
ROS Defense Systems and Terminal Oxidases in Bacteria



Abstract
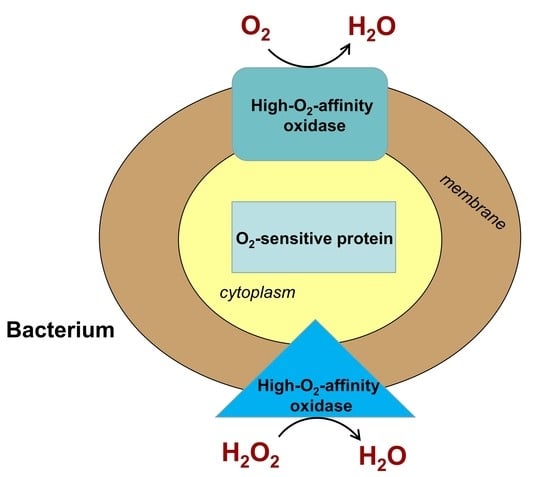
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. The bd-Type Oxidases by Fast O2 Scavenging Protect O2-Labile Enzymes from Oxidative Inactivation and Reduce Intracellular ROS Levels
3. Bacterial Mutants Devoid of Cytochrome bd Show Higher Sensitivity to H2O2. Cytochrome bd Expression Increases in the Presence of H2O2
4. Catalase-Like Activity of Cytochrome bd
5. Peroxidase-Like Activity of Cytochrome bd
6. ROS and Heme-Copper Oxidases
7. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| BNC | binuclear center |
| CO | carbon monoxide |
| COX | cytochrome c oxidase |
| dQH2 | decyl-ubiquinol |
| DTT | dithiothreitol |
| HQNO | 2-n-heptyl 4-hydroxyquinoline-N-oxide |
| NO | nitric oxide |
| Q1 | 2,3-dimethoxy-5-methyl-6-(3-methyl-2-butenyl)-1,4-benzoquinone |
References
- Mourenza, A.; Gil, J.A.; Mateos, L.M.; Letek, M. Oxidative stress-generating antimicrobials, a novel strategy to overcome antibacterial resistance. Antioxidants 2020, 9, 361. [Google Scholar] [CrossRef] [PubMed]
- Jakubovics, N.S.; Gill, S.R.; Vickerman, M.M.; Kolenbrander, P.E. Role of hydrogen peroxide in competition and cooperation between Streptococcus gordonii and Actinomyces naeslundii. FEMS Microbiol. Ecol. 2008, 66, 637–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seaver, L.C.; Imlay, J.A. Are respiratory enzymes the primary sources of intracellular hydrogen peroxide? J. Biol. Chem. 2004, 279, 48742–48750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vatansever, F.; de Melo, W.C.; Avci, P.; Vecchio, D.; Sadasivam, M.; Gupta, A.; Chandran, R.; Karimi, M.; Parizotto, N.A.; Yin, R.; et al. Antimicrobial strategies centered around reactive oxygen species—Bactericidal antibiotics, photodynamic therapy, and beyond. FEMS Microbiol. Rev. 2013, 37, 955–989. [Google Scholar] [CrossRef] [Green Version]
- Johnson, L.A.; Hug, L.A. Distribution of reactive oxygen species defense mechanisms across domain bacteria. Free Radic. Biol. Med. 2019, 140, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Loewen, P.C.; Switala, J.; Triggs-Raine, B.L. Catalases HPI and HPII in Escherichia coli are induced independently. Arch. Biochem. Biophys. 1985, 243, 144–149. [Google Scholar] [CrossRef]
- Imlay, J.A. Cellular defenses against superoxide and hydrogen peroxide. Annu. Rev. Biochem. 2008, 77, 755–776. [Google Scholar] [CrossRef] [Green Version]
- Khademian, M.; Imlay, J.A. Escherichia coli cytochrome c peroxidase is a respiratory oxidase that enables the use of hydrogen peroxide as a terminal electron acceptor. Proc. Natl. Acad. Sci. USA 2017, 114, E6922–E6931. [Google Scholar] [CrossRef] [Green Version]
- Nobrega, C.S.; Devreese, B.; Pauleta, S.R. YhjA—An Escherichia coli trihemic enzyme with quinol peroxidase activity. Biochim. Biophys. Acta Bioenerg. 2018, 1859, 411–422. [Google Scholar] [CrossRef]
- Malatesta, F.; Antonini, G.; Sarti, P.; Brunori, M. Structure and Function of a Molecular Machine: Cytochrome c Oxidase. Biophys. Chem. 1995, 54, 1–33. [Google Scholar] [CrossRef]
- Melo, A.M.; Teixeira, M. Supramolecular organization of bacterial aerobic respiratory chains: From cells and back. Biochim. Biophys. Acta 2016, 1857, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Pereira, M.M.; Sousa, F.L.; Verissimo, A.F.; Teixeira, M. Looking for the minimum common denominator in haem-copper oxygen reductases: Towards a unified catalytic mechanism. Biochim. Biophys. Acta 2008, 1777, 929–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papa, S.; Capitanio, N.; Capitanio, G.; Palese, L.L. Protonmotive cooperativity in cytochrome c oxidase. Biochim. Biophys. Acta 2004, 1658, 95–105. [Google Scholar] [CrossRef] [Green Version]
- Borisov, V.B.; Siletsky, S.A. Features of organization and mechanism of catalysis of two families of terminal oxidases: Heme-copper and bd-type. Biochemistry 2019, 84, 1390–1402. [Google Scholar] [CrossRef]
