
Potential Role of Antioxidant and Anti-Inflammatory Therapies to Prevent Severe SARS-Cov-2 Complications





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
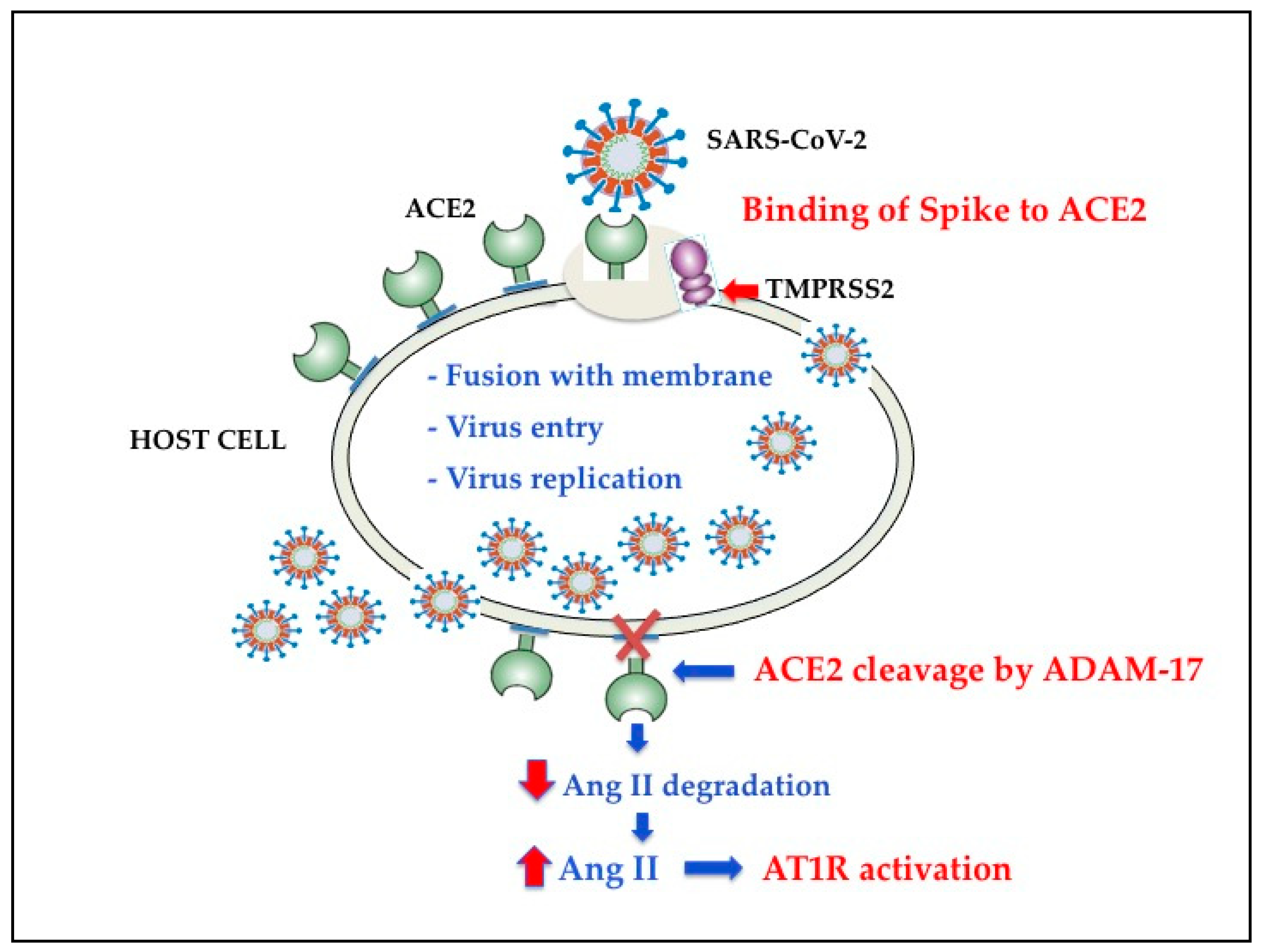
2. SARS-CoV-2 Cell Entry Mechanisms
2.1. SARS-CoV-2 Structural Basis
2.2. Structural Basis and Function of ACE2 Receptor
3. TMPRSS2 and SARS-CoV-2
4. Oxidative Stress and Inflammation Associated with SARS-CoV-2 Infection
4.1. Oxidative Stress (OS) in SARS-CoV-2 Infection
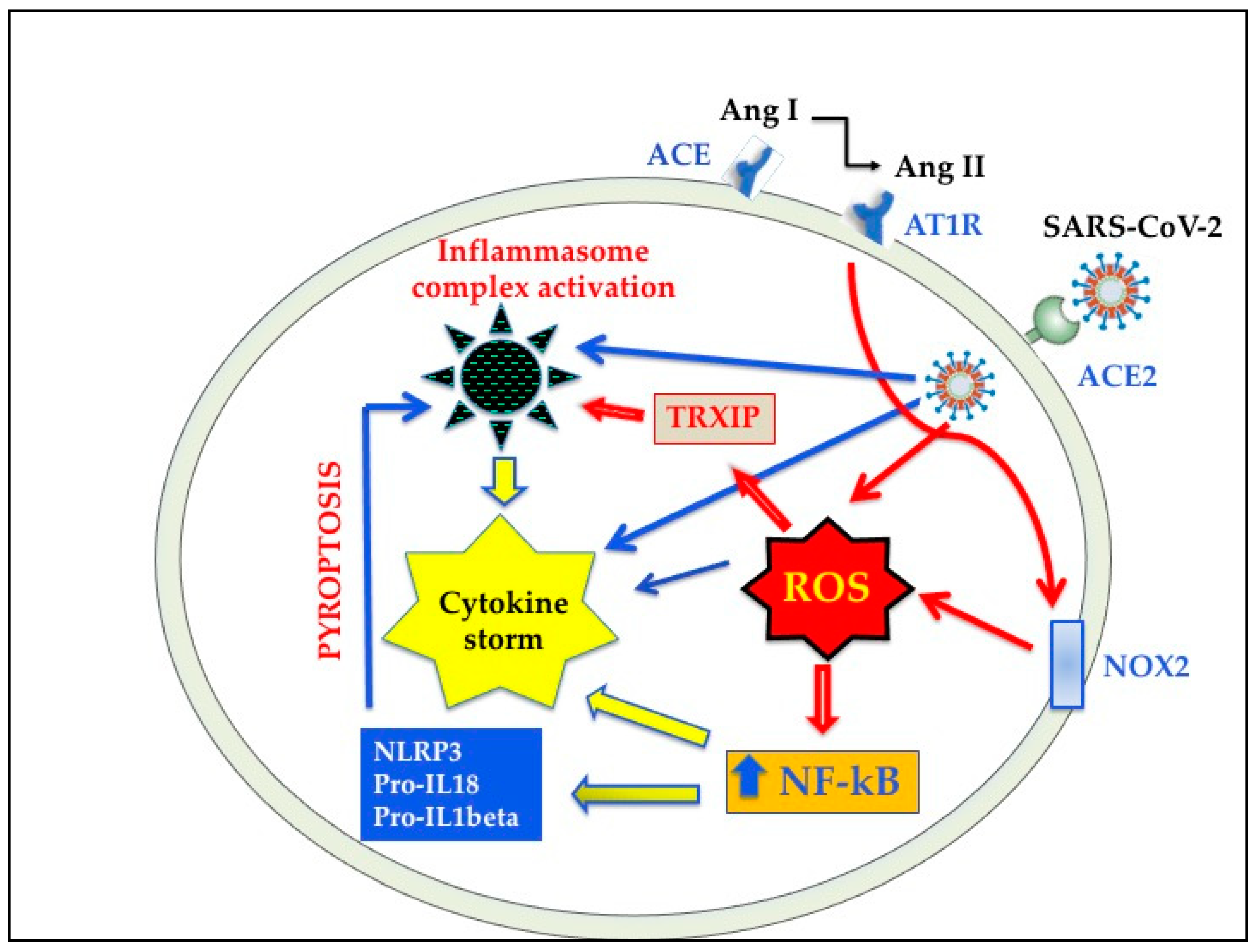
4.2. Cross Talks between Oxidative Stress and Inflammation in SARS-CoV-2 Infection
5. Rationale for Antioxidant and Anti-Inflammatory Therapies against COVID-19 Complications
5.1. Radical Scavengers
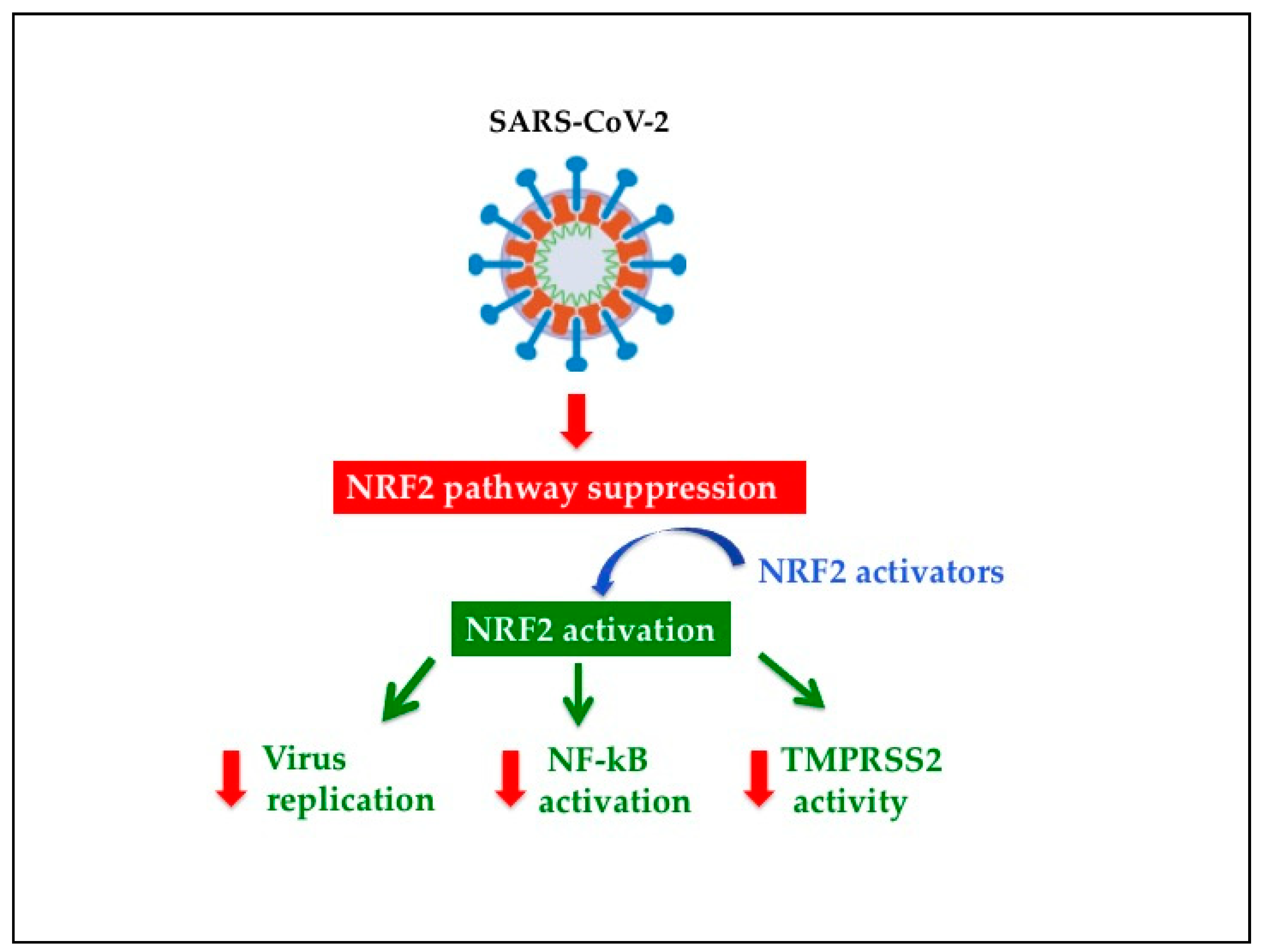
5.2. NRF2 Activators
5.3. Delivery of Soluble ACE2
5.4. Inhibitors of NLRP3 Inflammasome
5.5. Glucocorticoids (GCs) and Non-Steroidal Anti-Inflammatory Drugs (NSAIDs)
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACE | Angiotensin Converting Enzyme |
| Ang | Angiotensin |
| AT1R | Angiotensin II Type-1 Receptor |
| ADAM17 | A Disintegrin And Metalloproteinase17 |
| COX | cyclooxygenase |
| COVID-19 | Coronavirus disease 2019 |
| DAMPs | damage-associated molecular patterns |
| DMF | Dimethyl Fumarate |
| EMA | European Medicines Agency |
| FiO2 | Fraction Of Inspired Oxygen |
| FP | Fusion protein |
| GCs | Glucocorticoids |
| GSH | Glutathione |
| HO | Heme-Oxygenase |
| Hb | Hemoglobin |
| Keap-1 | Kelch-like enoyl-CoA hydratase-associated protein 1 |
| IFN | Interferon |
| IL | Interleukin |
| MAVS | Mitochondrial antiviral signaling system; |
| MCP1 | monocyte chemoattractant protein-1 |
| MEA | s-acetyl-mercaptoethylamine |
| MERS | Middle East respiratory syndrome |
| NAC | N-acetylcysteine |
| NETs | Neutrophil extracellular traps |
| NLRP3 | NOD-like receptors protein 3 |
| NRP1 | Neuropilin-1 |
| NOX | Nicotinamide adenine dinucleotide phosphate (NADPH) oxidases |
| NF-kB | Nuclear Factor-κB |
| NRF2 | Nuclear factor erythroid 2 p45-related factor2 |
| NSAIDs | non-steroidal anti-inflammatory drugs |
| 4-OI | 4-Octyl-Itaconate |
| Orf9b | Open reading frame-9b |
| OS | Oxidative stress |
| OxPLs | Oxidized phospholipids |
| PAI-1 | Plasminogen Activator Inhibitor-1 |
| PAMPs | Pathogen-associated molecular patterns |
| RAS | Renin-angiotensin system |
| RBD | Receptor-binding domain |
| rh | Recombinant Human |
| ROS | Reactive oxygen species |
| SARS-CoV-2 | Severe Acute Respiratory Syndrome (SARS)-like Coronavirus |
| S protein | Viral Membrane Spike protein |
| S1 | Receptor-binding subunit |
| S2 | Membrane fusion subunit |
| scRNA-seq | single cell RNA-sequence |
| TRXIP | Thioredoxin Interacting/Inhibiting Protein; |
| TOM | Translocase of Outer Membrane |
| TMPRSS2 | Transmembrane protease serine 2 |
| WHO | World Health Organization |
References
- Li, Q.; Guan, X.; Wu, P.; Wang, X.; Zhou, L.; Tong, Y.; Ren, R.; Leung, K.S.M.; Lau, E.H.Y.; Wong, J.Y.; et al. Early transmission dynamics in Wuhan, China, of novel coronavirus infected pneumonia. N. Engl. J. Med. 2020, 382, 1199–1207. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Qamar, M.T.; Alqahtani, S.M.; Alamri, M.A.; Chen, L.L. Structural basis of SARSCoV-2 3CLpro and anti-COVID-19 drug discovery from medicinal plants. J. Pharm. Anal. 2020, 4, 313–319. [Google Scholar] [CrossRef]
- Chan, J.W.; Ng, C.K.; Chan, Y.H.; Mok, T.Y.; Lee, S.; Chu, S.Y.; Law, W.L.; Lee, M.P.; Li, P.C. Short term outcome and risk factors for adverse clinical outcomes in adults with severe acute respiratory syndrome [SARS]. Thorax 2003, 58, 686–689. [Google Scholar] [CrossRef] [Green Version]
