxidative stress, gut leakiness, and liver injury in a rat model of alcoholic steatohepatitis. Alcohol 2009, 43, 163–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Liu, Y.; Kirpich, I.; Ma, Z.; Wang, C.; Zhang, M.; Suttles, J.; McClain, C.; Feng, W. Lactobacillus rhamnosus GG reduces hepatic TNFα production and inflammation in chronic alcohol-induced liver injury. J. Nutr. Biochem. 2013, 24, 1609–1615. [Google Scholar] [CrossRef] [Green Version]
- Samant, H.; Manatsathit, W.; Dies, D.; Shokouh-Amiri, H.; Zibari, G.; Boktor, M.; Alexander, J.S. Cholestatic liver diseases: An era of emerging therapies. World J. Clin. Cases 2019, 7, 1571–1581. [Google Scholar] [CrossRef] [PubMed]
- Delemos, A.S.; Friedman, L.S. Systemic Causes of Cholestasis. Clin. Liver Dis. 2013, 17, 301–317. [Google Scholar] [CrossRef] [PubMed]
- Sticova, E.; Jirsa, M.; Pawłowska, J. New Insights in Genetic Cholestasis: From Molecular Mechanisms to Clinical Implications. Can. J. Gastroenterol. Hepatol. 2018, 2018, 2313675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoda, J.; Kano, M.; Oda, K.; Kamiya, J.; Nimura, Y.; Suzuki, H.; Sugiyama, Y.; Miyazaki, H.; Todoroki, T.; Stengelin, S.; et al. The expression levels of plasma membrane transporters in the cholestatic liver of patients undergoing biliary drainage and their association with the impairment of biliary secretory function. Am. J. Gastroenterol. 2001, 96, 3368–3378. [Google Scholar] [CrossRef]
- Portincasa, P.; Grattagliano, I.; Testini, M.; Caruso, M.L.; Wang, D.Q.-H.; Moschetta, A.; Calamita, G.; Vacca, M.; Valentini, A.M.; Renna, G.; et al. Parallel intestinal and liver injury during early cholestasis in the rat: Modulation by bile salts and antioxidants. Free Radic. Biol. Med. 2007, 42, 1381–1391. [Google Scholar] [CrossRef] [PubMed]
- Sheen, J.-M.; Huang, L.-T.; Hsieh, C.-S.; Chen, C.-C.; Wang, J.-Y.; Tain, Y.-L. Bile duct ligation in developing rats: Temporal progression of liver, kidney, and brain damage. J. Pediatr. Surg. 2010, 45, 1650–1658. [Google Scholar] [CrossRef]
- Ljubuncic, P.; Tanne, Z.; Bomzon, A. Evidence of a systemic phenomenon for oxidative stress in cholestatic liver disease. Gut 2000, 47, 710–716. [Google Scholar] [CrossRef] [Green Version]
- Grintzalis, K.; Papapostolou, I.; Assimakopoulos, S.F.; Mavrakis, A.; Faropoulos, K.; Karageorgos, N.; Georgiou, C.; Chroni, E.; Konstantinou, D. Time-related alterations of superoxide radical levels in diverse organs of bile duct-ligated rats. Free Radic. Res. 2009, 43, 803–808. [Google Scholar] [CrossRef] [PubMed]
- Krähenbühl, S.; Talos, C.; Reichen, J. Mechanisms of impaired hepatic fatty acid metabolism in rats with long-term bile duct ligation. Hepatology 1994, 19, 1272–1281. [Google Scholar] [CrossRef] [PubMed]
- Sokol, R.J.; McKim, J.; Goff, M.C.; Ruyle, S.Z.; Devereaux, M.W.; Han, D.; Packer, L.; Everson, G. Vitamin E reduces oxidant injury to mitochondria and the hepatotoxicity of taurochenodeoxycholic acid in the rat. Gastroenterology 1998, 114, 164–174. [Google Scholar] [CrossRef]
- Krähenbühl, S.; Talos, C.; Lauterburg, B.H.; Reichen, J. Reduced antioxidative capacity in liver mitochondria from bile duct ligated rats. Hepatology 1995, 22, 607–612. [Google Scholar] [CrossRef]
- Neuschwander-Tetri, B.A.; Nicholson, C.; Wells, L.D.; Tracy, T.F. Cholestatic liver injury down-regulates hepatic glutathione synthesis. J. Surg. Res. 1996, 63, 447–451. [Google Scholar] [CrossRef]
- Gumpricht, E.; Devereaux, M.W.; Dahl, R.H.; Sokol, R.J. Glutathione status of isolated rat hepatocytes affects bile acid-induced cellular necrosis but not apoptosis. Toxicol. Appl. Pharmacol. 2000, 164, 102–111. [Google Scholar] [CrossRef]
- Serviddio, G.; Pereda, J.; Pallardó, F.V.; Carretero, J.; Borras, C.; Cutrin, J.; Vendemiale, G.; Poli, G.; Viña, J.; Sastre, J. Ursodeoxycholic Acid Protects against Secondary Biliary Cirrhosis in Rats by Preventing Mitochondrial Oxidative Stress. Hepatology 2004, 39, 711–720. [Google Scholar] [CrossRef]
- Mitsuyoshi, H.; Nakashima, T.; Sumida, Y.; Yoh, T.; Nakajima, Y.; Ishikawa, H.; Inaba, K.; Sakamoto, Y.; Okanoue, T.; Kashima, K. Ursodeoxycholic acid protects hepatocytes against oxidative injury via induction of antioxidants. Biochem. Biophys. Res. Commun. 1999, 263, 537–542. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Ramani, K.; Xia, M.; Ko, K.S.; Li, T.W.H.; Oh, P.; Li, J.; Lu, S.C. Dysregulation of glutathione synthesis during cholestasis in mice: Molecular mechanisms and therapeutic implications. Hepatology 2009, 49, 1982–1991. [Google Scholar] [CrossRef] [Green Version]
- Lu, S.C. Regulation of glutathione synthesis. Mol. Asp. Med. 2009, 30, 42–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Zeng, Y.; Lee, T.D.; Yang, Y.; Ou, X.; Chen, L.; Haque, M.; Rippe, R.; Lu, S.C. Role of AP-1 in the coordinate induction of rat glutamate-cysteine ligase and glutathione synthetase by tert-butylhydroquinone. J. Biol. Chem. 2002, 277, 35232–35239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Magilnick, N.; Ou, X.; Lu, S.C. Tumour necrosis factor α induces co-ordinated activation of rat GSH synthetic enzymes via nuclear factor κB and activator protein-1. Biochem. J. 2005, 391, 399–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benassi, B.; Fanciulli, M.; Fiorentino, F.; Porrello, A.; Chiorino, G.; Loda, M.; Zupi, G.; Biroccio, A. c-Myc phosphorylation is required for cellular response to oxidative stress. Mol. Cell 2006, 21, 509–519. [Google Scholar] [CrossRef]
