
Auranofin-Mediated NRF2 Induction Attenuates Interleukin 1 Beta Expression in Alveolar Macrophages

 , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
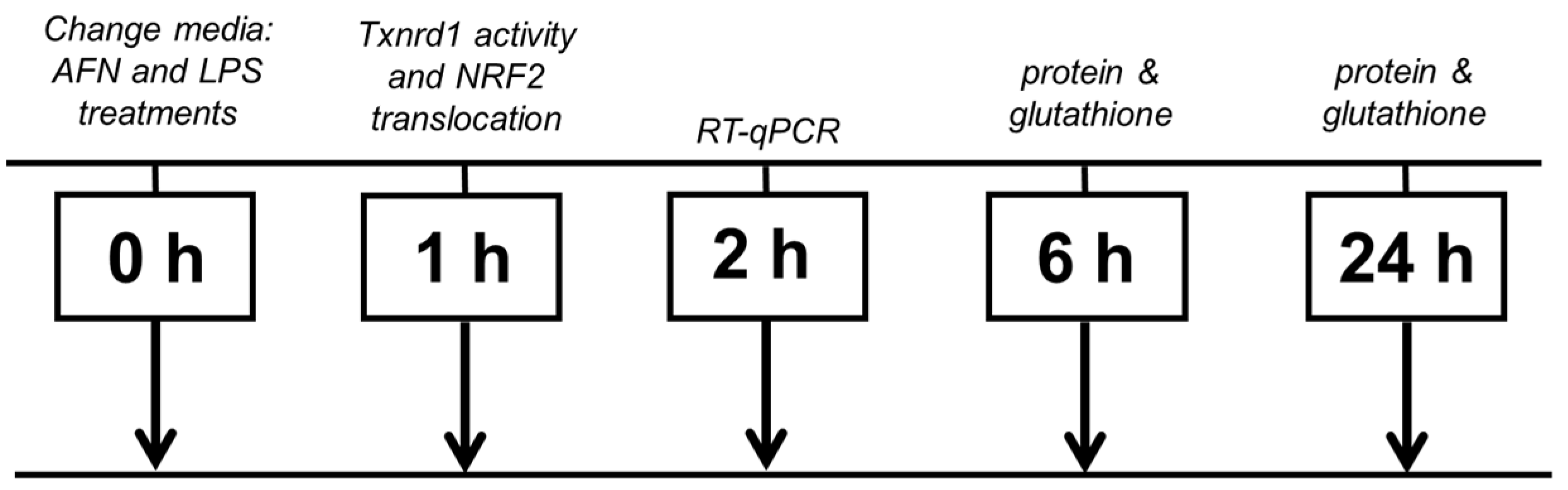
2. Materials and Methods
2.1. Cell Culture
2.2. IL-1β ELISA
2.3. Nuclear Fractionation
2.4. Immunoblot
2.5. Quantitative Real-Time RT-PCR
2.6. Glutathione Recycling Assay
2.7. Chromatin Immunoprecipitation (ChIP)
2.8. Statistics
3. Results
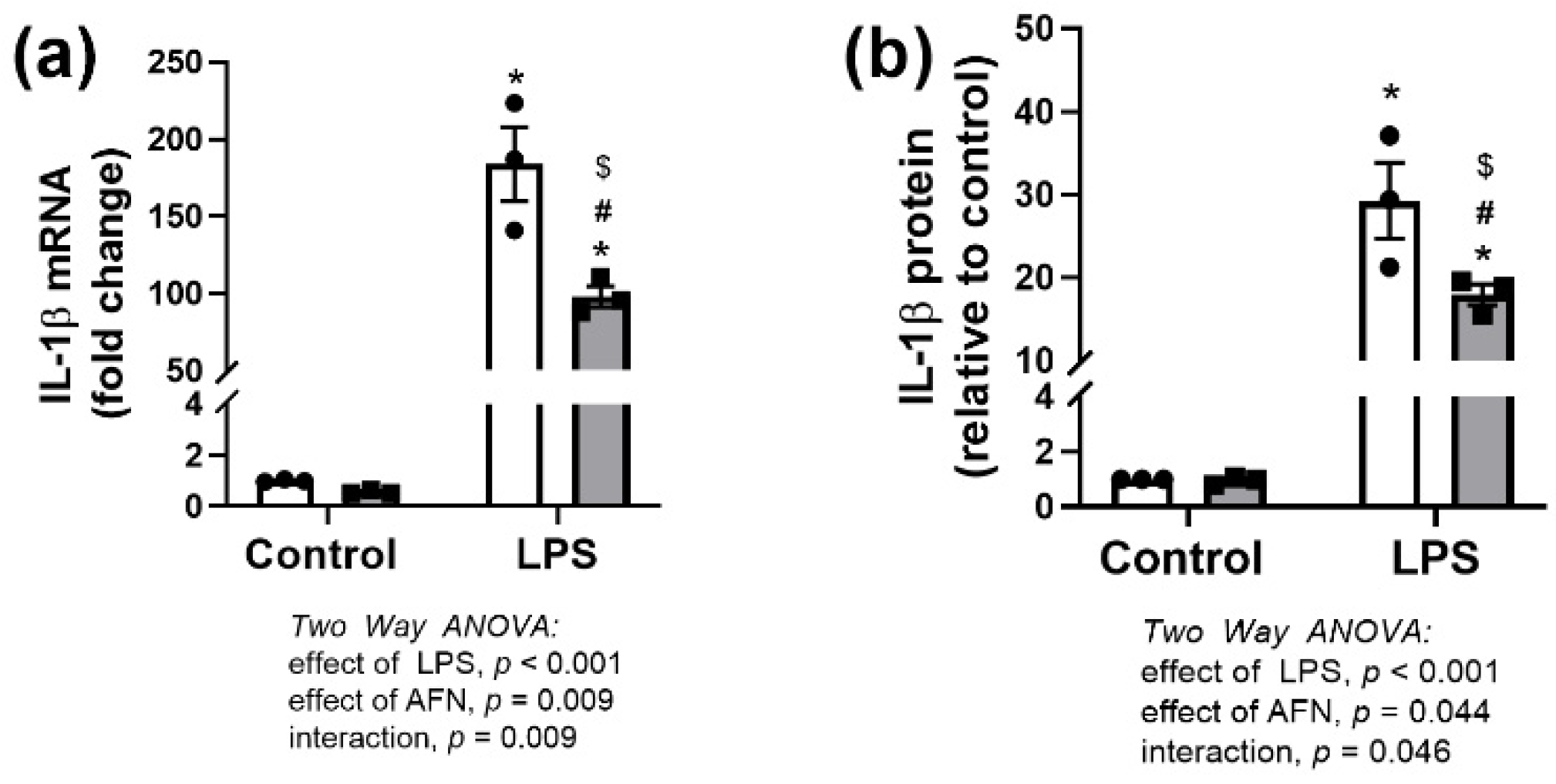
3.1. Effects of LPS and/or AFN on IL-1β Expression
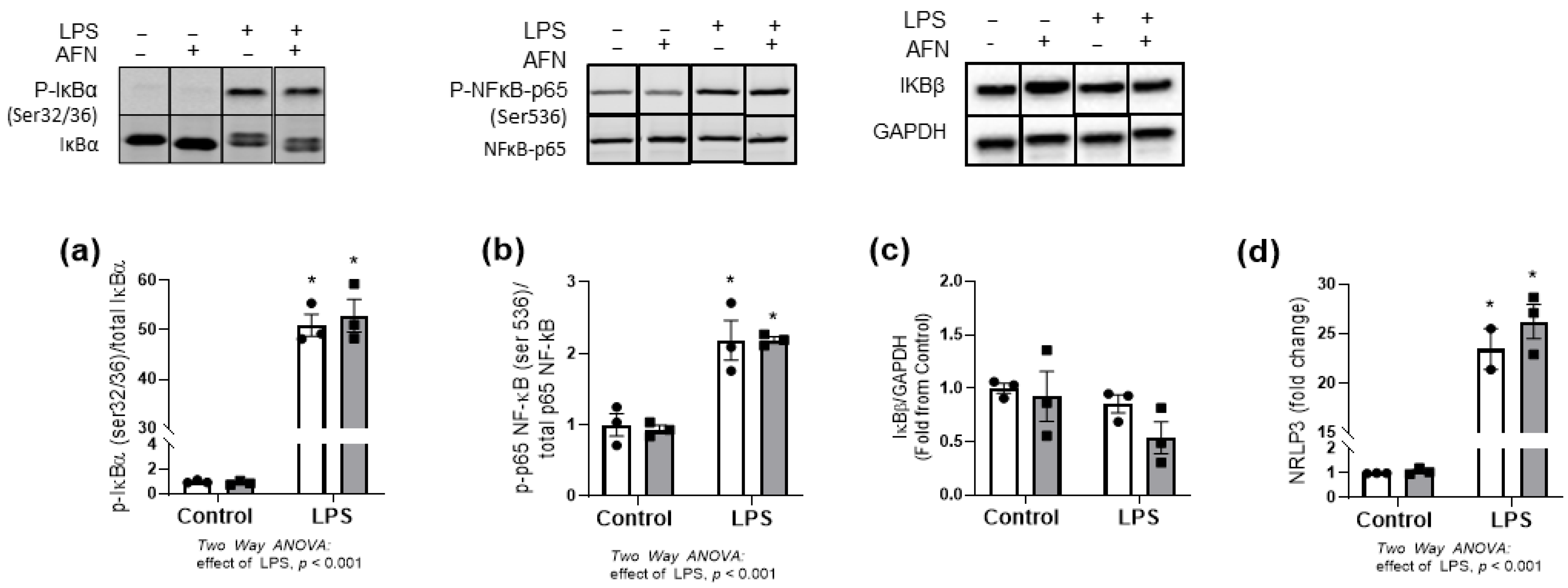
3.2. NFκB Pathway Responses to LPS and/or AFN
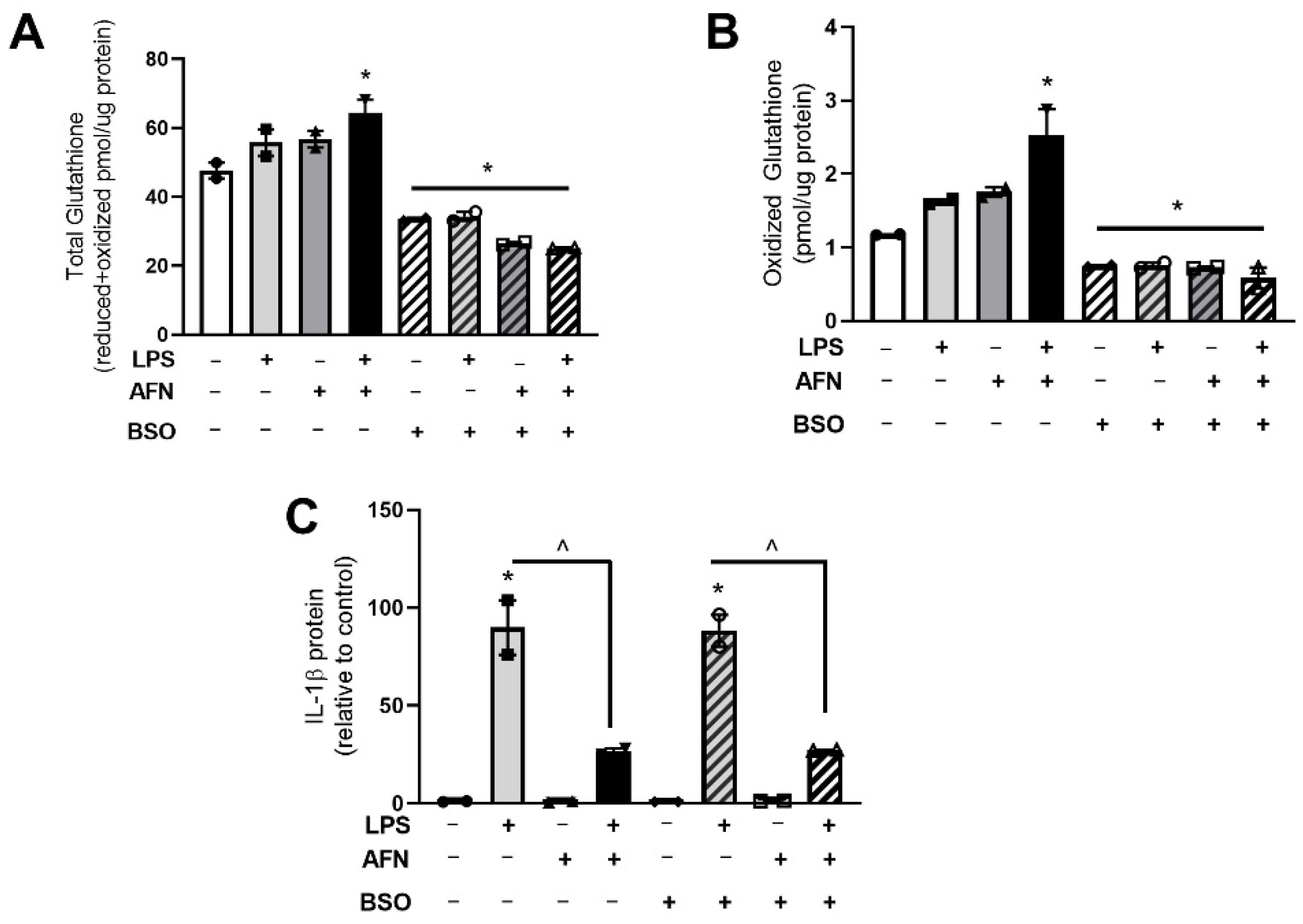
3.3. LPS and AFN Modulate Glutathione Levels
3.4. Decreases in Glutathione Do Not Affect IL-1β Expression
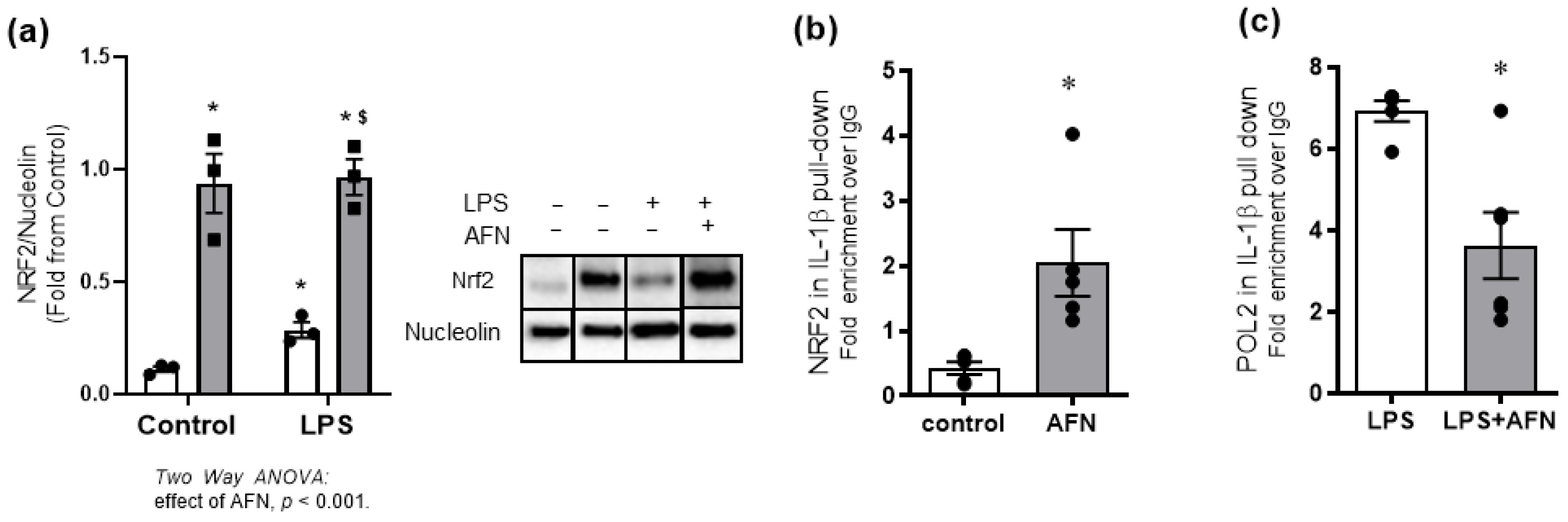
3.5. AFN-Mediated Attenuation of LPS-Induced IL-1β Expression Is Associated with NRF2 Binding to Il1β Promoter
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Fan, E.K.Y.; Fan, J. Regulation of alveolar macrophage death in acute lung inflammation. Respir. Res. 2018, 19, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Locy, M.L.; Rogers, L.K.; Prigge, J.R.; Schmidt, E.E.; Arnér, E.S.; Tipple, T.E. Thioredoxin Reductase Inhibition Elicits Nrf2-Mediated Responses in Clara Cells: Implications for Oxidant-Induced Lung Injury. Antioxid. Redox Signal. 2012, 17, 1407–1416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bondeson, J.; Sundler, R. Auranofin inhibits the induction of interleukin 1β and tumor necrosis factor α mRNA in macrophages. Biochem. Pharmacol. 1995, 50, 1753–1759. [Google Scholar] [CrossRef]