- Badawi, A.; Ryoo, S.G. Prevalence of comorbidities in the Middle East respiratory syndrome coronavirus [MERS-CoV]: A systematic review and meta-analysis. Int. J. Infect. Dis. 2016, 49, 129–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, N.; Zhou, M.; Dong, X.; Qu, J.; Gong, F.; Han, Y.; Qiu, Y.; Wang, J.; Liu, Y.; Wei, Y.; et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet 2020, 395, 507–513. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Fang, Y.Y.; Deng, Y.; Liu, W.; Wang, M.F.; Ma, J.P.; Xiao, W.; Wang, Y.N.; Zhong, M.H.; Li, C.H.; et al. Clinical characteristics of novel coronavirus cases in tertiary hospitals in Hubei Province. Chin. Med. J. 2020, 133, 1025–1031. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Hu, B.; Hu, C.; Zhu, F.; Liu, X.; Zhang, J.; Wang, B.; Xiang, H.; Cheng, Z.; Xiong, Y.; et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in Wuhan, China. JAMA 2020, 323, 1061–1069. [Google Scholar] [CrossRef]
- Xu, Z.; Shi, L.; Wang, Y.; Zhang, J.; Huang, L.; Zhang, C.; Liu, S.; Zhao, P.; Liu, H.; Zhu, L.; et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir. Med. 2020, 8, 420–422. [Google Scholar] [CrossRef]
- Han, H.; Xie, L.; Liu, R.; Yang, J.; Liu, F.; Wu, K.; Chen, L.; Hou, W.; Feng, Y.; Zhu, C. Analysis of heart injury laboratory parameters in 273 COVID-19 patients in one hospital in Wuhan, China. J. Med. Virol. 2020, 92, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult in patients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Guan, W.J.; Ni, Z.Y.; Hu, Y.; Liang, W.H.; Ou, C.Q.; He, J.X.; Liu, L.; Shan, H.; Lei, C.L.; Hui, D.; et al. Clinical characteristics of coronavirus disease 2019 in China. N. Engl. J. Med. 2020, 82, 1708–1720. [Google Scholar] [CrossRef]
- Li, Z.; Wu, M.; Yao, J.; Guo, J.; Liao, X.; Song, S.; Li, J.; Duan, G.; Zhou, Y.; Wu, X.; et al. Caution on kidney dysfunctions of COVID-19 patients. medRxiv 2020. [Google Scholar] [CrossRef]
- Mao, L.; Wang, M.; Chen, S.; He, Q.; Chang, J.; Hong, C.; Zhou, Y.; Wang, D.; Li, Y.; Jin, H.; et al. Neurologic manifestations of hospitalized patients with COVID-19 in Wuhan, China: A retrospective case series study. JAMA Neurol. 2020, 77, 683–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Xu, X.; Chen, Z.; Duan, J.; Hashimoto, K.; Yang, L.; Liu, C.; Yang, C. Nervous system involvement after infection with COVID-19 and other coronaviruses. Brain Behav. Immun. 2020, 87, 18–22. [Google Scholar] [CrossRef]
- Yao, X.H.; Li, T.Y.; He, Z.C.; Ping, Y.F.; Liu, H.W.; Yu, S.C.; Mou, H.M.; Wang, L.H.; Zhang, H.R.; Fu, W.J. A pathological report of three COVID-19 cases by minimally invasive autopsies. Zhonghua Bing Li Xue Za Zhi 2020, 8, 411–417. [Google Scholar]
- Xie, H.; Zhao, J.; Lian, N.; Lin, S.; Xie, Q.; Zhuo, H. Clinical characteristics of non-ICU hospitalized patients with coronavirus disease 2019 and liver injury: A Retrospective study. Liver Int. 2020, 40, 1321–1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Zheng, L.; Liu, L.; Zhao, M.; Xiao, J.; Zhao, Q. Liver impairment in COVID-19 patients: A retrospective analysis of 115 cases from a single center in Wuhan city, China. Liver Int. 2020, 40, 2095–2103. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Wu, D.; Guo, W.; Cao, Y.; Huang, D.; Wang, H.; Wang, T.; Zhang, X.; Chen, H.; Yu, H. Clinical and immunologic features in severe and moderate Coronavirus Disease 2019. J. Clin. Investig. 2020, 30, 2620–2629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, C.; Zhou, L.; Hu, Z.; Zhang, S.; Yang, S.; Tao, Y.; Xie, C.; Ma, K.; Shang, K.; Wang, W.; et al. Dysregulation of immune response in patients with COVID-19 in Wuhan, China. Clin. Infect. Dis. 2020, 71, 762–768. [Google Scholar] [CrossRef]
- Han, H.; Yang, L.; Liu, R.; Liu, F.; Wu, K.L.; Li, J.; Liu, X.H.; Zhu, C.L. Prominent changes in blood coagulation of patients with SARS-CoV-2 infection. Clin. Chem. Lab. Med. 2020, 58, 1116–1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, N.; Li, D.; Wang, X.; Sun, Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J. Thromb. Haemost. 2020, 18, 844–847. [Google Scholar] [CrossRef] [Green Version]
- Coronavirus WHO. COVID-19. WHO. 2020. Available online: https://who.sprinklr.com/ (accessed on 31 December 2020).