- Hederstedt, L. Molecular biology of Bacillus subtilis cytochromes anno 2020. Biochemistry 2021, 86, 8–21. [Google Scholar] [CrossRef]
- Sousa, F.L.; Alves, R.J.; Ribeiro, M.A.; Pereira-Leal, J.B.; Teixeira, M.; Pereira, M.M. The superfamily of heme-copper oxygen reductases: Types and evolutionary considerations. Biochim. Biophys. Acta 2012, 1817, 629–637. [Google Scholar] [CrossRef] [Green Version]
- Wikstrom, M.; Krab, K.; Sharma, V. Oxygen activation and energy conservation by cytochrome c oxidase. Chem. Rev. 2018, 118, 2469–2490. [Google Scholar] [CrossRef] [Green Version]
- Capitanio, N.; Palese, L.L.; Capitanio, G.; Martino, P.L.; Richter, O.M.; Ludwig, B.; Papa, S. Allosteric interactions and proton conducting pathways in proton pumping aa3 oxidases: Heme a as a key coupling element. Biochim. Biophys. Acta 2012, 1817, 558–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maneg, O.; Malatesta, F.; Ludwig, B.; Drosou, V. Interaction of cytochrome c with cytochrome oxidase: Two different docking scenarios. Biochim. Biophys. Acta 2004, 1655, 274–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Ballmoos, C.; Adelroth, P.; Gennis, R.B.; Brzezinski, P. Proton transfer in ba3 cytochrome c oxidase from Thermus thermophilus. Biochim. Biophys. Acta 2012, 1817, 650–657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rich, P.R. Mitochondrial cytochrome c oxidase: Catalysis, coupling and controversies. Biochem. Soc. Trans. 2017, 45, 813–829. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, S.; Shimada, A. Reaction mechanism of cytochrome c oxidase. Chem. Rev. 2015, 115, 1936–1989. [Google Scholar] [CrossRef] [PubMed]
- Siletsky, S.A. Steps of the coupled charge translocation in the catalytic cycle of cytochrome c oxidase. Front. Biosci. 2013, 18, 36–57. [Google Scholar] [CrossRef] [PubMed]
- Forte, E.; Giuffre, A.; Huang, L.S.; Berry, E.A.; Borisov, V.B. Nitric oxide does not inhibit but is metabolized by the cytochrome bcc-aa3 supercomplex. Int. J. Mol. Sci. 2020, 21, 8521. [Google Scholar] [CrossRef]
- Poole, R.K.; Cook, G.M. Redundancy of aerobic respiratory chains in bacteria? Routes, reasons and regulation. Adv. Microb. Physiol. 2000, 43, 165–224. [Google Scholar] [CrossRef] [PubMed]
- Ingledew, W.J.; Poole, R.K. The respiratory chains of Escherichia coli. Microbiol. Rev. 1984, 48, 222–271. [Google Scholar] [CrossRef]
- Safarian, S.; Rajendran, C.; Muller, H.; Preu, J.; Langer, J.D.; Ovchinnikov, S.; Hirose, T.; Kusumoto, T.; Sakamoto, J.; Michel, H. Structure of a bd oxidase indicates similar mechanisms for membrane-integrated oxygen reductases. Science 2016, 352, 583–586. [Google Scholar] [CrossRef] [Green Version]
- Safarian, S.; Hahn, A.; Mills, D.J.; Radloff, M.; Eisinger, M.L.; Nikolaev, A.; Meier-Credo, J.; Melin, F.; Miyoshi, H.; Gennis, R.B.; et al. Active site rearrangement and structural divergence in prokaryotic respiratory oxidases. Science 2019, 366, 100–104. [Google Scholar] [CrossRef]
- Theßeling, A.; Rasmussen, T.; Burschel, S.; Wohlwend, D.; Kagi, J.; Muller, R.; Bottcher, B.; Friedrich, T. Homologous bd oxidases share the same architecture but differ in mechanism. Nat. Commun. 2019, 10, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puustinen, A.; Finel, M.; Haltia, T.; Gennis, R.B.; Wikstrom, M. Properties of the two terminal oxidases of Escherichia coli. Biochemistry 1991, 30, 3936–3942. [Google Scholar] [CrossRef] [PubMed]
- Jasaitis, A.; Borisov, V.B.; Belevich, N.P.; Morgan, J.E.; Konstantinov, A.A.; Verkhovsky, M.I. Electrogenic reactions of cytochrome bd. Biochemistry 2000, 39, 13800–13809. [Google Scholar] [CrossRef] [PubMed]