- Jaiswal, A.K. Nrf2 signaling in coordinated activation of antioxidant gene expression. Free Radic. Biol. Med. 2004, 36, 1199–1207. [Google Scholar] [CrossRef]
- Dhakshinamoorthy, S.; Jaiswal, A.K. Small Maf (MafG and MafK) proteins negatively regulate antioxidant response element-mediated expression and antioxidant induction of the NAD(P)H:Quinone oxidoreductase1 gene. J. Biol. Chem. 2000, 275, 40134–40141. [Google Scholar] [CrossRef] [Green Version]
- Dhakshinamoorthy, S.; Jain, A.K.; Bloom, D.A.; Jaiswal, A.K. Bach1 competes with Nrf2 leading to negative regulation of the antioxidant response element (ARE)-mediated NAD(P)H:quinone oxidoreductase 1 gene expression and induction in response to antioxidants. J. Biol. Chem. 2005, 280, 16891–16900. [Google Scholar] [CrossRef] [Green Version]
- Roeb, E.; Purucker, E.; Gartung, C.; Geier, A.; Jansen, B.; Winograd, R.; Matern, S. Effect of glutathione depletion and hydrophilic bile acids on hepatic acute phase reaction in rats with extrahepatic cholestasis. Scand. J. Gastroenterol. 2003, 38, 878–885. [Google Scholar] [CrossRef]
- Ker, C.-G.; Huang, T.-J.; Sheen, P.-C. Electron Microscopic Assessment of Bile Regurgitation of Human in Extrahepatic Obstructive Jaundice. Eur. Surg. Res. 1985, 17, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Clichici, S.; David, L.; Moldovan, B.; Baldea, I.; Olteanu, D.; Filip, M.; Nagy, A.; Luca, V.; Crivii, C.; Mircea, P.; et al. Hepatoprotective effects of silymarin coated gold nanoparticles in experimental cholestasis. Mater. Sci. Eng. C 2020, 115, 111117. [Google Scholar] [CrossRef] [PubMed]
- Piletz, J.E.; Aricioglu, F.; Cheng, J.T.; Fairbanks, C.A.; Gilad, V.H.; Haenisch, B.; Halaris, A.; Hong, S.; Lee, J.E.; Li, J.; et al. Agmatine: Clinical applications after 100 years in translation. Drug Discov. Today 2013, 18, 880–893. [Google Scholar] [CrossRef] [PubMed]
- Ommati, M.M.; Farshad, O.; Mousavi, K.; Taghavi, R.; Farajvajari, S.; Azarpira, N.; Moezi, L.; Heidari, R. Agmatine alleviates hepatic and renal injury in a rat model of obstructive jaundice. PharmaNutrition 2020, 13, 100212. [Google Scholar] [CrossRef]
- Ommati, M.M.; Attari, H.; Siavashpour, A.; Shafaghat, M.; Azarpira, N.; Ghaffari, H.; Moezi, L.; Heidari, R. Mitigation of cholestasis-associated hepatic and renal injury by edaravone treatment: Evaluation of its effects on oxidative stress and mitochondrial function. Liver Res. 2020. [Google Scholar] [CrossRef]
- Yoshida, H.; Yanai, H.; Namiki, Y.; Fukatsu-Sasaki, K.; Furutani, N.; Tada, N. Neuroprotective effects of edaravone: A novel free radical scavenger in cerebrovascular injury. CNS Drug Rev. 2006, 12, 9–20. [Google Scholar] [CrossRef]
- Shinohara, Y.; Saito, I.; Kobayashi, S.; Uchiyama, S. Edaravone (Radical Scavenger) versus sodium ozagrel (Antiplatelet Agent) in acute noncardioembolic ischemic stroke (EDO Trial). Cerebrovasc. Dis. 2009, 27, 485–492. [Google Scholar] [CrossRef] [PubMed]
- Jackson, C.; Heiman-Patterson, T.; Kittrell, P.; Baranovsky, T.; McAnanama, G.; Bower, L.; Agnese, W.; Martin, M. Radicava (edaravone) for amyotrophic lateral sclerosis: US experience at 1 year after launch. Amyotroph. Lateral Scler. Front. Degener. 2019, 20, 605–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okatani, Y.; Wakatsuki, A.; Enzan, H.; Miyahara, Y. Edaravone protects against ischemia/reperfusion-induced oxidative damage to mitochondria in rat liver. Eur. J. Pharmacol. 2003, 465, 163–170. [Google Scholar] [CrossRef]
- Hassan, M.Q.; Akhtar, M.S.; Afzal, O.; Hussain, I.; Akhtar, M.; Haque, S.E.; Najmi, A.K. Edaravone and benidipine protect myocardial damage by regulating mitochondrial stress, apoptosis signalling and cardiac biomarkers against doxorubicin-induced cardiotoxicity. Clin. Exp. Hypertens. 2020, 42, 381–392. [Google Scholar] [CrossRef]
- Fukui, H.; Wiest, R. Changes of Intestinal Functions in Liver Cirrhosis. Inflamm. Intest. Dis. 2016, 1, 24–40. [Google Scholar] [CrossRef]
- Nolan, J.P. The role of intestinal endotoxin in liver injury: A long and evolving history. Hepatology 2010, 52, 1829–1835. [Google Scholar] [CrossRef] [PubMed]
- Ommati, M.M.; Farshad, O.; Niknahad, H.; Mousavi, K.; Moein, M.; Azarpira, N.; Mohammadi, H.; Jamshidzadeh, A.; Heidari, R. Oral administration of thiol-reducing agents mitigates gut barrier disintegrity and bacterial lipopolysaccharide translocation in a rat model of biliary obstruction. Curr. Res. Pharmacol. Drug Discov. 2020, 1, 10–18. [Google Scholar] [CrossRef]
- Assimakopoulos, S.F.; Vagianos, C.E.; Zervoudakis, G.; Filos, K.S.; Georgiou, C.; Nikolopoulou, V.; Scopa, C.D. Gut regulatory peptides bombesin and neurotensin reduce hepatic oxidative stress and histological alterations in bile duct ligated rats. Regul. Pept. 2004, 120, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.Y.; Wang, Y.Y.; Chen, W.Y.; Chuang, Y.H.; Pan, P.H.; Chen, C.J. Beneficial effect of quercetin on cholestatic liver injury. J. Nutr. Biochem. 2014, 25, 1183–1195. [Google Scholar] [CrossRef] [PubMed]
- Sadeghi, H.; Azarmehr, N.; Razmkhah, F.; Sadeghi, H.; Danaei, N.; Omidifar, N.; Vakilpour, H.; Pourghadamyari, H.; Doustimotlagh, A.H. The hydroalcoholic extract of watercress attenuates protein oxidation, oxidative stress, and liver damage after bile duct ligation in rats. J. Cell. Biochem. 2019, 120, 14875–14884. [Google Scholar] [CrossRef] [PubMed]