- Li, Y.; Zhou, W.; Yang, L.; You, R. Physiological and pathological regulation of ACE2, the SARS-CoV-2 receptor. Pharmacol. Res. 2020, 157, 104833. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Shang, J.; Graham, R.; Baric, R.S.; Li, F. Receptor recognition by novel coronavirus from Wuhan: An analysis based on decade-long structural studies of SARS. J. Virol. 2020, 94, e00127-20. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, J.H.; Li, W.; Choe, H.; Farzan, M. Angiotensin-converting enzyme 2: A functional receptor for SARS coronavirus. Cell. Mol. Life Sci. 2004, 61, 2738–2743. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Moore, M.J.; Vasilieva, N.; Sui, J.; Wong, S.K.; Berne, M.A.; Somasundaran, M.; Sullivan, J.L.; Luzuriaga, K.; Greenough, T.C.; et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003, 426, 450–454. [Google Scholar] [CrossRef] [Green Version]
- Kuba, K.; Imai, Y.; Rao, S.; Gao, H.; Guo, F.; Guan, B.; Huan, Y.; Yang, P.; Zhang, Y.; Deng, W.; et al. A crucial role of angiotensin converting enzyme 2 ACE2 in SARS coronavirus-induced lung injury. Nat. Med. 2005, 11, 875–879. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [Green Version]
- Jiang, S.; Hillyer, C.; Du, L. Neutralizing antibodies against SARS-CoV-2 and other human Coronaviruses. Trends Immunol. 2020, 41, 355–359. [Google Scholar] [CrossRef]
- Shang, J.; Wana, Y.; Luoa, C.; Yea, G.; Genga, Q.; Auerbacha, A.; Li, F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 2020, 181, 281–292. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [Green Version]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, Y.; Cao, D.; Zhang, Y.; Ma, J.; Qi, J.; Wang, Q.; Lu, G.; Wu, Y.; Yan, J.; Shi, Y.; et al. Cryo-EM structures of MERS-CoV and SARS-CoV spike glycoproteins reveal the dynamic receptor binding domains. Nat. Commun. 2017, 8, 15092. [Google Scholar] [CrossRef] [PubMed]
- Cantuti-Castelvetri, L.; Ojha, R.; Pedro, L.D.; Djannatian, M.; Franz, J.; Kuivanen, S.; van der Meer, F.; Kallio, K.; Kaya, T.; Anastasina, M.; et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science 2020, 370, 856–860. [Google Scholar] [CrossRef]
- Daly, J.L.; Simonetti, B.; Klein, K.; Chen, K.E.; Williamson, M.K.; Antón-Plágaro, C.; Deborah, K.; Shoemark, D.K.; Simón-Gracia, L.; Bauer, M.; et al. Neuropilin-1 is a host factor for SARS-CoV-2 infection. Science 2020, 370, 861–865. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, Q.; Guo, D.J. Emerging coronaviruses: Genome structure, replication, and pathogenesis. Med. Virol. 2020, 92, 418–423. [Google Scholar] [CrossRef]
- Chappell, M.C. Biochemical evaluation of the renin-angiotensin system: Then good, bad, and absolute? Am. J. Physiol. 2016, 310, 137–152. [Google Scholar] [CrossRef] [Green Version]
- Erdos, E.G. Conversion of angiotensin I to angiotensin II. Amer. J. Med. 1976, 60, 749–759. [Google Scholar] [CrossRef]
- Tikellis, C.; Thomas, M.C. Angiotensin-Converting Enzyme 2 [ACE2] is a key modulator of the Renin Angiotensin System in health and disease. Int. J. Pept. 2012, 25, 62–94. [Google Scholar] [CrossRef] [PubMed]
- Towler, P.; Staker, B.; Prasad, S.G.; Menon, S.; Tang, J.; Parsons, T.; Ryan, D.; Fisher, M.; Williams, D.; Natalie, A. ACE2 X-ray structures reveal a large hinge-bending motion important for inhibitor binding and catalysis. J. Biol. Chem. 2004, 279, 17996–18007. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Gheblawi, M.; Oudit, G.Y. Angiotensin converting enzyme 2: A double-edged sword. Circulation 2020, 142, 426–428. [Google Scholar] [CrossRef]
- Gheblawi, M.; Wang, K.; Viveiros, A.; Nguyen, Q.; Zhong, J.C.; Turner, A.J.; Raizada, M.K.; Grant, M.B.; Oudit, G.Y. Angiotensin-Converting Enzyme 2: SARS-CoV-2 Receptor and Regulator of the Renin-Angiotensin System: Celebrating the 20th Anniversary of the Discovery of ACE2. Circ. Res. 2020, 126, 1456–1474. [Google Scholar] [CrossRef]
- Burrell, L.M.; Johnston, C.I.; Tikellis, C.; Cooper, M.E. ACE2, a new regulator of the renin-angiotensin system. Trends Endocrinol. Metab. 2004, 15, 166–169. [Google Scholar] [CrossRef]
- Hikmet, F.; Méar, L.; Edvinsson, A.; Micke, P.; Uhlén, M.; Lindskog, C. The protein expression profile of ACE2 in human tissues. Mol. Syst. Biol. 2020, 16, e9610. [Google Scholar] [CrossRef]
- Zou, X.; Chen, K.; Zou, J.; Han, P.; Hao, J.; Han, Z. Single-cell RNA-seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to 2019-nCoV infection. Front. Med. 2020, 14, 185–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Datta, P.K.; Liu, F.; Fischer, T.; Rappaport, J.; Qin, X. SARS-CoV-2 pandemic and research gaps: Understanding SARS-CoV-2 interaction with the ACE2 receptor and implications for therapy. Theranostics 2020, 10, 7448–7464. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yang, Y.; Zhang, C.; Huang, F.; Wang, F.; Yuan, J.; Wang, Z.; Li, J.; Li, J.; Feng, C.; et al. Clinical and biochemical indexes from 2019-nCoV infected patients linked to viral loads and lung injury. Sci. China Life Sci. 2020, 63, 364–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, V.B.; Clarke, N.; Wang, Z.; Fan, D.; Parajuli, N.; Basu, R.; Putko, B.; Kassiri, Z.; Turner, A.J.; Oudit, G.Y. Angiotensin II induced proteolytic cleavage of myocardial ACE2 is mediated by TACE/ADAM-17: A positive feedback mechanism in the RAS. J. Mol. Cell. Cardiol. 2014, 66, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Derynck, R. Direct activation of TACE-mediated ectodomain shedding by p38 MAP kinase regulates EGF receptor-dependent cell proliferation. Mol. Cell. 2010, 37, 551–566. [Google Scholar] [CrossRef] [Green Version]
- Black, R.A.; Rauch, C.T.; Kozlosky, C.J.; Peschon, J.J.; Slack, J.L.; Wolfson, M.F.; Castner, B.J.; Stocking, K.L.; Reddy, P.; Srinivasan, S.; et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature 1997, 385, 729–733. [Google Scholar] [CrossRef]
- Bzowska, M.; Jura, N.; Lassak, A.; Black, R.A.; Bereta, J. Tumour necrosis factor-alpha stimulates expression of TNF-alpha converting enzyme in endothelial cells. Eur. J. Biochem. 2004, 271, 2808–2820. [Google Scholar] [CrossRef]
- Zablocki, D.; Sadoshima, J. Angiotensin II and Oxidative Stress in the Failing Heart. Antioxid. Redox Signal. 2013, 19, 1095–1109. [Google Scholar] [CrossRef]
- Dikalov, S.I.; Nazarewicz, R.R. Angiotensin II-Induced Production of Mitochondrial Reactive Oxygen Species: Potential Mechanisms and Relevance for Cardiovascular Disease. Antioxid. Redox Signal. 2013, 19, 1085–1094. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Vaarala, M.H.; Porvari, K.S.; Kellokumpu, S.; Kyllönen, A.P.; Vihko, P.T. Expression of transmembrane serine protease TMPRSS2 in mouse and human tissues. J. Pathol. 2001, 193, 134–140. [Google Scholar] [CrossRef]
- Metzger, E.; Willmann, D.; McMillan, J.; Forne, I.; Metzger, P.; Gerhardt, S.; Petroll, K.; von Maessenhausen, A.; Urban, S.; Schott, A.K.; et al. Assembly of methylated KDM1A and CHD1 drives androgen receptor-dependent transcription and translocation. Nat. Struct. Mol. Biol. 2016, 23, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Lukassen, S.; Chua, R.L.; Trefzer, T.; Kahn, N.C.; Schneider, M.; Muley, T.; Winter, H.; Meister, M.; Veith, C.; Boots, A.W.; et al. SARS-CoV-2 receptor ACE2 and TMPRSS2 are primarily expressed in bronchial transient secretory cells. EMBO J. 2020, 39, e105114. [Google Scholar] [CrossRef] [PubMed]
- Wrapp, N.D.; Wang, K.S.; Corbett, J.A.; Goldsmith, C.-L.; Hsieh, O.; Abiona, B.S.; Graham, J.S. McLellan, Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, M.; Kleine-Weber, H.; Pöhlmann, S. A multibasic cleavage site in the spike protein of SARS-CoV-2 is essential for infection of human lung cells. Mol. Cell 2020, 78, 779–784. [Google Scholar] [CrossRef]
- Lin, B.; Ferguson, C.; White, J.T.; Wang, S.; Vessella, R.; True, L.D.; Hood, L.; Nelson, P.S. Prostate-localized and androgen-regulated expression of the membrane bound serine protease TMPRSS2. Cancer Res. 1999, 59, 4180–4184. [Google Scholar]