- Siletsky, S.A.; Borisov, V.B.; Mamedov, M.D. Photosystem II and terminal respiratory oxidases: Molecular machines operating in opposite directions. Front. Biosci. 2017, 22, 1379–1426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belevich, I.; Borisov, V.B.; Zhang, J.; Yang, K.; Konstantinov, A.A.; Gennis, R.B.; Verkhovsky, M.I. Time-resolved electrometric and optical studies on cytochrome bd suggest a mechanism of electron-proton coupling in the di-heme active site. Proc. Natl. Acad. Sci. USA 2005, 102, 3657–3662. [Google Scholar] [CrossRef] [Green Version]
- Belevich, I.; Borisov, V.B.; Verkhovsky, M.I. Discovery of the true peroxy intermediate in the catalytic cycle of terminal oxidases by real-time measurement. J. Biol. Chem. 2007, 282, 28514–28519. [Google Scholar] [CrossRef] [Green Version]
- Borisov, V.B.; Belevich, I.; Bloch, D.A.; Mogi, T.; Verkhovsky, M.I. Glutamate 107 in subunit I of cytochrome bd from Escherichia coli is part of a transmembrane intraprotein pathway conducting protons from the cytoplasm to the heme b595/heme d active site. Biochemistry 2008, 47, 7907–7914. [Google Scholar] [CrossRef] [PubMed]
- Wikstrom, M.; Morgan, J.E.; Verkhovsky, M.I. Proton and electrical charge translocation by cytochrome c-oxidase. Biochim. Biophys. Acta 1997, 1318, 299–306. [Google Scholar] [CrossRef] [Green Version]
- Konstantinov, A.A.; Siletsky, S.; Mitchell, D.; Kaulen, A.; Gennis, R.B. The roles of the two proton input channels in cytochrome c oxidase from Rhodobacter sphaeroides probed by the effects of site-directed mutations on time-resolved electrogenic intraprotein proton transfer. Proc. Natl. Acad. Sci. USA 1997, 94, 9085–9090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borisov, V.B.; Murali, R.; Verkhovskaya, M.L.; Bloch, D.A.; Han, H.; Gennis, R.B.; Verkhovsky, M.I. Aerobic respiratory chain of Escherichia coli is not allowed to work in fully uncoupled mode. Proc. Natl. Acad. Sci. USA 2011, 108, 17320–17324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, M.M.; Santana, M.; Teixeira, M. A novel scenario for the evolution of haem-copper oxygen reductases. Biochim. Biophys. Acta 2001, 1505, 185–208. [Google Scholar] [CrossRef] [Green Version]
- Pereira, M.M.; Gomes, C.M.; Teixeira, M. Plasticity of proton pathways in haem-copper oxygen reductases. FEBS Lett. 2002, 522, 14–18. [Google Scholar] [CrossRef] [Green Version]
- Pereira, M.M.; Teixeira, M. Proton pathways, ligand binding and dynamics of the catalytic site in haem-copper oxygen reductases: A comparison between the three families. Biochim. Biophys. Acta 2004, 1655, 340–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borisov, V.B.; Gennis, R.B.; Hemp, J.; Verkhovsky, M.I. The cytochrome bd respiratory oxygen reductases. Biochim. Biophys. Acta 2011, 1807, 1398–1413. [Google Scholar] [CrossRef] [Green Version]
- Arutyunyan, A.M.; Sakamoto, J.; Inadome, M.; Kabashima, Y.; Borisov, V.B. Optical and magneto-optical activity of cytochrome bd from Geobacillus thermodenitrificans. Biochim. Biophys. Acta 2012, 1817, 2087–2094. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, V.; Rajendran, C.; Sousa, F.L.; Melo, A.M.; Saraiva, L.M.; Pereira, M.M.; Santana, M.; Teixeira, M.; Michel, H. Structure at 1.3 A resolution of Rhodothermus marinus caa3 cytochrome c domain. J. Mol. Biol. 2005, 345, 1047–1057. [Google Scholar] [CrossRef]
- Buschmann, S.; Warkentin, E.; Xie, H.; Langer, J.D.; Ermler, U.; Michel, H. The structure of cbb3 cytochrome oxidase provides insights into proton pumping. Science 2010, 329, 327–330. [Google Scholar] [CrossRef]
- Noor, M.R.; Soulimane, T. Structure of caa3 cytochrome c oxidase—A nature-made enzyme-substrate complex. Biol. Chem. 2013, 394, 579–591. [Google Scholar] [CrossRef]
- Azarkina, N.; Siletsky, S.; Borisov, V.; von Wachenfeldt, C.; Hederstedt, L.; Konstantinov, A.A. A cytochrome bb’-type quinol oxidase in Bacillus subtilis strain 168. J. Biol. Chem. 1999, 274, 32810–32817. [Google Scholar] [CrossRef] [Green Version]
- Verkhovsky, M.I.; Morgan, J.E.; Puustinen, A.; Wikstrom, M. Kinetic trapping of oxygen in cell respiration. Nature 1996, 380, 268–270. [Google Scholar] [CrossRef]