- Isakov, E.; Weisman-Shomer, P.; Benhar, M. Suppression of the pro-inflammatory NLRP3/interleukin-1β pathway in macrophages by the thioredoxin reductase inhibitor auranofin. Biochim. Biophys. Acta (BBA) Gen. Subj. 2014, 1840, 3153–3161. [Google Scholar] [CrossRef]
- Wall, S.B.; Wood, R.; Dunigan, K.; Li, Q.; Li, R.; Rogers, L.K.; Tipple, T.E. Thioredoxin Reductase-1 Inhibition Augments Endogenous Glutathione-Dependent Antioxidant Responses in Experimental Bronchopulmonary Dysplasia. Oxid. Med. Cell. Longev. 2019, 2019, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Staples, S.; Wall, S.B.; Li, R.; Tipple, T.E. Selenium-independent antioxidant and anti-inflammatory effects of thioredoxin reductase inhibition in alveolar macrophages. Life Sci. 2020, 259, 118285. [Google Scholar] [CrossRef]
- Britt, R.D.; Velten, M.; Locy, M.L.; Rogers, L.K.; Tipple, T.E. The thioredoxin reductase-1 inhibitor aurothioglucose attenuates lung injury and improves survival in a murine model of acute respiratory distress syndrome. Antioxid. Redox Signal. 2014, 20, 2681–2691. [Google Scholar] [CrossRef] [Green Version]
- McKenna, S.; Wright, C.J. Inhibiting IκBβ-NFκB signaling attenuates the expression of select pro-inflammatory genes. J. Cell Sci. 2015, 128, 2143–2155. [Google Scholar] [CrossRef] [Green Version]