- Yoo, S.; Pettersson, A.; Jordahl, K.M.; Lis, R.T.; Lindstrom, S.; Meisner, A.; Nuttall, E.J.; Stack, E.C.; Stampfer, M.J.; Kraft, K.; et al. Androgen receptor CAG repeat polymorphism and risk of TMPRSS2:ERG-positive prostate cancer. Cancer Epidemiol. Biomarkers Prev. 2014, 23, 2027–2031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikkonen, L.; Pihlajamaa, P.; Sahu, B.; Zhang, F.P.; Janne, O.A. Androgen receptor and androgen-dependent gene expression in lung. Mol. Cell Endocrinol. 2010, 317, 14–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richardson, S.; Hirsch, J.S.; Narasimhan, M.; Crawford, J.M.; McGinn, T.; Davidson, K.W.; Barnaby, D.P.; Becker, L.B.; Chelico, J.D.; Cohen, S.L.; et al. Presenting Characteristics, Comorbidities, and Outcomes among 5700 Patients Hospitalized with COVID-19 in the New York City Area. JAMA 2020, 323, 2052–2059. [Google Scholar] [CrossRef]
- Onder, G.; Rezza, G.; Brusaferro, S. Case-Fatality Rate and Characteristics of Patients Dying in Relation to COVID-19 in Italy. JAMA 2020, 323, 1775–1776. [Google Scholar] [CrossRef] [PubMed]
- Cattrini, C.; Bersanelli, M.; Latocca, M.M.; Conte, B.; Vallome, G.; Boccardo, F. Sex Hormones and Hormone Therapy during COVID-19 Pandemic: Implications for Patients with Cancer. Cancers 2020, 12, 2325. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Roche, L.; Mesta, F. Oxidative stress as key player in severe Acute Respiratory Syndrome Coronavirus (SARS-CoV) infection. Arch. Med. Res. 2020, 51, 384–387. [Google Scholar] [CrossRef]
- Khomich, O.A.; Kochetkov, S.N.; Bartosch, B.; Ivanov, A. Redox Biology of Respiratory viral infections. Viruses 2018, 10, 392. [Google Scholar] [CrossRef] [Green Version]
- Checconi, P.; De Angelis, M.; Marcocci, M.E.; Fraternale, A.; Magnani, M.; Palamara, A.T.; Nencioni, L. Redox-modulating agents in the treatment of viral Infections. Int. J. Mol. Sci. 2020, 21, 4084. [Google Scholar] [CrossRef]
- Cecchini, R.; Cecchini, A.L. SARS-CoV-2 infection pathogenesis is related to oxidative stress as a response to aggression. Med. Hypotheses 2020, 143, 101–102. [Google Scholar] [CrossRef] [PubMed]
- Merad, M.; Martin, J.C. Pathological inflammation in patients with COVID-19: A key role for monocytes and macrophages. Nat. Rev. Immunol. 2020, 20, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Chernyak, B.V.; Popova, E.N.; Prikhodko, A.S.; Grebenchikov, O.A.; Zinovkina, L.A.; Zinovkin, R.A. COVID-19 and oxidative stress. Biochemistry 2020, 85, 1543–1553. [Google Scholar]
- Fernandes, I.G.; de Brito, C.A.; Dos Reis, V.M.S.; Sato, M.N.; Pereira, N.Z. SARS-CoV-2 and other respiratory viruses: What does oxidative Stress have to do with it? Oxid. Med. Cell. Longev. 2020, 2020, 8844280. [Google Scholar] [CrossRef] [PubMed]
- Suhail, S.; Zajac, J.; Fossum, C.; Lowater, H.; McCracken, C.; Severson, N.; Laatsch, B.; Narkiewicz-Jodko, A.; Johnson, B.; Liebau, J.; et al. Role of oxidative Stress on SARS-CoV (SARS) and SARS-CoV-2 (COVID-19) Infection: A review. Protein J. 2020, 39, 644–656. [Google Scholar] [CrossRef]
- Violi, F.; Oliva, A.; Cangemi, R.; Ceccarelli, G.; Pignatelli, P.; Carnevale, R.; Cammisotto, V.; Lichtner, M.; Alessandri, F.; De Angelis, M.; et al. Nox2 activation in Covid-19. Redox Biol. 2020, 36, 101655. [Google Scholar] [CrossRef]
- Libby, P.; Luscher, T. COVID-19 is, in the end, an endothelial disease. Eur. Heart J. 2020, 41, 3038–3044. [Google Scholar] [CrossRef]
- Imai, Y.; Kuba, K.; Neely, G.G.; Yaghubian-Malhami, R.; Perkmann, T.; van Loo, G.; Ermolaeva, M.; Veldhuizen, R.; Leung, Y.H.; Wang, H.; et al. Identification of oxidative stress and Toll- like receptor 4 signaling as a key pathway of acute lung injury. Cell 2008, 133, 235–249. [Google Scholar] [CrossRef]
- Berliner, J.A.; Watson, A.D. A role for oxidized phospholipids in atherosclerosis. N. Engl. J. Med. 2005, 353, 9–11. [Google Scholar] [CrossRef]
- Owens, A.P.; Passam, F.H.; Antoniak, S.; Marshall, S.M.; McDaniel, A.L.; Rudel, L.; Williams, J.C.; Hubbard, B.K.; Dutton, J.A.; Wang, J.; et al. Monocyte tissue factor-dependent activation of coagulation in hypercholesterolemic mice and monkeys is inhibited by simvastatin. J. Clin. Investig. 2012, 122, 558–568. [Google Scholar]
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, J.; Sun, X.; Kang, R.; Tan, D. Ferroptosis: Process and function. Cell Death Differ. 2016, 23, 369–379. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Lai, C.L. SARS-CoV-2 infection: Can ferroptosis be a potential treatment target for multiple organ involvement. Cell Death Discov. 2020, 6, 130. [Google Scholar] [CrossRef] [PubMed]
- Edeas, M.; Saleh, J.; Peyssonnaux, C. Iron: Innocent bystander or vicious culprit in COVID-19 pathogenesis? Int. J. Infect. Dis. 2020, 97, 303–305. [Google Scholar] [CrossRef]
- Lin, C.W.; Lin, K.H.; Hsieh, T.H.; Shiu, S.Y.; Li, J.Y. Severe acute respiratory syndrome coronavirus 3C-like protease-induced apoptosis. FEMS Immunol. Med. Microbiol. 2006, 46, 375–380. [Google Scholar] [CrossRef] [Green Version]
- Collins, Y.; Chouchani, E.T.; James, A.M.; Menger, K.E.; Cocheme, H.M.; Murphy, M.P. Mitochondrial redox signalling at a glance. J. Cell Sci. 2012, 125, 801–806. [Google Scholar] [CrossRef] [Green Version]
- Dunn, J.D.; Alvarez, L.A.; Zhang, X.; Soldati, T. Reactive oxygen species and mitochondria: A nexus of cellular homeostasis. Redox Biol. 2015, 6, 472–485. [Google Scholar] [CrossRef]
- Anand, S.K.; Tikoo, S.K. Viruses as modulators of mitochondrial functions. Adv. Virol. 2013, 20, 738794. [Google Scholar] [CrossRef]
- de las Heras, N.; Giménez, V.M.M.; Ferder, L.; Manucha, W.; Lahera, V. Implications of Oxidative Stress and Potential Role of Mitochondrial Dysfunction in COVID-19: Therapeutic Efects of Vitamin D. Antioxidants 2020, 9, 897. [Google Scholar] [CrossRef]
- Burtscher, J.; Cappellano, G.; Omori, A.; Koshiba, T.; Millet, G.P. Mitochondria: In the Cross fire of SARS-CoV-2 and immunity. Science 2020, 23, 101631. [Google Scholar]
- Shi, C.S.; Qi, H.Y.; Boularan, C.; Huang, N.N.; Abu-Asab, M.; Shelhamer, J.H.; Kehrl, J.H. SARS-CoV ORF-9b suppresses innate immunity by targeting mitochondria and the MAVS/TRAF3/TRAF6 signalosome. J. Immunol. 2014, 193, 3080–3089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McBride, R.; Fielding, B.C. The role of severe acute respiratory syndrome (SARS)-coronavirus accessory proteins in virus pathogenesis. Viruses 2013, 4, 2902–2923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scherz-Shouval, R.; Elazar, Z. Regulation of autophagy by ROS: Physiology and pathology. Trends Biochem. Sci. 2011, 36, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Danielle, L. A SARS-CoV-2 protein interaction map reveals targets for drug-repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Zhang, H.; Meng, Q.; Xie, J.; Li, Y.; Chen, H.; Zheng, Y.; Wang, X.; Qi, H.; Zhang, J.; et al. SARS-CoV-2 Orf9b suppresses type I interferon responses by targeting TOM70. Cell. Mol. Immunol. 2020, 17, 998–1000. [Google Scholar] [CrossRef]
- Filadi, R.; Leal, N.S.; Schreiner, B.; Rossi, A.; Dentoni, G.; Pinho, C.M.; Wiehager, B.; Cieri, D.; Calì, T.; Pizzo, P. TOM70 sustains cell bioenergetics by promoting IP3R3-Mediated ER to mitochondria Ca(2+) transfer. Curr. Biol. 2018, 28, 369–382. [Google Scholar] [CrossRef] [Green Version]
- Sgarbanti, R.; Nencioni, L.; Amatore, D.; Coluccio, P.; Fraternale, A.; Sale, P.; Mammola, C.L.; Carpino, G.; Gaudio, E.; Magnani, M.; et al. Redox regulation of the influenza hemagglutinin maturation process: A new cell-mediated strategy for anti-influenza therapy. Antioxid. Redox Signal. 2011, 15, 593–606. [Google Scholar] [CrossRef]
- Cao, M.; Zhang, D.; Wang, Y.; Lu, Y.; Zhu, X.; Li, Y.; Xue, H.; Lin, Y.; Zhang, M.; Sun, Y.; et al. Clinical features of patients infected with the 2019 novel coronavirus [COVID-19] in Shanghai, China. medRxiv 2020. preprint. [Google Scholar] [CrossRef] [Green Version]