- D’mello, R.; Hill, S.; Poole, R.K. The cytochrome bd quinol oxidase in Escherichia coli has an extremely high oxygen affinity and two-oxygen-binding haems: Implicaitons for regulation of activity in vivo by oxygen inihibition. Microbiology 1996, 142, 755–763. [Google Scholar] [CrossRef] [Green Version]
- Belevich, I.; Borisov, V.B.; Konstantinov, A.A.; Verkhovsky, M.I. Oxygenated complex of cytochrome bd from Escherichia coli: Stability and photolability. FEBS Lett. 2005, 579, 4567–4570. [Google Scholar] [CrossRef] [Green Version]
- Belevich, I.; Borisov, V.B.; Bloch, D.A.; Konstantinov, A.A.; Verkhovsky, M.I. Cytochrome bd from Azotobacter vinelandii: Evidence for high-affinity oxygen binding. Biochemistry 2007, 46, 11177–11184. [Google Scholar] [CrossRef]
- Forte, E.; Borisov, V.B.; Konstantinov, A.A.; Brunori, M.; Giuffre, A.; Sarti, P. Cytochrome bd, a key oxidase in bacterial survival and tolerance to nitrosative stress. Ital. J. Biochem. 2007, 56, 265–269. [Google Scholar]
- Borisov, V.B.; Forte, E.; Siletsky, S.A.; Arese, M.; Davletshin, A.I.; Sarti, P.; Giuffre, A. Cytochrome bd protects bacteria against oxidative and nitrosative stress: A potential target for next-generation antimicrobial agents. Biochemistry 2015, 80, 565–575. [Google Scholar] [CrossRef]
- Giuffre, A.; Borisov, V.B.; Mastronicola, D.; Sarti, P.; Forte, E. Cytochrome bd oxidase and nitric oxide: From reaction mechanisms to bacterial physiology. FEBS Lett. 2012, 586, 622–629. [Google Scholar] [CrossRef]
- Giuffre, A.; Borisov, V.B.; Arese, M.; Sarti, P.; Forte, E. Cytochrome bd oxidase and bacterial tolerance to oxidative and nitrosative stress. Biochim. Biophys. Acta 2014, 1837, 1178–1187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forte, E.; Borisov, V.B.; Vicente, J.B.; Giuffre, A. Cytochrome bd and gaseous ligands in bacterial physiology. Adv. Microb. Physiol. 2017, 71, 171–234. [Google Scholar] [CrossRef] [PubMed]
- Borisov, V.B.; Forte, E.; Konstantinov, A.A.; Poole, R.K.; Sarti, P.; Giuffre, A. Interaction of the bacterial terminal oxidase cytochrome bd with nitric oxide. FEBS Lett. 2004, 576, 201–204. [Google Scholar] [CrossRef] [Green Version]
- Borisov, V.B.; Forte, E.; Sarti, P.; Brunori, M.; Konstantinov, A.A.; Giuffre, A. Nitric oxide reacts with the ferryl-oxo catalytic intermediate of the CuB-lacking cytochrome bd terminal oxidase. FEBS Lett. 2006, 580, 4823–4826. [Google Scholar] [CrossRef] [Green Version]
- Borisov, V.B.; Forte, E.; Sarti, P.; Brunori, M.; Konstantinov, A.A.; Giuffre, A. Redox control of fast ligand dissociation from Escherichia coli cytochrome bd. Biochem. Biophys. Res. Commun. 2007, 355, 97–102. [Google Scholar] [CrossRef]
- Mason, M.G.; Shepherd, M.; Nicholls, P.; Dobbin, P.S.; Dodsworth, K.S.; Poole, R.K.; Cooper, C.E. Cytochrome bd confers nitric oxide resistance to Escherichia coli. Nat. Chem. Biol. 2009, 5, 94–96. [Google Scholar] [CrossRef]
- Borisov, V.B.; Forte, E.; Giuffre, A.; Konstantinov, A.; Sarti, P. Reaction of nitric oxide with the oxidized di-heme and heme-copper oxygen-reducing centers of terminal oxidases: Different reaction pathways and end-products. J. Inorg. Biochem. 2009, 103, 1185–1187. [Google Scholar] [CrossRef]
- Shepherd, M.; Achard, M.E.; Idris, A.; Totsika, M.; Phan, M.D.; Peters, K.M.; Sarkar, S.; Ribeiro, C.A.; Holyoake, L.V.; Ladakis, D.; et al. The cytochrome bd-I respiratory oxidase augments survival of multidrug-resistant Escherichia coli during infection. Sci. Rep. 2016, 6, 35285. [Google Scholar] [CrossRef] [PubMed]
- Holyoake, L.V.; Hunt, S.; Sanguinetti, G.; Cook, G.M.; Howard, M.J.; Rowe, M.L.; Poole, R.K.; Shepherd, M. CydDC-mediated reductant export in Escherichia coli controls the transcriptional wiring of energy metabolism and combats nitrosative stress. Biochem. J. 2016, 473, 693–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones-Carson, J.; Husain, M.; Liu, L.; Orlicky, D.J.; Vazquez-Torres, A. Cytochrome bd-dependent bioenergetics and antinitrosative defenses in Salmonella pathogenesis. mBio 2016, 7, e02052–16. [Google Scholar] [CrossRef] [Green Version]