- Ferrigno, A.; Di Pasqua, L.G.; Palladini, G.; Berardo, C.; Verta, R.; Richelmi, P.; Perlini, S.; Collotta, D.; Collino, M.; Vairetti, M. Transient expression of reck under hepatic ischemia/reperfusion conditions is associated with mapk signaling pathways. Biomolecules 2020, 10, 747. [Google Scholar] [CrossRef]
- Lemasters, J.J.V. Necrapoptosis and the mitochondrial permeability transition: Shared pathways to necrosis and apoptosis. Am. J. Physiol. 1999, 276, G1–G6. [Google Scholar] [CrossRef] [PubMed]
- Klaassen, C.D.; Reisman, S.A. Nrf2 the rescue: Effects of the antioxidative/electrophilic response on the liver. Toxicol. Appl. Pharmacol. 2010, 244, 57–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kensler, T.W.; Wakabayashi, N.; Biswal, S. Cell Survival Responses to Environmental Stresses Via the Keap1-Nrf2-ARE Pathway. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 89–116. [Google Scholar] [CrossRef] [PubMed]
- Baird, L.; Dinkova-Kostova, A.T. The cytoprotective role of the Keap1-Nrf2 pathway. Arch. Toxicol. 2011, 85, 241–272. [Google Scholar] [CrossRef]
- Kudoh, K.; Uchinami, H.; Yoshioka, M.; Seki, E.; Yamamoto, Y. Nrf2 activation protects the liver from ischemia/reperfusion injury in Mice. Ann. Surg. 2014, 260, 118–127. [Google Scholar] [CrossRef] [Green Version]
- Jaeschke, H.; Farhood, A. Neutrophil and Kupffer cell-induced oxidant stress and ischemia-reperfusion injury in rat liver. Am. J. Physiol. Gastrointest. Liver Physiol. 1991, 260, G355–G362. [Google Scholar] [CrossRef]
- Schauer, R.J.; Kalmuk, S.; Gerbes, A.L.; Leiderer, R.; Meissner, H.; Schildberg, F.W.; Messmer, K.; Bilzer, M. Intravenous administration of glutathione protects parechymal and non-paranchymal liver cells against reperfusion injury following rat liver transplantation. World J. Gastroenterol. 2004, 10, 864–870. [Google Scholar] [CrossRef]
- Schauer, R.J.; Gerbes, A.L.; Vonier, D.; Meissner, H.; Michl, P.; Leiderer, R.; Schildberg, F.W.; Messmer, K.; Bilzer, M. Glutathione Protects the Rat Liver Against Reperfusion Injury after Prolonged Warm Ischemia. Ann. Surg. 2004, 239, 220–231. [Google Scholar] [CrossRef]
- Bilzer, M.; Paumgartner, G.; Gerbes, A.L. Glutathione protects the rat liver against reperfusion injury after hypothermic preservation. Gastroenterology 1999, 117, 200–210. [Google Scholar] [CrossRef] [Green Version]
- Smyrniotis, V.; Arkadopoulos, N.; Kostopanagiotou, G.; Theodoropoulos, T.; Theodoraki, K.; Farantos, C.; Kairi, E.; Paphiti, A. Attenuation of ischemic injury by N-acetylcysteine preconditioning of the liver. J. Surg. Res. 2005, 129, 31–37. [Google Scholar] [CrossRef]
- Nagasaki, H.; Nakano, H.; Boudjema, K.; Jaeck, D.; Alexandre, E.; Baek, Y.; Kitamura, N.; Yamaguchi, M.; Kumada, K. Efficacy of preconditioning with n-acetylcysteine against reperfusion injury after prolonged cold ischaemia in rats liver in which glutathione had been reduced by buthionine sulphoximine. Eur. J. Surg. 1998, 164, 139–146. [Google Scholar] [CrossRef]
- Sener, G.; Tosun, O.; Şehirli, A.Ö.; Kaçmaz, A.; Arbak, S.; Ersoy, Y.; Ayanoǧlu-Dülger, G. Melatonin and N-acetylcysteine have beneficial effects during hepatic ischemia and reperfusion. Life Sci. 2003, 72, 2707–2718. [Google Scholar] [CrossRef]
- Saito, C.; Zwingmann, C.; Jaeschke, H. Novel mechanisms of protection against acetaminophen hepatotoxicity in mice by glutathione and N-acetylcysteine. Hepatology 2010, 51, 246–254. [Google Scholar] [CrossRef] [Green Version]
- D’Amico, F.; Vitale, A.; Piovan, D.; Bertacco, A.; Ramirez Morales, R.; Chiara Frigo, A.; Bassi, D.; Bonsignore, P.; Gringeri, E.; Valmasoni, M.; et al. Use of N-acetylcysteine during liver procurement: A prospective randomized controlled study. Liver Transplant. 2013, 19, 135–144. [Google Scholar] [CrossRef]
- Wendel, A.; Cikryt, P. The level and half-life of glutathione in human plasma. FEBS Lett. 1980, 120, 209–211. [Google Scholar] [CrossRef] [Green Version]
- Zwacka, R.M.; Zhou, W.; Zhang, Y.; Darby, C.J.; Dudus, L.; Halldorson, J.; Oberley, L.; Engelhardt, J.F. Redox gene therapy for ischemia/reperfusion injury of the liver reduces AP1 and NF-κB activation. Nat. Med. 1998, 4, 698–704. [Google Scholar] [CrossRef]
- Lehmann, T.G.; Wheeler, M.D.; Froh, M.; Schwabe, R.F.; Bunzendahl, H.; Samulski, J.R.; Lemasters, J.J.; Brenner, D.A.; Thurman, R.G. Effects of three superoxide dismutase genes delivered with an adenovirus on graft function after transplantation of fatty livers in the rat1. Transplantation 2003, 76, 28–37. [Google Scholar] [CrossRef]
- Shau, H.; Merino, A.; Chen, L.; Shih, C.C.-Y.; Colquhoun, S.D. Induction of Peroxiredoxins in Transplanted Livers and Demonstration of Their In Vitro Cytoprotection Activity. Antioxid. Redox Signal. 2000, 2, 347–354. [Google Scholar] [CrossRef]
- Glantzounis, G.K.; Salacinski, H.J.; Yang, W.; Davidson, B.R.; Seifalian, A.M. The contemporary role of antioxidant therapy in attenuating liver ischemia-reperfusion injury: A review. Liver Transplant. 2005, 11, 1031–1047. [Google Scholar] [CrossRef]
- Bayramoglu, G.; Bayramoglu, A.; Engur, S.; Senturk, H.; Ozturk, N.; Colak, S. The hepatoprotective effects of Hypericum perforatum L. on hepatic ischemia/reperfusion injury in rats. Cytotechnology 2014, 66, 443–448. [Google Scholar] [CrossRef] [Green Version]
- Ramalho, L.N.; Pasta, Â.A.; Terra, V.A.; Augusto, M.; Sanches, S.C.; Souza-Neto, F.P.; Cecchini, R.; Gulin, F.; Ramalho, F.S. Rosmarinic acid attenuates hepatic ischemia and reperfusion injury in rats. Food Chem. Toxicol. 2014, 74, 270–278. [Google Scholar] [CrossRef]