- Michaelis, K.A.; Agboke, F.; Liu, T.; Han, K.; Muthu, M.; Galambos, C.; Yang, G.; Dennery, P.A.; Wright, C.J. IκBβ-Mediated NF-κB Activation Confers Protection Against Hyperoxic Lung Injury. Am. J. Respir. Cell Mol. Biol. 2013, 50, 429–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, P.; Hayden, M.S.; Long, M.; Scott, M.L.; West, A.P.; Zhang, D.; Oeckinghaus, A.; Lynch, C.; Hoffmann, A.; Baltimore, D.; et al. IκBβ acts to inhibit and activate gene expression during the inflammatory response. Nat. Cell Biol. 2010, 466, 1115–1119. [Google Scholar] [CrossRef] [Green Version]
- Cuadrado, A.; Martín-Moldes, Z.; Ye, J.; Lastres-Becker, I. Transcription Factors NRF2 and NF-κB Are Coordinated Effectors of the Rho Family, GTP-binding Protein RAC1 during Inflammation. J. Biol. Chem. 2014, 289, 15244–15258. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Hayashi, M.; Sekine, H.; Tanaka, N.; Moriguchi, T.; Motohashi, H.; Nakayama, K.; et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat. Commun. 2016, 7, 11624. [Google Scholar] [CrossRef] [PubMed]
- Michaelson, D.; Abidi, W.; Guardavaccaro, D.; Zhou, M.; Ahearn, I.; Pagano, M.; Philips, M.R. Rac1 accumulates in the nucleus during the G2 phase of the cell cycle and promotes cell division. J. Cell Biol. 2008, 181, 485–496. [Google Scholar] [CrossRef] [Green Version]