- Meng, Q.; Yin, J.; Jin, M.; Gao, H. Distinct nitrite and nitric oxide physiologies in Escherichia coli and Shewanella oneidensis. Appl. Environ. Microbiol. 2018, 84, e00559–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beebout, C.J.; Eberly, A.R.; Werby, S.H.; Reasoner, S.A.; Brannon, J.R.; De, S.; Fitzgerald, M.J.; Huggins, M.M.; Clayton, D.B.; Cegelski, L.; et al. Respiratory heterogeneity shapes biofilm formation and host colonization in uropathogenic Escherichia coli. mBio 2019, 10, e02400–18. [Google Scholar] [CrossRef] [Green Version]
- Borisov, V.B.; Forte, E.; Siletsky, S.A.; Sarti, P.; Giuffre, A. Cytochrome bd from Escherichia coli catalyzes peroxynitrite decomposition. Biochim. Biophys. Acta 2015, 1847, 182–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forte, E.; Borisov, V.B.; Falabella, M.; Colaco, H.G.; Tinajero-Trejo, M.; Poole, R.K.; Vicente, J.B.; Sarti, P.; Giuffre, A. The terminal oxidase cytochrome bd promotes sulfide-resistant bacterial respiration and growth. Sci. Rep. 2016, 6, 23788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korshunov, S.; Imlay, K.R.; Imlay, J.A. The cytochrome bd oxidase of Escherichia coli prevents respiratory inhibition by endogenous and exogenous hydrogen sulfide. Mol. Microbiol. 2016, 101, 62–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forte, E.; Giuffre, A. How bacteria breathe in hydrogen sulphide-rich environments. Biochemist 2016, 38, 8–11. [Google Scholar] [CrossRef]
- Borisov, V.B.; Forte, E. Terminal oxidase cytochrome bd protects bacteria against hydrogen sulfide toxicity. Biochemistry 2021, 86, 22–32. [Google Scholar] [CrossRef]
- Forte, E.; Siletsky, S.A.; Borisov, V.B. In Escherichia coli ammonia inhibits cytochrome bo3 but activates cytochrome bd-I. Antioxidants 2021, 10, 13. [Google Scholar] [CrossRef] [PubMed]
- Kita, K.; Konishi, K.; Anraku, Y. Terminal oxidases of Escherichia coli aerobic respiratory chain. II. Purification and properties of cytochrome b558-d complex from cells grown with limited oxygen and evidence of branched electron-carrying systems. J. Biol. Chem. 1984, 259, 3375–3381. [Google Scholar] [CrossRef]
- Sakamoto, J.; Koga, E.; Mizuta, T.; Sato, C.; Noguchi, S.; Sone, N. Gene structure and quinol oxidase activity of a cytochrome bd-type oxidase from Bacillus stearothermophilus. Biochim. Biophys. Acta 1999, 1411, 147–158. [Google Scholar] [CrossRef] [Green Version]
- Borisov, V.B.; Siletsky, S.A.; Paiardini, A.; Hoogewijs, D.; Forte, E.; Giuffre, A.; Poole, R.K. Bacterial oxidases of the cytochrome bd family: Redox enzymes of unique structure, function and utility as drug targets. Antioxid. Redox Signal. 2021, 34, 1280–1318. [Google Scholar] [CrossRef] [PubMed]
- Mascolo, L.; Bald, D. Cytochrome bd in Mycobacterium tuberculosis: A respiratory chain protein involved in the defense against antibacterials. Prog. Biophys. Mol. Biol. 2020, 152, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.S.; Sviriaeva, E.; Pethe, K. Targeting the cytochrome oxidases for drug development in mycobacteria. Prog. Biophys. Mol. Biol. 2020, 152, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Cook, G.M.; Hards, K.; Dunn, E.; Heikal, A.; Nakatani, Y.; Greening, C.; Crick, D.C.; Fontes, F.L.; Pethe, K.; Hasenoehrl, E.; et al. Oxidative phosphorylation as a target space for tuberculosis: Success, caution, and future directions. Microbiol. Spectr. 2017, 5, 295–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bald, D.; Villellas, C.; Lu, P.; Koul, A. Targeting energy metabolism in Mycobacterium tuberculosis, a new paradigm in antimycobacterial drug discovery. mBio 2017, 8, e00272–17. [Google Scholar] [CrossRef] [Green Version]