- Xing, H.-C.; Li, L.-J.; Xu, K.-J.; Shen, T.; Chen, Y.-B.; Sheng, J.-F.; Chen, Y.; Fu, S.-Z.; Chen, C.-L.; Wang, J.-G.; et al. Protective role of supplement with foreign Bifidobacterium and Lactobacillus in experimental hepatic ischemia-reperfusion injury. J. Gastroenterol. Hepatol. 2006, 21, 647–656. [Google Scholar] [CrossRef]
- Westbrook, R.H.; Dusheiko, G. Natural history of hepatitis C. J. Hepatol. 2014, 61, S58–S68. [Google Scholar] [CrossRef] [Green Version]
- Hoofnagle, J.H. Course and outcome of hepatitis C. Hepatology 2002, 36, s21–s29. [Google Scholar]
- Ruggieri, A.; Anticoli, S.; Nencioni, L.; Sgarbanti, R.; Garaci, E. Interplay between Hepatitis C Virus and Redox Cell Signaling. Int. J. Mol. Sci. 2013, 14, 4705–4721. [Google Scholar] [CrossRef] [Green Version]
- Paracha, U.Z.; Fatima, K.; Alqahtani, M.; Chaudhary, A.; Abuzenadah, A. Oxidative stress and hepatitis C virus. Virol. J. 2013, 10, 1. [Google Scholar] [CrossRef] [Green Version]
- Roe, B.; Kensicki, E.; Mohney, R.; Hall, W.W. Metabolomic Profile of Hepatitis C Virus-Infected Hepatocytes. PLoS ONE 2011, 6, 1–8. [Google Scholar] [CrossRef]
- De Mochel, N.S.R.; Seronello, S.; Wang, S.H.; Ito, C.; Zheng, J.X.; Liang, T.J.; Lambeth, J.D.; Choi, J. Hepatocyte NAD(P)H oxidases as an endogenous source of reactive oxygen species during hepatitis C virus infection. Hepatology 2011, 52, 209–228. [Google Scholar] [CrossRef] [Green Version]
- Paul, D.; Madan, V.; Bartenschlager, R. Hepatitis C Virus RNA Replication and Assembly: Living on the Fat of the Land. Cell Host Microbe 2014, 16, 569–579. [Google Scholar] [CrossRef] [Green Version]
- Medvedev, R.; Ploen, D.; Hildt, E. HCV and Oxidative Stress: Implications for HCV Life Cycle and HCV-Associated Pathogenesis. Oxid. Med. Cell. Longev. 2016, 2016, 13. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Sun, L.; Seth, R.B.; Pineda, G.; Chen, Z.J. Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc. Natl. Acad. Sci. USA 2005, 102, 17717–17722. [Google Scholar] [CrossRef] [Green Version]
- Korenaga, M.; Showalter, L.; Chan, T.; Sun, J. Hepatitis C Virus Core Protein Inhibits Mitochondrial Electron Transport and Hepatitis C Virus Core Protein Inhibits Mitochondrial Electron (ROS) Production. J. Biol. Chem. 2005, 280, 37481–37488. [Google Scholar] [CrossRef] [Green Version]
- Abdalla, M.Y.; Ahmad, I.M.; Spitz, D.R.; Schmidt, W.N.; Britigan, B.E. Hepatitis C Virus-Core and Non Structural Proteins Lead to Different Effects on Cellular Antioxidant Defenses. J. Med. Virol. 2005, 497, 489–497. [Google Scholar] [CrossRef]
- Bender, D.; Hildt, E. Effect of Hepatitis Viruses on the Nrf2 / Keap1-Signaling Pathway and Its Impact on Viral Replication and Pathogenesis. Int. J. Mol. Sci. 2019, 18, 4659. [Google Scholar] [CrossRef] [Green Version]
- Carvajal-Yepes, M.; Himmelsbach, K.; Schaedler, S.; Ploen, D.; Krause, J.; Ludwig, L.; Weiss, T.; Klingel, K.; Hildt, E. Hepatitis C Virus Impairs the Induction of Cytoprotective Nrf2 Target Genes by Delocalization of Small Maf Proteins. J. Biol. Chem. 2011, 286, 8941–8951. [Google Scholar] [CrossRef] [Green Version]
- Dodson, M.; Castro-Portuguez, R.; Zhang, D.D. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 2019, 23, 101107. [Google Scholar] [CrossRef]
- Brault, C.; Lévy, P.; Duponchel, S.; Michelet, M.; Sallé, A.; Pécheur, E.; Plissonnier, M.; Parent, R.; Véricel, E.; Ivanov, A.V.; et al. Glutathione peroxidase 4 is reversibly induced by HCV to control lipid peroxidation and to increase virion infectivity. Gut 2016, 65, 144–154. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.; Corder, N.L.B.; Koduru, B.; Wang, Y. Oxidative stress and hepatic Nox proteins in chronic hepatitis C and hepatocellular carcinoma. Free Radic. Biol. Med. 2014, 72, 267–284. [Google Scholar] [CrossRef] [Green Version]
- Barbaro, G.; Di Lorenzo, G.; Ribersani, M.; Soldini, M.; Giancaspro, G.; Bellomo, G.; Belloni, G.; Grisorio, B.; Barbarini, G. Serum ferritin and hepatic glutathione concentrations in chronic hepatitis C patients related to the hepatitis C virus genotype. J. Hepatol. 1999, 30, 174–182. [Google Scholar] [CrossRef]
- Kerins, M.J.; Ooi, A. The Roles of NRF2 in Modulating Cellular Iron Homeostasis. Antioxid. Redox Signal. 2018, 29, 1756–1773. [Google Scholar] [CrossRef] [Green Version]
- Ansari, M.K.H.; Omrani, M.; Kheradmand, F. Oxidative Stress Response in Patients Infected by Diverse Hepatitis C Virus Genotypes. Hepat. Mon. 2015, 15, e22069. [Google Scholar] [CrossRef] [Green Version]
- Razzaq, Z.; Malik, A. Viral load is associated with abnormal serum levels of micronutrients and glutathione and glutathione-dependent enzymes in genotype 3 HCV patients. BBA Clin. 2014, 2, 72–78. [Google Scholar] [CrossRef] [Green Version]
- Farias, M.S.; Budni, P.; Ribeiro, C.M.; Parisotto, E.B.; Santos, C.E.I.; Ferraz Dias, J.; Dalmarco, E.M.; Frode, S.T.; Pedrosa, R.C.; Filho, W.D. Antioxidant supplementation attenuates oxidative stress in chronic hepatitis C patients. Gastroenterol. Hepatol. 2012, 35, 386–394. [Google Scholar] [CrossRef]
- Revill, P.A.; Penicaud, C.; Brechot, C.; Zoulim, F. Meeting the Challenge of Eliminating Chronic Hepatitis b Infection. Genes 2019, 10, 260. [Google Scholar] [CrossRef] [Green Version]