- Tipple, T.E.; Rogers, L.K. Methods for the Determination of Plasma or Tissue Glutathione Levels. Methods Mol. Biol. 2012, 889, 315–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffith, O.W. Determination of glutathione and glutathione disulfide using glutathione reductase and 2-vinylpyridine. Anal. Biochem. 1980, 106, 207–212. [Google Scholar] [CrossRef]
- Gromer, S.; Arscott, L.D.; Williams, C.H., Jr.; Schirmer, R.H.; Becker, K. Human placenta thioredoxin reductase. Isolation of the selenoenzyme, steady state kinetics, and inhibition by therapeutic gold compounds. J. Biol. Chem. 1998, 273, 20096–20101. [Google Scholar] [CrossRef] [Green Version]
- Yue, S.; Luo, M.; Liu, H.; Wei, S. Recent Advances of Gold Compounds in Anticancer Immunity. Front. Chem. 2020, 8, 543. [Google Scholar] [CrossRef]
- Saha, S.; Buttari, B.; Panieri, E.; Profumo, E.; Saso, L. An Overview of Nrf2 Signaling Pathway and Its Role in Inflammation. Molecules 2020, 25, 5474. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wall, S.B.; Ren, C.; Velten, M.; Hill, C.L.; Locy, M.L.; Rogers, L.K.; Tipple, T.E. Thioredoxin Reductase Inhibition Attenuates Neonatal Hyperoxic Lung Injury and Enhances Nuclear Factor E2–Related Factor 2 Activation. Am. J. Respir. Cell Mol. Biol. 2016, 55, 419–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tipple, T.E.; Welty, S.E.; Rogers, L.K.; Hansen, T.N.; Choi, Y.-E.; Kehrer, J.P.; Smith, C.V. Thioredoxin-Related Mechanisms in Hyperoxic Lung Injury in Mice. Am. J. Respir. Cell Mol. Biol. 2007, 37, 405–413. [Google Scholar] [CrossRef] [Green Version]