- Silvagno, F.; Vernone, A.; Pescarmona, G.P. The Role of Glutathione in Protecting against the Severe Inflammatory Response Triggered by COVID-19. Antioxidants 2020, 9, 624. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Chen, X.; Cai, Y.; Xia, J.; Zhou, X.; Xu, S.; Huang, H.; Zhang, L.; Zhou, X.; Du, C.; et al. Risk factors associated with acute respiratory distress syndrome and death in patients with coronavirus disease 2019 pneumonia in Wuhan, China. JAMA Intern. Med. 2020, 180, 934–943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruan, Q.; Yang, K.; Wang, W.; Jiang, L.; Song, J. Clinical predictors of mortality due to COVID-19 based on an analysis of data of 150 patients from Wuhan, China. Intensive Care Med. 2020, 46, 846–848. [Google Scholar] [CrossRef] [Green Version]
- Gong, J.; Dong, H.; Xia, Q.; Huang, Z.; Wang, D.; Zhao, Y.; Liu, W.; Tu, S.; Zhang, M.; Wang, Q.; et al. Correlation analysis between disease severity and inflammation-related parameters in patients with COVID-19 pneumonia. medRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Martinon, F. Detection of immune danger signals by NALP3. J. Leukoc. Biol. 2008, 83, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Siu, K.; Yuen, K.; Castano-Rodriguez, C.; Ye, Z.; Yeung, M.; Fung, S.; Yuan, S.; Chan, C.; Yuen, K.; Enjuanes, L.; et al. Severe acute respiratory syndrome Coronavirus ORF3a protein activates the NLRP3 inflammasome by promoting TRAF3-dependent ubiquitination of ASC. FASEB J. 2019, 33, 8865–8877. [Google Scholar] [CrossRef] [PubMed]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Fox, S.E.; Akmatbekov, A.; Harbert, J.L.; Li, G.; Brown, J.Q.; Vander Heide, R.S. Pulmonary and cardiac pathology in Covid-19: The first autopsy series from New Orleans. medRxiv 2020. [Google Scholar] [CrossRef]
- Tian, S.; Hu, W.; Niu, L.; Liu, H.; Xu, H.; Xiao, S.Y. Pulmonary pathology of early phase 2019 novel coronavirus [COVID-19] pneumonia in two patients with lung cancer. J. Thorac. Oncol. 2020, 15, 700–704. [Google Scholar] [CrossRef]
- Yang, M. Cell pyroptosis, a potential pathogenic mechanism of 2019-nCoV Infection, SSRN. Electron. J. 2020, 3527420. [Google Scholar]
- Cookson, B.T.; Brennan, M.A. Pro-inflammatory programmed cell death. Trends Microbiol. 2001, 9, 113–114. [Google Scholar] [CrossRef]
- Fink, S.L.; Cookson, B.T. Apoptosis, pyroptosis, and necrosis: Mechanistic description of dead and dying eukaryotic cells. Infect. Immun. 2005, 73, 1907–1916. [Google Scholar] [CrossRef] [Green Version]
- Faust, H.; Mangalmurti, N.S. Collateral damage: Necroptosis in the development of lung injury. Am. J. Phys. Lung Cell. Mol. Phys. 2020, 318, 215–225. [Google Scholar] [CrossRef]
- Sauler, M.; Bazan, I.S.; Lee, P.J. Cell death in the lung: The apoptosis-necroptosis axis. Annu. Rev. Physiol. 2019, 81, 375–402. [Google Scholar] [CrossRef]
- Ueno, H.; Matsuda, T.; Hashimoto, S.; Amaya, F.; Kitamura, Y.; Tanaka, M.; Kobayashi, A.; Maruyama, I.; Yamada, S.; Hasegawa, N.; et al. Contributions of high mobility group box protein in experimental and clinical acute lung injury. Am. J. Respir. Crit. Care Med. 2004, 170, 1310–1316. [Google Scholar]
- Liao, M.; Liu, Y.; Yuan, J.; Wen, Y.; Xu, G.; Zhao, J.; Cheng, L.; Li, J.; Wang, X.; Wang, F.; et al. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat. Med. 2020, 26, 842–844. [Google Scholar] [CrossRef] [PubMed]
- Mason, R.J. Pathogenesis of COVID-19 from a cell biologic perspective. Eur. Respir. J. 2020, 55, 2000607. [Google Scholar] [CrossRef] [Green Version]
- Aberdein, J.D.; Cole, J.; Bewley, M.A.; Marriott, H.M.; Dockrell, D.H. Alveolar macrophages in pulmonary host defence the unrecognized role of apoptosis as a mechanism of intracellular bacterial killing. Clin. Exp. Immunol. 2013, 174, 193–202. [Google Scholar]
- Losa García, J.E.; Rodríguez, F.M.; Martín de Cabo, M.R.; García Salgado, M.J.; Losada, J.P.; Villarón, L.G.; López, A.J.; Arellano, J.L. Evaluation of inflammatory cytokine secretion by human alveolar macrophages. Mediat. Inflamm. 1999, 8, 43–51. [Google Scholar] [CrossRef] [Green Version]
- Han, S.; Mallampalli, R.K. The acute respiratory distress syndrome: From mechanism to translation. J. Immunol. 2015, 194, 855–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frantzeskaki, F.; Armaganidis, A.; Orfanos, S.E. Immunothrombosis in acute respiratory distress syndrome: Cross talks between inflammation and coagulation. Respiration 2017, 93, 212–225. [Google Scholar] [CrossRef] [PubMed]
- Haouari, M. Platelet oxidative stress and its relationship with cardiovascular diseases in type 2 diabetes mellitus patients. Curr. Med. Chem. 2019, 26, 4145–4165. [Google Scholar] [CrossRef]
- Freedman, J.E. Oxidative stress and platelets. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 11–16. [Google Scholar] [CrossRef] [Green Version]
- Øynebråten, I.; Barois, N.; Bergeland, T.; Küchler, A.M.; Bakke, O.; Haraldsen, G. Oligomerized, filamentous surface presentation of RANTES/CCL5 on vascular endothelial cells. Sci. Rep. 2015, 6, 9261. [Google Scholar] [CrossRef] [Green Version]
- Sonmez, O.; Sonmez, M. Role of platelets in immune system and inflammation. Porto Biomed. J. 2017, 2, 311–314. [Google Scholar] [CrossRef]
- Finsterbusch, M.; Schrottmaier, W.; Kral-Pointner, J.B.; Salzmann, M.; Asinger, A. Measuring and interpreting platelet-leukocyte aggregates. Platelets 2018, 29, 677–685. [Google Scholar] [CrossRef]
- Sreeramkumar, V.; Adrover, J.M.; Ballesteros, I.; Cuartero, M.I.; Rossaint, J.; Bilbao, I.; Nácher, M.; Pitaval, C.; Radovanovic, I.; Fukui, Y.; et al. Neutrophils scan for activated platelets to initiate inflammation. Science 2014, 346, 1234–1238. [Google Scholar] [CrossRef] [Green Version]
- Engelmann, B.; Massberg, S. Thrombosis as an intravascular effector of innate Immunity. Nat. Rev. Immunol. 2013, 13, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Kimball, A.S.; Obi, A.T.; Diaz, J.A.; Henke, P.K. The emerging role of NETs in venous thrombosis and immunothrombosis. Front. Immunol. 2016, 7, 236. [Google Scholar] [CrossRef] [Green Version]
- Pfeiler, S.; Massberg, S.; Engelmann, B. Biological basis and pathological relevance of microvascular thrombosis. Thromb. Res. 2014, 133, 35–37. [Google Scholar] [CrossRef] [PubMed]
- Prabhakaran, P.; Ware, L.B.; White, K.E.; Cros, M.T.; Matthay, M.A.; Olman, M.A. Elevated levels of plasminogen activator inhibitor-1 in pulmonary edema fluid are associated with mortality in acute lung injury. Am. J. Phys. Lung Cell. Mol. Phys. 2003, 285, 20–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sapru, A.; Curley, M.A.Q.; Brady, S.; Matthay, M.A.; Flori, H. Elevated PAI-1 is associated with poor clinical outcomes in pediatric patients with acute lung Injury. Intens. Care Med. 2010, 36, 157–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, M.; Sun, Z.; Shao, M.; Yin, J.; Deng, Z.; Zhang, J.; Xing, L.; Yang, X.; Chen, B.L.; Dong, Z.; et al. Diagnostic and prognostic utility of tissue factor for severe sepsis and sepsis-induced acute lung injury. J. Transl. Med. 2015, 13, 172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadav, H.; Kor, D.J. Platelets in the pathogenesis of acute respiratory distress Syndrome. Amer. J. Physiol. Lung Cell. Mol. Physiol. 2015, 309, 915–923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gando, S.; Otomo, Y. Local hemostasis, immunothrombosis, and systemic disseminated intravascular coagulation in trauma and traumatic shock. Crit. Care 2015, 19, 72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuo, Y.; Yalavarthi, S.; Shi, H.; Gockman, K.; Zuo, M.; Madison, J.A.; Blair, C.; Weber, A.; Barnes, B.J.; Egeblad, M.; et al. Neutrophil extracellular traps in COVID-19. JCI Insight 2020, e138999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gattinoni, L.; Coppola, S.; Cressoni, M.; Busana, M.; Rossi, S.; Chiumello, S.D. Covid-19 does not lead to a “typical” acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 2020, 201, 1299–1300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, Y.; Liu, X.; Tan, X.Z.; Jiang, C.X.; Chen, S.W.; Liang, G.N.; He, X.M.; Wu, J.; Chen, T.; Xu, Y. ROS-induced NLRP3 inflammasome priming and activation mediate PCB 118-induced pyroptosis in endothelial cells. Ecotoxicol. Environ. Saf. 2020, 189, 109937. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Shi, P.; Chen, Q.; Huang, Z.; Zou, D.; Zhang, J.; Gao, X.; Lin, Z. Mitochondrial ROS promote macrophage pyroptosis by inducing GSDMD oxidation. J. Mol. Cell Biol. 2019, 11, 1069–1082. [Google Scholar] [CrossRef] [Green Version]