- Ivanov, A.V.; Valuev-Elliston, V.T.; Tyurina, D.A.; Ivanova, O.N.; Kochetkov, S.N.; Bartosch, B.; Isaguliants, M.G. Oxidative stress, a trigger of hepatitis C and B virus-induced liver carcinogenesis. Oncotarget 2017, 8, 3895–3932. [Google Scholar] [CrossRef] [Green Version]
- Liang, T.J. Hepatitis B: The virus and disease. Hepatology 2009, 49 (Suppl. S5), S13–S21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shim, H.Y.; Quan, X.; Yi, Y.S.; Jung, G. Heat shock protein 90 facilitates formation of the HBV capsid via interacting with the HBV core protein dimers. Virology 2011, 410, 161–169. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.S.; Seo, H.W.; Jung, G. Reactive oxygen species promote heat shock protein 90-mediated HBV capsid assembly. Biochem. Biophys. Res. Commun. 2015, 457, 328–333. [Google Scholar] [CrossRef]
- Wang, Q.; Na, B.; Ou, J. hsiung J.; Pulliam, L.; Yen, T.S.B. Hepatitis B virus alters the antioxidant system in transgenic mice and sensitizes hepatocytes to fas signaling. PLoS ONE 2012, 7, e36818. [Google Scholar] [CrossRef] [Green Version]
- Peiffer, K.H.; Akhras, S.; Himmelsbach, K.; Hassemer, M.; Finkernagel, M.; Carra, G.; Nuebling, M.; Chudy, M.; Niekamp, H.; Glebe, D.; et al. Intracellular accumulation of subviral HBsAg particles and diminished Nrf2 activation in HBV genotype G expressing cells lead to an increased ROI level. J. Hepatol. 2015, 62, 791–798. [Google Scholar] [CrossRef]
- Schaedler, S.; Krause, J.; Himmelsbach, K.; Carvajal-Yepes, M.; Lieder, F.; Klingel, K.; Nassal, M.; Weiss, T.S.; Werner, S.; Hildt, E. Hepatitis B virus induces expression of antioxidant response element-regulated genes by activation of Nrf2. J. Biol. Chem. 2010, 285, 41074–41086. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Fang, M.; He, Z.; Cui, D.; Jia, S.; Lin, X.; Xu, X.; Zhou, T.; Liu, W. Hepatitis B virus stimulates G6PD expression through HBx-mediated Nrf2 activation. Cell Death Dis. 2015, 6, e1980. [Google Scholar] [CrossRef]
- Li, H.; Zhu, W.; Zhang, L.; Lei, H.; Wu, X.; Guo, L.; Chen, X.; Wang, Y.; Tang, H. The metabolic responses to hepatitis B virus infection shed new light on pathogenesis and targets for treatment. Sci. Rep. 2015, 5, 8421. [Google Scholar] [CrossRef] [Green Version]
- Severi, T.; Ying, C.; Vermeesch, J.R.; Cassiman, D.; Cnops, L.; Verslype, C.; Fevery, J.; Arckens, L.; Neyts, J.; van Pelt, J.F. Hepatitis B virus replication causes oxidative stress in HepAD38 liver cells. Mol. Cell. Biochem. 2006, 290, 79–85. [Google Scholar] [CrossRef]
- Wu, Y.L.; Wang, D.; Peng, X.E.; Chen, Y.L.; Zheng, D.L.; Chen, W.N.; Lin, X. Epigenetic silencing of NAD(P)H:quinone oxidoreductase 1 by hepatitis B virus X protein increases mitochondrial injury and cellular susceptibility to oxidative stress in hepatoma cells. Free Radic. Biol. Med. 2013, 65, 632–644. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.-P.; Ji, X.-F.; Li, X.-Y.; Gao, S.; Fan, Y.-C.; Wang, K. Methylation of the Glutathione-S-Transferase P1 Gene Promoter Is Associated with Oxidative Stress in Patients with Chronic Hepatitis B. Tohoku J. Exp. Med. 2016, 238, 57–64. [Google Scholar] [CrossRef] [Green Version]
- Tong, A.; Wu, L.; Lin, Q.; Quek, C.L.; Zhao, X.; Li, J.; Chen, P.; Chen, L.; Tang, H.; Huang, C.; et al. Proteomic analysis of cellular protein alterations using a hepatitis B virus-producing cellular model. Proteomics 2008, 8, 2012–2023. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.C.; Cui, J.Y.; Klaassen, C.D. Effect of graded nrf2 activation on phase-i and -ii drug metabolizing enzymes and transporters in mouse liver. PLoS ONE 2012, 7, e39006. [Google Scholar] [CrossRef]
- Board, P.G.; Coggan, M.; Chelvanayagam, G.; Easteal, S.; Jermiin, L.S.; Schulte, G.K.; Danley, D.E.; Hoth, L.R.; Griffor, M.C.; Kamath, A.V.; et al. Identification, characterization, and crystal structure of the omega class glutathione transferases. J. Biol. Chem. 2000, 275, 24798–24806. [Google Scholar] [CrossRef] [Green Version]
- Hughes, M.M.; Hooftman, A.; Angiari, S.; Tummala, P.; Zaslona, Z.; Runtsch, M.C.; McGettrick, A.F.; Sutton, C.E.; Diskin, C.; Rooke, M.; et al. Glutathione Transferase Omega-1 Regulates NLRP3 Inflammasome Activation through NEK7 Deglutathionylation. Cell Rep. 2019, 29, 151–161.e5. [Google Scholar] [CrossRef] [Green Version]
- Ding, C.; Wei, H.; Sun, R.; Zhang, J.; Tian, Z. Hepatocytes proteomic alteration and seroproteome analysis of HBV-transgenic mice. Proteomics 2009, 9, 87–105. [Google Scholar] [CrossRef] [PubMed]
- Cho, I.J.; Ki, S.H.; Brooks, C.; Kim, S.G. Role of hepatitis B virus X repression of C/EBPβ activity in the down-regulation of glutathione S-transferase A2 gene: Implications in other phase II detoxifying enzyme expression. Xenobiotica 2009, 39, 182–192. [Google Scholar] [CrossRef]
- Yi, Y.S.; Park, S.G.; Byeon, S.M.; Kwon, Y.G.; Jung, G. Hepatitis B virus X protein induces TNF-α expression via down-regulation of selenoprotein P in human hepatoma cell line, HepG2. Biochim. Biophys. Acta Mol. Basis Dis. 2003, 1638, 249–256. [Google Scholar] [CrossRef] [Green Version]
- Himoto, T.; Masaki, T. Current trends of essential trace elements in patients with chronic liver diseases. Nutrients 2020, 12, 2084. [Google Scholar] [CrossRef]