- Britt, R.D., Jr.; Locy, M.L.; Tipple, T.E.; Nelin, L.D.; Rogers, L.K. Lipopolysaccharide-induced Cyclooxygenase-2 Expression in Mouse Transformed Clara Cells. Cell. Physiol. Biochem. 2012, 29, 213–222. [Google Scholar] [CrossRef]
- Roder, C.; Thomson, M.J. Auranofin: Repurposing an Old Drug for a Golden New Age. Drugs R D 2015, 15, 13–20. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Gao, Y.; Ci, X. Role of Nrf2 and Its Activators in Respiratory Diseases. Oxidat. Med. Cell. Longev. 2019, 2019, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Sun, Z.; Niu, Z.; Wu, S.; Shan, S. Protective mechanism of sulforaphane in Nrf2 and anti-lung injury in ARDS rabbits. Exp. Ther. Med. 2018, 15, 4911–4915. [Google Scholar] [CrossRef] [PubMed]
- Thimmulappa, R.K.; Lee, H.; Rangasamy, T.; Reddy, S.P.; Yamamoto, M.; Kensler, T.W.; Biswal, S. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J. Clin. Investig. 2006, 116, 984–995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamatam, C.M.; Reddy, N.M.; Potteti, H.R.; AnkiReddy, A.; Noone, P.M.; Yamamoto, M.; Kensler, T.W.; Reddy, S.P. Preconditioning the immature lung with enhanced Nrf2 activity protects against oxidant-induced hypoalveolarization in mice. Sci. Rep. 2020, 10, 1–13. [Google Scholar] [CrossRef]