- Hards, K.; Cook, G.M. Targeting bacterial energetics to produce new antimicrobials. Drug Resist. Update 2018, 36, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Winstedt, L.; Frankenberg, L.; Hederstedt, L.; von Wachenfeldt, C. Enterococcus faecalis V583 contains a cytochrome bd-type respiratory oxidase. J. Bacteriol. 2000, 182, 3863–3866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Junemann, S.; Butterworth, P.J.; Wrigglesworth, J.M. A suggested mechanism for the catalytic cycle of cytochrome bd terminal oxidase based on kinetic analysis. Biochemistry 1995, 34, 14861–14867. [Google Scholar] [CrossRef] [PubMed]
- Poole, R.K.; Hill, S. Respiratory protection of nitrogenase activity in Azotobacter vinelandii—Roles of the terminal oxidases. Biosci. Rep. 1997, 17, 307–317. [Google Scholar] [CrossRef]
- Kaminski, P.A.; Kitts, C.L.; Zimmerman, Z.; Ludwig, R.A. Azorhizobium caulinodans uses both cytochrome bd (quinol) and cytochrome cbb3 (cytochrome c) terminal oxidases for symbiotic N2 fixation. J. Bacteriol. 1996, 178, 5989–5994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juty, N.S.; Moshiri, F.; Merrick, M.; Anthony, C.; Hill, S. The Klebsiella pneumoniae cytochrome bd’ terminal oxidase complex and its role in microaerobic nitrogen fixation. Microbiology 1997, 143, 2673–2683. [Google Scholar] [CrossRef] [Green Version]
- Kelly, M.J.S.; Poole, R.K.; Yates, M.G.; Kennedy, C. Cloning and mutagenesis of genes encoding the cytochrome bd terminal oxidase complex in Azotobacter vinelandii: Mutants deficient in the cytochrome d complex are unable to fix nitrogen in air. J. Bacteriol. 1990, 172, 6010–6019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baughn, A.D.; Malamy, M.H. The strict anaerobe Bacteroides fragilis grows in and benefits from nanomolar concentrations of oxygen. Nature 2004, 427, 441–444. [Google Scholar] [CrossRef] [PubMed]
- Lemos, R.S.; Gomes, C.M.; Santana, M.; LeGall, J.; Xavier, A.V.; Teixeira, M. The ‘strict’ anaerobe Desulfovibrio gigas contains a membrane-bound oxygen-reducing respiratory chain. FEBS Lett. 2001, 496, 40–43. [Google Scholar] [CrossRef] [Green Version]
- Machado, P.; Felix, R.; Rodrigues, R.; Oliveira, S.; Rodrigues-Pousada, C. Characterization and expression analysis of the cytochrome bd oxidase operon from Desulfovibrio gigas. Curr. Microbiol. 2006, 52, 274–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassani, B.K.; Steunou, A.S.; Liotenberg, S.; Reiss-Husson, F.; Astier, C.; Ouchane, S. Adaptation to oxygen: Role of terminal oxidases in photosynthesis initiation in the purple photosynthetic bacterium, Rubrivivax gelatinosus. J. Biol. Chem. 2010, 285, 19891–19899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korshunov, S.; Imlay, J.A. Two sources of endogenous hydrogen peroxide in Escherichia coli. Mol. Microbiol. 2010, 75, 1389–1401. [Google Scholar] [CrossRef] [Green Version]
- Wall, D.; Delaney, J.M.; Fayet, O.; Lipinska, B.; Yamamoto, T.; Georgopoulos, C. arc-Dependent thermal regulation and extragenic suppression of the Escherichia coli cytochrome d operon. J. Bacteriol. 1992, 174, 6554–6562. [Google Scholar] [CrossRef] [Green Version]
- Goldman, B.S.; Gabbert, K.K.; Kranz, R.G. The temperature-sensitive growth and survival phenotypes of Escherichia coli cydDC and cydAB strains are due to deficiencies in cytochrome bd and are corrected by exogenous catalase and reducing agents. J. Bacteriol. 1996, 178, 6348–6351. [Google Scholar] [CrossRef] [Green Version]
- Lindqvist, A.; Membrillo-Hernandez, J.; Poole, R.K.; Cook, G.M. Roles of respiratory oxidases in protecting Escherichia coli K12 from oxidative stress. Antonie Leeuwenhoek 2000, 78, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Edwards, S.E.; Loder, C.S.; Wu, G.; Corker, H.; Bainbridge, B.W.; Hill, S.; Poole, R.K. Mutation of cytochrome bd quinol oxidase results in reduced stationary phase survival, iron deprivation, metal toxicity and oxidative stress in Azotobacter vinelandii. FEMS Microbiol. Lett. 2000, 185, 71–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, X.; Wu, S.; Li, L.; Xu, B.; Wang, G. The cytochrome bd complex is essential for chromate and sulfide resistance and is regulated by a GbsR-type regulator, CydE, in Alishewanella sp. WH16-1. Front. Microbiol. 2018, 9, 1849. [Google Scholar] [CrossRef]