- Niu, D.; Zhang, J.; Ren, Y.; Feng, H.; Chen, W.N. HBx genotype D represses GSTP1 expression and increases the oxidative level and apoptosis in HepG2 cells. Mol. Oncol. 2009, 3, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Alavian, S.M.; Showraki, A. Hepatitis B and its relationship with oxidative stress. Hepat. Mon. 2016, 16, e37973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Fan, Y.C.; Sun, F.K.; Zhao, Z.H.; Wang, L.Y.; Hu, L.H.; Yin, Y.P.; Li, T.; Gao, S.; Wang, K. Peripheral type i interferon receptor correlated with oxidative stress in chronic hepatitis b virus infection. J. Interf. Cytokine Res. 2013, 33, 405–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisgin, N.T.; Aydin, B.K.; Sarikaya, H.; Tanyel, E.; Esen, S.; Sunbul, M.; Leblebicioglu, H. Oxidative stress and antioxidant defense in patients with chronic hepatitis B. Clin. Lab. 2012, 58, 273–280. [Google Scholar]
- Shaban, N.Z.; Salem, H.H.; Elsadany, M.A.; Ali, B.A.; Hassona, E.M.; Mogahed, F.A.K. Alterations in Lipid Peroxidation and Antioxidants in Patients’ with Different Stages of Hepatitis B Virus Infection in Egypt. Life Sci. J. 2014, 11, 960–967. [Google Scholar]
- Qian, L.; Wang, W.; Zhou, Y.; Ma, J. Effects of reduced glutathione therapy on chronic hepatitis B. Cent. Eur. J. Immunol. 2017, 42, 2015–2018. [Google Scholar] [CrossRef] [Green Version]
- Melhem, A.; Stern, M.; Shibolet, O.; Israeli, E.; Ackerman, Z.; Pappo, O.; Hemed, N.; Rowe, M.; Ohana, H.; Zabrecky, G.; et al. Treatment of chronic hepatitis C virus infection via antioxidants: Results of a phase I clinical trial. J. Clin. Gastroenterol. 2005, 39, 737–742. [Google Scholar] [CrossRef]
- Barakat, E.M.F.; El Wakeel, L.M.; Hagag, R.S. Effects of Nigella sativa on outcome of hepatitis C in Egypt. World J. Gastroenterol. 2013, 19, 2529–2536. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.; Zahoor, A.; Ibrahim, M.; Younus, M.; Nawaz, S.; Naseer, R.; Akram, Q.; Deng, C.L.; Ojha, S.C. Enhanced Efficacy of Direct-Acting Antivirals in Hepatitis C Patients by Coadministration of Black Cumin and Ascorbate as Antioxidant Adjuvants. Oxid. Med. Cell. Longev. 2020, 2020, 7087921. [Google Scholar] [CrossRef] [PubMed]
- Gunduz, H.; Karabay, O.; Tamer, A.; Özaras, R.; Mert, A.; Tabak, Ö.F. N-acetyl cysteine therapy in acute viral hepatitis. World J. Gastroenterol. 2003, 9, 2698–2700. [Google Scholar] [CrossRef]
- Grant, P.R.; Black, A.; Garcia, N.; Prieto, J.; Garson, J.A. Combination therapy with interferon-alpha plus N-acetyl cysteine for chronic hepatitis C: A placebo controlled double-blind multicentre study. J. Med. Virol. 2000, 442, 439–442. [Google Scholar] [CrossRef]
- Shohrati, M.; Dermanaki, F.; Babaei, F.; Alavian, S.M. Evaluation of the effects of Oral N-Acetylcysteine and a Placebo in Paraclinical and Oxidative Stress Parameters of Patients with Chronic Hepatitis B. Hepat. Mon. 2010, 10, 95–100. [Google Scholar]
- Look, M.P.; Gerard, A.; Rao, G.S.; Sudhop, T.; Fischer, H.P.; Sauerbruch, T.; Spengler, U. Interferon/antioxidant combination therapy for chronic hepatitis C—A controlled pilot trial. Antivir. Res. 1999, 43, 113–122. [Google Scholar] [CrossRef]
- Wagoner, J.; Negash, A.; Kane, O.J.; Martinez, L.E.; Nahmias, Y.; Bourne, N.; Owen, D.M.; Grove, J.; Brimacombe, C.; McKeating, J.A.; et al. Multiple effects of silymarin on the hepatitis C virus lifecycle. Hepatology 2010, 51, 1912–1921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umetsu, T.; Inoue, J.; Kogure, T.; Kakazu, E.; Ninomiya, M.; Iwata, T.; Takai, S.; Nakamura, T.; Sano, A.; Shimosegawa, T. Inhibitory effect of silibinin on hepatitis B virus entry. Biochem. Biophys. Rep. 2018, 14, 20–25. [Google Scholar] [CrossRef]
- Wu, Y.F.; Fu, S.L.; Kao, C.H.; Yang, C.W.; Lin, C.H.; Hsu, M.T.; Tsai, T.F. Chemopreventive effect of silymarin on liver pathology in HBV X protein transgenic mice. Cancer Res. 2008, 68, 2033–2042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polyak, S.J.; Ferenci, P.; Pawlotsky, J.M. Hepatoprotective and antiviral functions of silymarin components in hepatitis C virus infection. Hepatology 2013, 57, 1262–1271. [Google Scholar] [CrossRef] [Green Version]
- Gordon, A.; Hobbs, D.A.; Bowden, D.S.; Bailey, M.J.; Mitchell, J.; Francis, A.J.P.; Roberts, S.K. Effects of Silybum marianum on serum hepatitis C virus RNA, alanine aminotransferase levels and well-being in patients with chronic hepatitis C. J. Gastroenterol. Hepatol. 2006, 21, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Hawke, R.L.; Schrieber, S.J.; Soule, T.A.; Wen, Z.; Smith, P.C.; Reddy, K.R.; Wahed, A.S.; Belle, S.H.; Afdhal, N.H.; Navarro, V.J.; et al. Silymarin ascending multiple oral dosing phase I study in noncirrhotic patients with chronic hepatitis C. J. Clin. Pharmacol. 2010, 50, 434–449. [Google Scholar] [CrossRef] [PubMed]
- Fried, M.W.; Navarro, V.J.; Afdhal, N.; Belle, S.H.; Wahed, A.S.; Hawke, R.L.; Doo, E.; Meyers, C.M.; Reddy, K.R. Effect of silymarin (milk thistle) on liver disease in patients with chronic hepatitis C unsuccessfully treated with interferon therapy: A randomized controlled trial. JAMA 2012, 308, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Wei, F.; Liu, S.K.; Liu, X.Y.; Li, Z.J.; Li, B.; Zhou, Y.L.; Zhang, H.Y.; Li, Y.W. Meta-analysis: Silymarin and its combination therapy for the treatment of chronic hepatitis B. Eur. J. Clin. Microbiol. Infect. Dis. 2013, 32, 657–669. [Google Scholar] [CrossRef]
- Mohamadkhani, A. On the potential role of intestinal microbial community in hepatocarcinogenesis in chronic hepatitis B. Cancer Med. 2018, 7, 3095–3100. [Google Scholar] [CrossRef] [Green Version]