- Cho, H.-Y.; Wang, X.; Li, J.; Bell, D.A.; Kleeberger, S.R. Potential therapeutic targets in Nrf2-dependent protection against neonatal respiratory distress disease predicted by cDNA microarray analysis and bioinformatics tools. Curr. Opin. Toxicol. 2016, 1, 125–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, N.M.; Kleeberger, S.R.; Kensler, T.W.; Yamamoto, M.; Hassoun, P.M.; Reddy, S.P. Disruption of Nrf2 Impairs the Resolution of Hyperoxia-Induced Acute Lung Injury and Inflammation in Mice. J. Immunol. 2009, 182, 7264–7271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wardyn, J.D.; Ponsford, A.H.; Sanderson, C.M. Dissecting molecular cross-talk between Nrf2 and NF-κB response pathways. Biochem. Soc. Trans. 2015, 43, 621–626. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Zhang, X.; Ding, Y.; Zhou, W.; Tao, L.; Lu, P.; Wang, Y.; Hu, R. Nuclear Factor E2-Related Factor-2 Negatively Regulates NLRP3 Inflammasome Activity by Inhibiting Reactive Oxygen Species-Induced NLRP3 Priming. Antioxid. Redox Signal. 2017, 26, 28–43. [Google Scholar] [CrossRef] [Green Version]
- Marzec, J.M.; Christie, J.D.; Reddy, S.P.; Jedlicka, A.E.; Vuong, H.; Lanken, P.N.; Aplenc, R.; Yamamoto, T.; Yamamoto, M.; Cho, H.-Y.; et al. Functional polymorphisms in the transcription factor NRF2 in humans increase the risk of acute lung injury. FASEB J. 2007, 21, 2237–2246. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.-Y.; Jedlicka, A.E.; Reddy, S.P.M.; Zhang, L.