- Endley, S.; McMurray, D.; Ficht, T.A. Interruption of the cydB locus in Brucella abortus attenuates intracellular survival and virulence in the mouse model of infection. J. Bacteriol. 2001, 183, 2454–2462. [Google Scholar] [CrossRef] [Green Version]
- Leclerc, J.; Rosenfeld, E.; Trainini, M.; Martin, B.; Meuric, V.; Bonnaure-Mallet, M.; Baysse, C. The cytochrome bd oxidase of Porphyromonas gingivalis contributes to oxidative stress resistance and dioxygen tolerance. PLoS ONE 2015, 10, e0143808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Small, J.L.; Park, S.W.; Kana, B.D.; Ioerger, T.R.; Sacchettini, J.C.; Ehrt, S. Perturbation of cytochrome c maturation reveals adaptability of the respiratory chain in Mycobacterium tuberculosis. mBio 2013, 4, e00475–13. [Google Scholar] [CrossRef] [Green Version]
- Lu, P.; Heineke, M.H.; Koul, A.; Andries, K.; Cook, G.M.; Lill, H.; van Spanning, R.; Bald, D. The cytochrome bd-type quinol oxidase is important for survival of Mycobacterium smegmatis under peroxide and antibiotic-induced stress. Sci. Rep. 2015, 5, 10333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, W.; Small, D.A.; Toghrol, F.; Bentley, W.E. Global transcriptome analysis of Staphylococcus aureus response to hydrogen peroxide. J. Bacteriol. 2006, 188, 1648–1659. [Google Scholar] [CrossRef] [Green Version]
- Borisov, V.B.; Forte, E.; Davletshin, A.; Mastronicola, D.; Sarti, P.; Giuffre, A. Cytochrome bd oxidase from Escherichia coli displays high catalase activity: An additional defense against oxidative stress. FEBS Lett. 2013, 587, 2214–2218. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.C. Reversible binding and inhibition of catalase by nitric oxide. Eur. J. Biochem. 1995, 232, 188–191. [Google Scholar] [CrossRef] [PubMed]
- Deisseroth, A.; Dounce, A.L. Catalase: Physical and chemical properties, mechanism of catalysis, and physiological role. Physiol. Rev. 1970, 50, 319–375. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Panmanee, W.; Wilson, J.J.; Mahtani, H.K.; Li, Q.; Vanderwielen, B.D.; Makris, T.M.; Rogers, M.; McDaniel, C.; Lipscomb, J.D.; et al. Catalase (KatA) plays a role in protection against anaerobic nitric oxide in Pseudomonas aeruginosa. PLoS ONE 2014, 9, e91813. [Google Scholar] [CrossRef]
- Al-Attar, S.; Yu, Y.; Pinkse, M.; Hoeser, J.; Friedrich, T.; Bald, D.; de Vries, S. Cytochrome bd displays significant quinol peroxidase activity. Sci. Rep. 2016, 6, 27631. [Google Scholar] [CrossRef] [Green Version]
- Meunier, B.; Madgwick, S.A.; Reil, E.; Oettmeier, W.; Rich, P.R. New inhibitors of the quinol oxidation sites of bacterial cytochromes bo and bd. Biochemistry 1995, 34, 1076–1083. [Google Scholar] [CrossRef]
- Reisz, J.A.; Bechtold, E.; King, S.B.; Poole, L.B.; Furdui, C.M. Thiol-blocking electrophiles interfere with labeling and detection of protein sulfenic acids. FEBS J. 2013, 280, 6150–6161. [Google Scholar] [CrossRef] [Green Version]
- Forte, E.; Borisov, V.B.; Siletsky, S.A.; Petrosino, M.; Giuffre, A. In the respiratory chain of Escherichia coli cytochromes bd-I and bd-II are more sensitive to carbon monoxide inhibition than cytochrome bo3. Biochim. Biophys. Acta Bioenerg. 2019, 1860, 148088. [Google Scholar] [CrossRef]
- Borisov, V.; Arutyunyan, A.M.; Osborne, J.P.; Gennis, R.B.; Konstantinov, A.A. Magnetic circular dichroism used to examine the interaction of Escherichia coli cytochrome bd with ligands. Biochemistry 1999, 38, 740–750. [Google Scholar] [CrossRef]
- Borisov, V.B. Effect of membrane environment on ligand-binding properties of the terminal oxidase cytochrome bd-I from Escherichia coli. Biochemistry 2020, 85, 1603–1612. [Google Scholar] [CrossRef]