- Fooladi, A.A.I.; Hosseini, H.M.; Nourani, M.R.; Khani, S.; Alavian, S.M. Probiotic as a novel treatment strategy against liver disease. Hepat. Mon. 2013, 13, e7521. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Yu, X.; Lu, H.; Xiao, D.; Mao, W.; Li, L. Antioxidant protective effects of lactitol against endotoxemia in patients with chronic viral hepatitis. Mol. Med. Rep. 2013, 7, 401–405. [Google Scholar] [CrossRef]
- Zeng, Y.; Chen, S.; Fu, Y.; Wu, W.; Chen, T.; Chen, J.; Yang, B.; Ou, Q. Gut microbiota dysbiosis in patients with hepatitis B virus–induced chronic liver disease covering chronic hepatitis, liver cirrhosis and hepatocellular carcinoma. J. Viral Hepat. 2020, 27, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Sehgal, R.; Bedi, O.; Trehanpati, N. Role of Microbiota in Pathogenesis and Management of Viral Hepatitis. Front. Cell. Infect. Microbiol. 2020, 10, 341. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marra, M.; Sordelli, I.M.; Lombardi, A.; Lamberti, M.; Tarantino, L.; Giudice, A.; Stiuso, P.; Abbruzzese, A.; Sperlongano, R.; Accardo, M.; et al. Molecular targets and oxidative stress biomarkers in hepatocellular carcinoma: An overview. J. Transl. Med. 2011, 9, 171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janevska, D.; Chaloska-ivanova, V.; Janevski, V. Hepatocellular Treatment Carcinoma: Risk Diagnosis and Treatment. Open Access Maced. J. Med. Sci. 2015, 3, 732–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haque, E.; Rezaul Karim, M.; Teeli, A.S.; Śmiech, M.; Leszczynski, P.; Winiarczyk, D.; Parvanov, E.D.; Atanasov, A.G.; Taniguchi, H. Molecular mechanisms underlying hepatocellular carcinoma induction by aberrant nrf2 activation-mediated transcription networks: Interaction of nrf2-keap1 controls the fate of hepatocarcinogenesis. Int. J. Mol. Sci. 2020, 21, 5378. [Google Scholar] [CrossRef]
- Sun, J.; Zhou, C.; Ma, Q.; Chen, W.; Atyah, M.; Yin, Y.; Fu, P.; Liu, S.; Hu, B.; Ren, N.; et al. High GCLC level in tumor tissues is associated with poor prognosis of hepatocellular carcinoma after curative resection. J. Cancer 2019, 10, 3333–3343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Ou, Z.; Chen, R.; Niu, X.; Chen, D.; Kang, R.; Tang, D. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 2016, 63, 173–184. [Google Scholar] [CrossRef]
- Shao, S.; Duan, W.; Xu, Q.; Li, X.; Han, L.; Li, W.; Zhang, D.; Wang, Z.; Lei, J. Curcumin suppresses hepatic stellate cell-induced hepatocarcinoma angiogenesis and invasion through downregulating CTGF. Oxid. Med. Cell. Longev. 2019, 2019, 8148510. [Google Scholar] [CrossRef]
- Cheng, C.-S.; Wang, N.; Feng, Y. Selenium in the Prevention and Treatment of Hepatocellular Carcinoma: From Biomedical Investigation to Clinical Application. In Importance of Selenium in the Environment and Human Health; IntechOpen: London, UK, 2020. [Google Scholar]
- Brigelius-Flohé, R.; Kipp, A. Glutathione peroxidases in different stages of carcinogenesis. Biochim. Biophys. Acta Gen. Subj. 2009, 1790, 1555–1568. [Google Scholar] [CrossRef]
- Liu, T.; Kan, X.F.; Ma, C.; Chen, L.L.; Cheng, T.T.; Zou, Z.W.; Li, Y.; Cao, F.J.; Zhang, W.J.; Yao, J.; et al. GPX2 overexpression indicates poor prognosis in patients with hepatocellular carcinoma. Tumor Biol. 2017, 39, 28635398. [Google Scholar] [CrossRef] [Green Version]
- Guerriero, E.; Capone, F.; Accardo, M.; Sorice, A.; Costantini, M.; Colonna, G.; Castello, G.; Costantini, S. GPX4 and GPX7 over-expression in human hepatocellular carcinoma tissues. Eur. J. Histochem. 2015, 59, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Ghalia, A.H.A.; Fouad, I.M. Glutathione and Its Metabolizing Enzymes in Patients with Different Benign and Malignant Diseases. Clin. Biochem. 2001, 33, 657–662. [Google Scholar] [CrossRef]
- Czeczot, H.; Ścibior, D.; Skrzycki, M.; Podsiad, M. Glutathione and GSH-dependent enzymes in patients with liver cirrhosis and hepatocellular carcinoma. Acta Biochim. Pol. 2006, 53, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Zhao, X.P.; Wang, L.Y.; Gao, S.; Zhao, J.; Fan, Y.C.; Wang, K. Glutathione S-transferase P1 correlated with oxidative stress in hepatocellular carcinoma. Int. J. Med. Sci. 2013, 10, 683–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Cai, Y.; Chen, H.; Mao, L. Clinical significance and association of GSTP1 hypermethylation with hepatocellular carcinoma: A meta-analysis. J. Cancer Res. Ther. 2018, 14, S486–S489. [Google Scholar] [CrossRef]
- Huang, Z.Z.; Chen, C.; Zeng, Z.; Yang, H.; Oh, J.; Chen, L.; Lu, S.C. Mechanism and significance of increased glutathione level in human hepatocellular carcinoma and liver regeneration. FASEB J. 2011, 15, 19–21. [Google Scholar] [CrossRef]
- Diaz Vivancos, P.; Wolff, T.; Markovic, J.; Pallardò, F.V.; Foyer, C.H. A nuclear glutathione cycle within the cell cycle. Biochem. J. 2010, 178, 169–178. [Google Scholar] [CrossRef] [Green Version]
- Cheng, S.; Liu, H.; Chen, S.; Lin, P.; Lai, C.; Huang, C. Changes of Oxidative Stress, Glutathione, and Its Dependent Antioxidant Enzyme Activities in Patients with Hepatocellular Carcinoma before and after Tumor Resection. PLoS ONE 2017, 12, e0170016. [Google Scholar] [CrossRef] [PubMed]
- Baulies, A.; Montero, J.; Matías, N.; Insausti, N.; Terrones, O.; Basañez, G.; Vallejo, C.; de La Rosa, L.C.; Martinez, L.; Robles, D.; et al. The 2-oxoglutarate carrier promotes liver cancer by sustaining mitochondrial GSH despite cholesterol loading. Redox Biol. 2018, 14, 164–177. [Google Scholar] [CrossRef] [PubMed]