- Abais, J.M.; Xia, M.; Zhang, Y.; Boini, K.M.; Li, P.L. Redox regulation of NLRP3 inflammasomes: ROS as trigger or effector? Antioxid. Redox Signal. 2015, 22, 1111–1129. [Google Scholar] [CrossRef] [Green Version]
- Martinon, F. Signaling by ROS drives inflammasome activation. Eur. J. Immunol. 2010, 40, 616–619. [Google Scholar] [CrossRef]
- Boaru, S.G.; Borkham-Kamphorst, E.; Van de Leur, E.; Lehnen, E.; Liedtke, C.; Weiskirchen, R. NLRP3 inflammasome expression is driven by NF-kB in cultured hepatocytes. Biochem. Biophys. Res. Commun. 2015, 458, 700–706. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Ratajczak, M.Z.; Kucia, M. SARS-CoV-2 infection and overactivation of Nlrp3 inflammasome as a trigger of cytokine “storm” and risk factor for damage of hematopoietic stem cells. Leukemia 2020, 34, 1726–1729. [Google Scholar] [CrossRef]
- Donath, M.Y.; Böni-Schnetzler, M.; Ellingsgaard, H.; Halban, P.A.; Ehses, J.A. Cytokine production by islets in health and diabetes: Cellular origin, regulation and function. Trends Endocrinol. Metab. 2010, 21, 261–267. [Google Scholar] [CrossRef]
- Qiao, Y.; Wang, P.; Qi, J.; Zhang, L.; Gao, C. TLR-induced NF-kB activation regulates NLRP3 expression in murine macrophages. FEBS Lett. 2012, 586, 1022–1026. [Google Scholar] [CrossRef] [Green Version]
- Place, D.E.; Kanneganti, T.D. Recent advances in inflammasome biology. Curr. Opin. Immunol. 2018, 50, 32–38. [Google Scholar] [CrossRef]
- Zhao, M.; Bai, M.; Ding, G.; Zhang, Y.; Huang, S.; Jia, Z.; Zhang, A. Angiotensin II stimulates the NLRP3 inflammasome to induce podocyte injury and mitochondrial dysfunction. Kidney Dis. 2018, 4, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Van den Berg, D.F.; Te Velde, A.A. Severe COVID-19: NLRP3 inflammasome dysregulated. Front. Immunol. 2020, 11, 1580. [Google Scholar] [CrossRef] [PubMed]
- De Flora, S.; Grassi, C.; Carati, L. Attenuation of influenza-like symptomatology and improvement of cell-mediated immunity with long-term N-acetylcysteine treatment. Eur. Respir. J. 1997, 10, 1535–1541. [Google Scholar] [CrossRef]
- McCarty, M.F.; DiNicolantonio, J.J. Nutraceuticals have potential for boosting the type 1 interferon response to RNA viruses including influenza and coronavirus. Prog. Cardiovasc. Dis. 2020, 63, 383–385. [Google Scholar] [CrossRef] [PubMed]
- De Flora, S.; Balansky, R.; La Maestra, S. Rationale for the use of N acetylcysteine in both prevention and adjuvant therapy of COVID-19. FASEB J. 2020, 34, 13185–13193. [Google Scholar] [CrossRef]
- Hati, S.; Bhattacharyya, S. Impact of thiol-disulfide balance on the binding of Covid-19 spike protein with angiotensin converting enzyme 2 receptor. CS Omega 2020, 5, 16292–16298. [Google Scholar] [CrossRef] [PubMed]
- Geiler, J.; Michaelis, M.; Naczk, P.; Leutz, A.; Langer, K.; Doerr, H.W.; Cinatl, J. N-acetyl-L-cysteine (NAC) inhibits virus replication and expression of pro-inflammatory molecules in A549 cells infected with highly pathogenic H5N1 influenza A virus. Bioch. Pharmacol. 2010, 79, 413–420. [Google Scholar] [CrossRef] [Green Version]
- Mata, M.; Morcillo, E.; Gimeno, C.; Cortijo, J. N-acetyl-L-cysteine (NAC) inhibit mucin syn-thesis and pro-inflammatory mediators in alveolar type II epithelial cells infected with influenza virus A and B and with respiratory syncytial virus (RSV). Bioch Pharmacol. 2011, 82, 548–555. [Google Scholar] [CrossRef] [Green Version]
- Bauer, S.R.; Kapoor, A.; Rath, M.; Thomas, S.A. What is the role of supplementation with ascorbic acid, zinc, vitamin D, or N-acetylcysteine for prevention or treatment of COVID-19? Cleve Clin. J. Med. 2020. [Google Scholar] [CrossRef]
- Šalamon, Š.; Kramar, B.; Marolt, T.P.; Poljšak, B.; Milisav, I. Medical and dietary uses of N-acetylcysteine. Antioxidants 2019, 8, 111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guglielmetti, G.; Quaglia, M.; Sainaghi, P.P.; Castello, L.M.; Vaschetto, R.; Pirisi, M.; Corte, F.D.; Avanzi, G.C.; Stratta, P.; Cantaluppi, V. “War to the knife”against thromboinflammation to protect endothelial function of COVID-19 patients. Crit. Care. 2020, 24, 365. [Google Scholar] [CrossRef] [PubMed]
- Andreou, A.; Trantza, S.; Filippou, D.; Sipsas, N.; Tsiodras, S. COVID- 19: The potential role of copper and N-acetylcysteine [NAC] in a combination of candidate antiviral treatments against SARS-CoV-2. In Vivo 2020, 34, 1567–1588. [Google Scholar] [CrossRef]
- Jorge-Aarón, R.M.; Rosa-Ester, M.P. N-acetylcysteine as a potential treatment for novel coronavirus disease 2019 [published online ahead of print, 2020 Jul 14]. Future Microbiol. 2020, 5, 959–962. [Google Scholar] [CrossRef] [PubMed]
- Poe, F.L.; Corn, J. N-Acetylcysteine: A potential therapeutic agent for SARS-CoV-2. Med. Hypotheses 2020, 143, 109862. [Google Scholar] [CrossRef]
- Memorial Sloan Kettering Cancer Center. A Study of N-Acetylcysteine in Patients with COVID-19 Infection 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT04374461 (accessed on 31 December 2020).
- Kensler, T.W.; Wakabayashi, N.; Biswal, S. Cell survival responses to environmental stresses via the Keap1-NRF2-ARE pathway. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 89–116. [Google Scholar] [CrossRef]
- Sadowska, A.M. N-Acetylcysteine mucolysis in the management of chronic obstructive pulmonary disease. Ther. Adv. Respir. Dis. 2012, 6, 127–135. [Google Scholar] [CrossRef] [Green Version]
- Hui, D.S.C.; Lee, N. Adjunctive therapies and immunomodulating agents for severe influenza. Influenza Other Respir. Viruses 2013, 7, 52–59. [Google Scholar] [CrossRef]
- Crinelli, R.; Zara, C.; Smietana, M.; Retini, M.; Magnani, M.; Fraternale, A. Boosting GSH using the Co-drug approach: I-152, a conjugate of N-acetyl-cysteine and mercaptoethylamine. Nutrients 2019, 11, 1291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horowitz, R.I.; Freeman, P.M.; Bruzzese, J. Efficacy of gluthahione therapy in relieving dyspnea associated with COVID-19 pneumonia. A report of 2 cases. Respir. Med. Case Rep. 2020, 30, 101063. [Google Scholar]
- Marik, P.E. Vitamin C for the treatment of sepsis: The scientific rationale. Pharmacol. Ther. 2018, 189, 63–70. [Google Scholar] [CrossRef]
- Mohammed, B.M.; Fisher, B.J.; Kraskauskas, D.; Farkas, D.; Brophy, D.F.; Fowler, A.A., III; Natarajan, R. Vitamin C: A novel regulator of neutrophil extracellular trap formation. Nutrients 2013, 5, 3131–3151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuchs, T.A.; Abed, U.; Goosmann, C.; Hurwitz, R.; Schulze, I.; Wahn, V. Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol. 2007, 176, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Radermecker, C.; Detrembleur, N.; Guiot, J.; Cavalier, E.; Henket, M. Neutrophil extracellular traps infiltrate the lung airway, interstitial, and vascular compartments in severe COVID-19. J Exp Med. 2020, 217, e20201012. [Google Scholar] [CrossRef] [PubMed]
- Abobaker, A.; Alzwi, A.; Alraied, A.H.A. Overview of the possible role of vitamin C in management of COVID-19. Pharmacol. Rep. 2020, 72, 1517–1528. [Google Scholar] [CrossRef]
- Marik, P.E.; Kory, P.; Varon, J.; Iglesias, J.; Meduri, G.U. MATH+ protocol for the treatment of SARS-CoV-2 infection: The scientific rationale. Expert Rev. Anti Infect. Ther. 2020, 10, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Domain-Specific Appendix: VITAMIN, C. REMAP-CAP: Randomized, Embedded, Multifactorial Adaptive Platform Trial for Community-Acquired Pneumonia 2020. Available online: https://static1.squarespace.com/static/5cde3c7d9a69340001d79e/t/5f1bba732cda7f10310643fe/1595652735252/REMAP-CAP+Vitamin+C+Domain+Specific+Appendix+V2+-08+June+2020_WM.pdf (accessed on 31 December 2020).