-Y.; Kensler, T.W.; Kleeberger, S.R. Linkage analysis of susceptibility to hyperoxia. Nrf2 is a candidate gene. Am. J. Respir. Cell Mol. Biol. 2002, 26, 42–51. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wall, S.B.; Li, R.; Butler, B.; Burg, A.R.; Tse, H.M.; Larson-Casey, J.L.; Carter, A.B.; Wright, C.J.; Rogers, L.K.; Tipple, T.E. Auranofin-Mediated NRF2 Induction Attenuates Interleukin 1 Beta Expression in Alveolar Macrophages. Antioxidants 2021, 10, 632. https://doi.org/10.3390/antiox10050632
Wall SB, Li R, Butler B, Burg AR, Tse HM, Larson-Casey JL, Carter AB, Wright CJ, Rogers LK, Tipple TE. Auranofin-Mediated NRF2 Induction Attenuates Interleukin 1 Beta Expression in Alveolar Macrophages. Antioxidants. 2021; 10(5):632. https://doi.org/10.3390/antiox10050632
Chicago/Turabian StyleWall, Stephanie B., Rui Li, Brittany Butler, Ashley R. Burg, Hubert M. Tse, Jennifer L. Larson-Casey, A. Brent Carter, Clyde J. Wright, Lynette K. Rogers, and Trent E. Tipple. 2021. "Auranofin-Mediated NRF2 Induction Attenuates Interleukin 1 Beta Expression in Alveolar Macrophages" Antioxidants 10, no. 5: 632. https://doi.org/10.3390/antiox10050632
APA StyleWall, S. B., Li, R., Butler, B., Burg, A. R., Tse, H. M., Larson-Casey, J. L., Carter, A. B., Wright, C. J., Rogers, L. K., & Tipple, T. E. (2021). Auranofin-Mediated NRF2 Induction Attenuates Interleukin 1 Beta Expression in Alveolar Macrophages. Antioxidants, 10(5), 632. https://doi.org/10.3390/antiox10050632