- Miyanishi, K.; Hoki, T.; Tanaka, S.; Kato, J. Prevention of hepatocellular carcinoma: Focusing on antioxidant therapy. World J. Hepatol. 2015, 7, 593–599. [Google Scholar] [CrossRef]
- Brandon-Warner, E.; Sugg, J.A.; Schrum, L.W.; McKillop, I.H. Silibinin inhibits ethanol metabolism and ethanol-dependent cell proliferation in an in vitro model of hepatocellular carcinoma. Cancer Lett. 2010, 291, 120–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramakrishnan, G.; Lo Muzio, L.; Elinos-Báez, C.M.; Jagan, S.; Augustine, T.A.; Kamaraj, S.; Anandakumar, P.; Devaki, T. Silymarin inhibited proliferation and induced apoptosis in hepatic cancer cells. Cell Prolif. 2009, 42, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, G.; Raghavendran, H.R.B.; Vinodhkumar, R.; Devaki, T. Suppression of N-nitrosodiethylamine induced hepatocarcinogenesis by silymarin in rats. Chem. Biol. Interact. 2006, 161, 104–114. [Google Scholar] [CrossRef]
- Stagos, D.; Amoutzias, G.D.; Matakos, A.; Spyrou, A.; Tsatsakis, A.M.; Kouretas, D. Chemoprevention of liver cancer by plant polyphenols. Food Chem. Toxicol. 2012, 50, 2155–2170. [Google Scholar] [CrossRef] [PubMed]
- Vásquez-Garzón, V.; Arellanes-Robledo, J.; García-Román, R.; Aparicio-Rautista, D.; Villa-Treviño, S. Inhibition of reactive oxygen species and pre-neoplastic lesions by quercetin through an antioxidant defense mechanism. Free Radic. Res. 2009, 43, 128–137. [Google Scholar] [CrossRef]
- Wang, L.; Zhan, J.; Huang, W. Grape seed proanthocyanidins induce apoptosis and cell cycle arrest of hepg2 cells accompanied by induction of the MAPK pathway and NAG-1. Antioxidants 2020, 9, 1200. [Google Scholar] [CrossRef]
- Fatima, N.; Akhtar, T.; Sheikh, N. Prebiotics: A Novel Approach to Treat Hepatocellular Carcinoma. Can. J. Gastroenterol. Hepatol. 2017, 2017, 6238106. [Google Scholar] [PubMed]
- Traverso, N.; Ricciarelli, R.; Nitti, M.; Marengo, B.; Furfaro, A.L.; Pronzato, M.A.; Marinari, U.M.; Domenicotti, C. Role of glutathione in cancer progression and chemoresistance. Oxid. Med. Cell. Longev. 2013, 2013, 972913. [Google Scholar] [CrossRef] [Green Version]
- Estrela, J.M.; Ortega, A.; Obrador, E. Glutathione in cancer biology and therapy. Crit. Rev. Clin. Lab. Sci. 2006, 43, 143–181. [Google Scholar] [CrossRef]
- Duan, B.; Huang, C.; Bai, J.; Zhang, Y.L.; Wang, X.; Yang, J.; Li, J. Multidrug Resistance in Hepatocellular Carcinoma. In Hepatocellular Carcinoma; Codon Publications: Brisbane, Australia, 2019; pp. 141–158. [Google Scholar]
- Lohitesh, K.; Chowdhury, R.; Mukherjee, S. Resistance a major hindrance to chemotherapy in hepatocellular carcinoma: An insight. Cancer Cell Int. 2018, 18, 44. [Google Scholar] [CrossRef] [PubMed]
- Tompkins, S.C.; Sheldon, R.D.; Rauckhorst, A.J.; Noterman, M.F.; Solst, S.R.; Buchanan, J.L.; Mapuskar, K.A.; Pewa, A.D.; Gray, L.R.; Oonthonpan, L.; et al. Disrupting Mitochondrial Pyruvate Uptake Directs Glutamine into the TCA Cycle away from Glutathione Synthesis and Impairs Hepatocellular Tumorigenesis. Cell Rep. 2019, 28, 2608–2619.e6. [Google Scholar] [CrossRef] [PubMed]
- Duval, A.P.; Troquier, L.; Silva, O.D.S.; Demartines, N.; Dormond, O. Diclofenac potentiates sorafenib-based treatments of hepatocellular carcinoma by enhancing oxidative stress. Cancers 2019, 11, 1453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Vijgh, W.J.F. Protection of Normal Tissues from the Cytotoxic Effects of Chemotherapy and Radiation by Amifostine (WR-2721): Preclinical Aspects. Eur. J. Cancer Part A 1995, 31, S1–S7. [Google Scholar] [CrossRef]
- Sidi, V.; Arsos, G.; Papakonstantinou, E.; Hatzipantelis, E.; Fragandrea, I.; Gombakis, N.; Koliouskas, E. Use of amifostine in the treatment of recurrent solid tumours in children. Hippokratia 2007, 11, 25–29. [Google Scholar] [PubMed]
- Feng, M.; Smith, D.E.; Normolle, D.P.; Knol, J.A.; Pan, C.C.; Ben-Josef, E.; Lu, Z.; Feng, M.R.; Chen, J.; Ensminger, W.; et al. A phase i clinical and pharmacology study using amifostine as a radioprotector in dose-escalated whole liver radiation therapy. Int. J. Radiat. Oncol. Biol. Phys. 2012, 83, 1441–1447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Symon, Z.; Levi, M.; Ensminger, W.D.; Smith, D.E.; Lawrence, T.S. Selective radioprotection of hepatocytes by systemic and portal vein infusions of amifostine in a rat liver tumor model. Int. J. Radiat. Oncol. Biol. Phys. 2001, 50, 473–478. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vairetti, M.; Di Pasqua, L.G.; Cagna, M.; Richelmi, P.; Ferrigno, A.; Berardo, C. Changes in Glutathione Content in Liver Diseases: An Update. Antioxidants 2021, 10, 364. https://doi.org/10.3390/antiox10030364
Vairetti M, Di Pasqua LG, Cagna M, Richelmi P, Ferrigno A, Berardo C. Changes in Glutathione Content in Liver Diseases: An Update. Antioxidants. 2021; 10(3):364. https://doi.org/10.3390/antiox10030364
Chicago/Turabian StyleVairetti, Mariapia, Laura Giuseppina Di Pasqua, Marta Cagna, Plinio Richelmi, Andrea Ferrigno, and Clarissa Berardo. 2021. "Changes in Glutathione Content in Liver Diseases: An Update" Antioxidants 10, no. 3: 364. https://doi.org/10.3390/antiox10030364
APA StyleVairetti, M., Di Pasqua, L. G., Cagna, M., Richelmi, P., Ferrigno, A., & Berardo, C. (2021). Changes in Glutathione Content in Liver Diseases: An Update. Antioxidants, 10(3), 364. https://doi.org/10.3390/antiox10030364