- Hiedra, R.; Lo, K.B.; Elbashabsheh, M.; Gul, F.; Wright, R.M.; Albano, J.; Azmaiparashvili, Z.; Patarroyo Aponte, G. The use of IV vitamin C for patients with COVID-19: A case series. Expert Rev. Anti Infect. Ther. 2020, 18, 1259–1261. [Google Scholar] [CrossRef]
- Komaravelli, N.; Casola, A. Respiratory Viral Infections and Subversion of Cellular Antioxidant Defenses. J. Pharm. Pharm. 2014, 5, 1000141. [Google Scholar]
- Wyler, E.; Franke, V.; Menegatti, J.; Kocks, C.; Boltengagen, A.; Praktiknjo, S.; Walch-Rückheim, B.; Bosse, J.; Rajewsky, N.; Grässer, F.; et al. Single-cell RNA-sequencing of herpes simplexvirus 1-infected cells connects NRF2 activation to an antiviral program. Nat. Commun. 2019, 10, 4878. [Google Scholar] [CrossRef] [Green Version]
- Halder, U.C.; Bagchi, P.; Chattopadhyay, S.; Dutta, D.; Chawla-Sarkar, M. Cell death regulation during influenza A virus infection by matrix [M1] protein: A model of viral control over the cellular survival pathway. Cell Death Dis. 2011, 2, e197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olagnier, D.; Farahani, E.; Thyrsted, J.; Cadanet, J.B.; Herengt, A.; Idorn, M.; Hait, A.; Hernaez, B.; Knudsen, A.; Iversen, M.B.; et al. Identification of SARSCoV2-mediated suppression of NRF2 signaling reveals a potent antiviral and anti-inflammatory activity of 4-octyl-itaconate and dimethyl fumarate. Nat. Commun. 2020, 11, 4938. [Google Scholar] [CrossRef]
- Cuadrado, A.; Martín-Moldes, Z.; Ye, J.; Lastres-Becker, I. Transcription factors NRF2 and NF-kappaB are coordinated effectors of the Rho family, GTP binding protein RAC1 during inflammation. J. Biol. Chem. 2014, 289, 15244–15258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lastres-Becker, I.; Innamorato, N.G.; Jaworski, T.; Rábano, A.; Kügler, S.; Van Leuven, F.; Cuadrado, A. Fractalkine activates NRF2/NFE2L2 and heme oxygenase 1 to restrain tauopathy induced microgliosis. Brain 2014, 137, 78–91. [Google Scholar] [CrossRef] [PubMed]
- Boutten, A.; Goven, D.; Artaud-Macari, E.; Boczkowski, J.; Bonay, M. NRF2 targeting: A promising therapeutic strategy in chronic obstructive pulmonary disease. Trends Mol. Med. 2011, 17, 363–371. [Google Scholar] [CrossRef]
- DeDiego, M.L.; Nieto-Torres, J.L.; Regla-Nava, J.A.; Jimenez-Guardeño, J.M.; Fernandez-Delgado, R.; Fett, C.; Castaño-Rodriguez, C.; Perlman, S.; Enjuanes, L. Inhibition of NF-kappaB-mediated inflammation in severe acute respiratory syndrome coronavirus-infected mice increases survival. J. Virol. 2014, 88, 913–924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dosch, S.F.; Mahajan, S.D.; Collins, A.R. SARS coronavirus spike protein-induced innate immune response occurs via activation of the NF-kappaB pathway in human monocyte macrophages in vitro. Virus Res. 2009, 142, 19–27. [Google Scholar] [CrossRef]
- Cuadrado, A.; Pajares, M.; Benito, C.; Jiménez-Villegas, J.; Escoll, M.; Fernández-Ginés, R.; Garcia Yagüe, A.J.; Lastra, D.; Manda, G.; Ana, I.; et al. Can activation of NRF2 be a strategy against COVID-19? Trends Pharmacol. Sci. 2020, 41, 598–610. [Google Scholar] [CrossRef]
- Espinoza, J.A.; González, P.A.; Kalergis, A.M. Modulation of antiviral immunity by heme oxygenase-1. Am. J. Pathol. 2017, 187, 487–493. [Google Scholar] [CrossRef] [Green Version]
- Wagener, F.A.D.T.G.; Pickkers, P.; Peterson, S.J.; Immenschuh, S.; Abraham, N.G. Targeting the heme-heme oxygenase system to prevent severe complications following COVID-19 infections. Antioxidants 2020, 9, 540. [Google Scholar] [CrossRef]
- McCord, J.M.; Hybertson, B.M.; Cota-Gomez, A.; Geraci, K.P.; Gao, B. NRF2 Activator PB125 as a Potential Therapeutic Agent against COVID-19. Antioxidants 2020, 9, 518. [Google Scholar] [CrossRef] [PubMed]
- Dittmann, M.; Hoffmann, H.H.; Bieniasz, P.D.; Rice, C.M. A Serpin Shapes the Extracellular Environment to Prevent Influenza A Virus Maturation. Cell 2015, 160, 631–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mrityunjaya, M.; Pavithra, V.; Neelam, R.; Janhavi, P.; Halami, P.M.; Ravindra, P.V. Immune-Boosting, Antioxidant and Anti-inflammatory Food Supplements Targeting Pathogenesis of COVID-19. Front. Immunol. 2020, 11, 570122. [Google Scholar] [CrossRef] [PubMed]
- Lucas, J.M.; Heinlein, C.; Kim, T.; Hernandez, S.A.; Malik, M.S.; True, L.D.; Morrissey, C.; Corey, E.; Montgomery, B.; Mostaghel, E.; et al. The androgen-regulated protease TMPRSS2 activates a proteolytic cascade involving components of the tumor microenvironment and promotes prostate cancer metastasis. Cancer Discov. 2014, 4, 1310–1325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Jiang, J.; Ren, Q.; Jia, Y.; Shen, J.; Shen, H.; Lin, X.; Lu, H.; Xie, Q. Ambroxol inhalation ameliorates LPS-induced airway inflammation and mucus secretion through the extracellular signal-regulated kinase 1/2 signaling pathway. Eur. J. Pharmacol. 2016, 775, 138–148. [Google Scholar] [CrossRef] [PubMed]
- Sunkari, S.; Thatikonda, S.; Pooladanda, V.; Challa, V.S.; Godugu, C. Protective effects of ambroxol in psoriasis like skin inflammation: Exploration of possible mechanisms. Intern. Immunopharmacol. 2019, 71, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Ansarin, K.; Tolouian, R.; Ardalan, M.; Taghizadieh, A.; Varshochi, M.; Teimouri, S.; Vaezi, T.; Valizadeh, H.; Saleh, P.; Safiri, S.; et al. Effect of bromhexine on clinical outcomes and mortality in COVID-19 patients: A randomized clinical trial. BioImpacts 2020, 10, 209–215. [Google Scholar] [CrossRef]
- Montes Diaz, G.; Hupperts, R.; Fraussen, J.; Somers, V. Dimethyl fumarate treatment in multiple sclerosis: Recent advances in clinical and immunological studies. Autoimmun. Rev. 2018, 17, 1240–1250. [Google Scholar] [CrossRef]
- Meng, Y.; Li, T.; Zhou, G.S.; Chen, Y.; Yu, C.H.; Pang, M.X.; Li, W.; Li, Y.; Zhang, W.Y.; Li, X. The angiotensin-converting enzyme 2/angiotensin [1–7]/mas axis protects against lung fibroblast migration and lung fibrosis by inhibiting the NOX4-derived ROS-mediated RhoA/Rho kinase pathway. Antioxid. Redox. Signal. 2015, 22, 241–258. [Google Scholar] [CrossRef]
- Lei, C.; Qian, K.; Li, T.; Zhang, S.; Fu, W.; Ding, M.; Hu, S. Neutralization of SARS-CoV-2 spike pseudotyped virus by recombinant ACE2-Ig. Nat. Commun. 2020, 11, 2070. [Google Scholar] [CrossRef] [Green Version]
- Monteil, V.; Kwon, H.; Prado, P.; Hagelkrüys, A.; Wimmer, R.A.; Stahl, M.; Leopoldi, A.; Garreta, E.; Hurtado Del Pozo, C.; Prosper, F.; et al. Inhibition of SARS-CoV-2 Infections in Engineered Human Tissues Using Clinical-Grade Soluble Human ACE2. Cell 2020, 181, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Zoufaly, A.; Poglitsch, M.; Aberle, J.H.; Hoepler, W.; Seitz, T.; Traugott, M.; Grieb, A.; Pawelka, E.; Laferl, H.; Wenisch, C.; et al. Human recombinant soluble ACE2 in severe COVID-19. Lancet Respir. Med. 2020, 8, 1154–1158. [Google Scholar] [CrossRef]
- Shah, A. Novel Coronavirus-Induced NLRP3 Inflammasome Activation: A Potential Drug Target in the Treatment of COVID-19. Front. Immunol. 2020, 11, 1021. [Google Scholar] [CrossRef]
- He, H.; Jiang, H.; Chen, Y.; Ye, J.; Wang, A.; Wang, C.; Liu, Q.; Liang, G.; Deng, X.; Jiang, W.; et al. Oridonin is a covalent NLRP3 inhibitor with strong anti-inflammasome activity. Nat. Commun. 2018, 9, 2550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juliana, C.; Fernandes-Alnemri, T.; Wu, J.; Datta, P.; Solorzano, L.; Yu, J.W.; Meng, R.; Quong, A.A.; Latz, E.; Scott, C.P.; et al. Anti-inflammatory compounds parthenolide and bay 11-7082 are direct inhibitors of the inflammasome. J. Biol. Chem. 2010, 285, 9792–9802. [Google Scholar] [CrossRef] [Green Version]
- Costa, L.S.; Outlioua, A.; Anginot, A.; Akarid, K.; Arnoult, D. RNA viruses promote activation of the NLRP3 inflammasome through cytopathogenic effect-induced potassium efflux. Cell Death Dis. 2019, 10, 346. [Google Scholar] [CrossRef]
- Lamkanfi, M.; Mueller, J.L.; Vitari, A.C.; Misaghi, S.; Fedorova, A.; Deshayes, K.; Lee, W.P.; Hoffman, H.M.; Dixit, V.M. Glyburide inhibits the Cryopyrin/Nalp3 inflammasome. J. Cell Biol. 2009, 187, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Jiang, H.; Chen, Y.; Wang, X.; Yang, Y.; Tao, J.; Deng, X.; Liang, G.; Zhang, H.; Jiang, W.; et al. Tranilast directly targets NLRP 3 to treat inflammasome-driven diseases. EMBO Mol. Med. 2018, 10, e8689. [Google Scholar] [CrossRef]
- Leung, Y.Y.; Yao Hui, L.L.; Kraus, V.B. Colchicine—update on mechanisms of action and therapeutic uses. Semin. Arthritis Rheum. 2015, 45, 341–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misawa, T.; Takahama, M.; Kozaki, T.; Lee, H.; Zou, J.; Saitoh, T.; Akira, S. Microtubule driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat. Immunol. 2013, 14, 454–460. [Google Scholar] [CrossRef]
- Daniels, M.J.; Rivers-Auty, J.; Schilling, T.; Spencer, N.G.; Watremez, W.; Fasolino, V.; Booth, S.J.; White, C.S.; Baldwin, A.G.; Freeman, S.; et al. Fenamate NSAIDs inhibit the NLRP3 inflammasome and protect against Alzheimer’s disease in rodent models. Nat Commun. 2016, 7, 12504. [Google Scholar] [CrossRef] [Green Version]
- Rothan, H.A.; Bahrani, H.; Abdulrahman, A.Y.; Mohamed, Z.; Teoh, T.C.; Othman, S.; Rashid, N.N.; Rahman, N.A.; Yusof, R. Mefenamic acid in combination with ribavirin shows significant effects in reducing chikungunya virus infection in vitro and in vivo. Antiviral. Res. 2016, 127, 50–56. [Google Scholar] [CrossRef]
- Cain, D.W.; Cidlowski, J.A. Immune regulation by glucocorticoids. Nat Rev. Immunol 2017, 17, 233–247. [Google Scholar] [CrossRef] [PubMed]
- Busillo, J.M.; Cidlowski, J.A. The five Rs of glucocorticoid action during inflammation: Ready, reinforce, repress, resolve, and restore. Trends Endocrinol. Metab. 2013, 24, 109–119. [Google Scholar] [CrossRef] [Green Version]
- Quatrini, L.; Ugolini, S. New insights into the cell- and tissue-specificity of glucocorticoid actions. Cell. Mol. Immunol. 2020, 18, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Zhang, W.; Shi, X.M. Glucocorticoid-induced leucine zipper (GILZ) mediates glucocorticoid action and inhibits inflammatory cytokine-induced COX-2 expression. J. Cell. Biochem. 2008, 103, 1760–1771. [Google Scholar] [CrossRef] [PubMed]
- Mitroulis, I.; Alexaki, V.I.; Kourtzelis, I.; Ziogas, A.; Hajishengallis, G.; Chavakis, T. Leukocyte integrins: Role in leukocyte recruitment and as therapeutic targets in inflammatory disease. Pharmacol. Ther. 2015, 147, 123–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zielinska, K.A.; Van Moortel, L.; Opdenakker, G.; De Bosscher, K.; Van den Steen, P.E. Endothelial response to glucocorticoids in inflammatory diseases. Front Immunol 2016, 7, 592. [Google Scholar] [CrossRef]
- Busillo, J.M.; Ehrchen, J.; Steinmuller, L.; Barczyk, K. Glucocorticoids induce differentiation of a specifically activated, anti-inflammatory subtype of human monocytes. Blood 2007, 109, 1265–1274. [Google Scholar]
- Meers, G.K.; Bohnenberger, H.; Reichardt, H.M.; Luhder, F.; Reichardt, S.T. Impaired resolution of DSS-induced colitis in mice lacking the glucocorticoid receptor inmyeloid cells. PLoS ONE 2018, 13, e0190846. [Google Scholar] [CrossRef] [PubMed]
- Vallelian, F.; Schaer, C.A.; Kaempfer, T.; Ghering, P.; Duerst, E.; Schoedon, G.; Schaer, D.J. Glucocorticoid treatment skews human monocyte differentiation into a hemoglobin-clearance phenotype with enhanced heme-iron recycling and antioxidant capacity. Blood 2010, 116, 5347–5356. [Google Scholar] [CrossRef] [Green Version]
- RECOVERY Collaborative Group; Horby, P.; Lim, W.S.; Emberson, J.R.; Mafham, M.; Bell, J.L.; Linsell, L.; Staplin, N.; Brightling, C.; Ustianowski, A.; et al. Dexamethasone in Hospitalized Patients with Covid-19 - Preliminary Report. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- The WHO Rapid Evidence Appraisal for COVID-19 Therapies (REACT) Working Group. Association between administration of systemic corticosteroids and mortality among critically ill Patients with COVID-19. A Meta-analysis. JAMA 2020, 324, 1330–1341. [Google Scholar]
- Vane, J.R.; Botting, R.M. Mechanism of action of nonsteroidal anti-inflammatory drugs. Am. J. Med. 1998, 104, 2S–8S. [Google Scholar] [CrossRef]
- Day, M. Covid-19: Ibuprofen should not be used for managing symptoms, say doctors and scientists. BMJ 2020, 368, m1086. [Google Scholar] [CrossRef] [Green Version]
- FitzGerald, G.A. Misguided drug advice for COVID-19. Science 2020, 367, 1434. [Google Scholar] [PubMed] [Green Version]
- Little, P. Non-steroidal anti-inflammatory drugs and covid-19. BMJ 2020, 368, m1185. [Google Scholar] [CrossRef] [Green Version]
- Huh, K.; Ji, W.; Kang, M.; Hong, J.; Bae, G.H.; Lee, R.; Na, Y.; Jung, J. Association of prescribed medications with the risk of COVID-19 infection and severity among adults in South Korea. Int. J. Infect. Dis. 2020, 104, 7–14. [Google Scholar] [CrossRef]
- Freites Nuñez, D.D.; Leon, L.; Mucientes, A.; Rodriguez-Rodriguez, L.; Font Urgelles, J.; Madrid García, A.; Colomer, J.I.; Jover, J.A.; Fernandez-Gutierrez, B.; Abasolo, L. Risk factors for hospital admissions related to COVID-19 in patients with autoimmune inflammatory rheumatic diseases. Ann. Rheum. Dis. 2020, 79, 1393–1399. [Google Scholar] [CrossRef]
- Imam, Z.; Odish, F.; Gill, I.; O’Connor, D.; Armstrong, J.; Vanood, A.; Ibironke, O.; Hanna, A.; Ranski, A.; Halalau, A. Older age and comorbidity are independent mortality predictors in a large cohort of 1305 COVID-19 patients in Michigan, United States. J. Intern. Med. 2020, 288, 469–476. [Google Scholar] [CrossRef]
- Hong, W.; Chen, Y.; You, K.; Tan, S.; Wu, F.; Tao, J.; Chen, X.; Zhang, J.; Xiong, Y.; Yuan, F. Celebrex Adjuvant Therapy on Coronavirus Disease 2019: An Experimental Study. Front Pharmacol. 2020, 11, 561674. [Google Scholar] [CrossRef]
- EMA advice on the use of NSAIDs for Covid-19. Drug Ther. Bull. 2020, 58, 69. [CrossRef] [PubMed]
- Robb, C.T.; Goepp, M.; Rossi, A.G.; Yao, C. Non-steroidal anti-inflammatory drugs, prostaglandins, and COVID-19. Br. J Pharmacol. 2020, 177, 4899–4920. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fratta Pasini, A.M.; Stranieri, C.; Cominacini, L.; Mozzini, C. Potential Role of Antioxidant and Anti-Inflammatory Therapies to Prevent Severe SARS-Cov-2 Complications. Antioxidants 2021, 10, 272. https://doi.org/10.3390/antiox10020272
Fratta Pasini AM, Stranieri C, Cominacini L, Mozzini C. Potential Role of Antioxidant and Anti-Inflammatory Therapies to Prevent Severe SARS-Cov-2 Complications. Antioxidants. 2021; 10(2):272. https://doi.org/10.3390/antiox10020272
Chicago/Turabian StyleFratta Pasini, Anna M., Chiara Stranieri, Luciano Cominacini, and Chiara Mozzini. 2021. "Potential Role of Antioxidant and Anti-Inflammatory Therapies to Prevent Severe SARS-Cov-2 Complications" Antioxidants 10, no. 2: 272. https://doi.org/10.3390/antiox10020272
APA StyleFratta Pasini, A. M., Stranieri, C., Cominacini, L., & Mozzini, C. (2021). Potential Role of Antioxidant and Anti-Inflammatory Therapies to Prevent Severe SARS-Cov-2 Complications. Antioxidants, 10(2), 272. https://doi.org/10.3390/antiox10020272