- Chanin, R.B.; Winter, M.G.; Spiga, L.; Hughes, E.R.; Zhu, W.; Taylor, S.J.; Arenales, A.; Gillis, C.C.; Buttner, L.; Jimenez, A.G.; et al. Epithelial-derived reactive oxygen species enable AppBCX-mediated aerobic respiration of Escherichia coli during intestinal inflammation. Cell Host Microbe 2020, 28, 780–788. [Google Scholar] [CrossRef] [PubMed]
- Forte, E.; Borisov, V.B.; Davletshin, A.; Mastronicola, D.; Sarti, P.; Giuffre, A. Cytochrome bd oxidase and hydrogen peroxide resistance in Mycobacterium tuberculosis. mBio 2013, 4, e01006–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borisov, V.B.; Davletshin, A.I.; Konstantinov, A.A. Peroxidase activity of cytochrome bd from Escherichia coli. Biochemistry 2010, 75, 428–436. [Google Scholar] [CrossRef] [PubMed]
- Siletsky, S.A.; Konstantinov, A.A. Cytochrome c oxidase: Charge translocation coupled to single-electron partial steps of the catalytic cycle. Biochim. Biophys. Acta 2012, 1817, 476–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaila, V.R.I.; Wikstrom, M. Architecture of bacterial respiratory chains. Nat. Rev. Microbiol. 2021, 19, 319–330. [Google Scholar] [CrossRef]
- Siletsky, S.A.; Belevich, I.; Jasaitis, A.; Konstantinov, A.A.; Wikstrom, M.; Soulimane, T.; Verkhovsky, M.I. Time-resolved single-turnover of ba3 oxidase from Thermus thermophilus. Biochim. Biophys. Acta 2007, 1767, 1383–1392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siletskiy, S.; Soulimane, T.; Azarkina, N.; Vygodina, T.V.; Buse, G.; Kaulen, A.; Konstantinov, A. Time-resolved generation of a membrane potential by ba3 cytochrome c oxidase from Thermus thermophilus. Evidence for reduction-induced opening of the binuclear center. FEBS Lett. 1999, 457, 98–102. [Google Scholar] [CrossRef] [Green Version]
- Blomberg, M.R.A. Role of the two metals in the active sites of heme copper oxidases-A study of NO reduction in cbb3 cytochrome c oxidase. Inorg. Chem. 2020, 59, 11542–11553. [Google Scholar] [CrossRef]
- Orii, Y. The cytochrome c peroxidase activity of cytochrome oxidase. J. Biol. Chem. 1982, 257, 9246–9248. [Google Scholar] [CrossRef]
- Orii, Y.; Okunuki, K. Studies on cytochrome a. X. Effect of hydrogen peroxide on absorption spectra of cytochrome a. J. Biochem. 1963, 54, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Bolshakov, I.A.; Vygodina, T.V.; Gennis, R.; Karyakin, A.A.; Konstantinov, A.A. Catalase activity of cytochrome c oxidase assayed with hydrogen peroxide-sensitive electrode microsensor. Biochemistry 2010, 75, 1352–1360. [Google Scholar] [CrossRef] [PubMed]
- Sedlak, E.; Fabian, M.; Robinson, N.C.; Musatov, A. Ferricytochrome c protects mitochondrial cytochrome c oxidase against hydrogen peroxide-induced oxidative damage. Free Radic. Biol. Med. 2010, 49, 1574–1581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ksenzenko, M.Y.; Berka, V.; Vygodina, T.V.; Ruuge, E.K.; Konstantinov, A.A. Cytochrome oxidase-catalyzed superoxide generation from hydrogen peroxide. FEBS Lett. 1992, 297, 63–66. [Google Scholar] [CrossRef] [Green Version]
- Jancura, D.; Stanicova, J.; Palmer, G.; Fabian, M. How hydrogen peroxide is metabolized by oxidized cytochrome c oxidase. Biochemistry 2014, 53, 3564–3575. [Google Scholar] [CrossRef]
- Pereverzev, M.O.; Vygodina, T.V.; Konstantinov, A.A.; Skulachev, V.P. Cytochrome c, an ideal antioxidant. Biochem. Soc. Trans. 2003, 31, 1312–1315. [Google Scholar] [CrossRef]
- Ramzan, R.; Rhiel, A.; Weber, P.; Kadenbach, B.; Vogt, S. Reversible dimerization of cytochrome c oxidase regulates mitochondrial respiration. Mitochondrion 2019, 49, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Hilbers, F.; von der Hocht, I.; Ludwig, B.; Michel, H. True wild type and recombinant wild type cytochrome c oxidase from Paracoccus denitrificans show a 20-fold difference in their catalase activity. Biochim. Biophys. Acta 2013, 1827, 319–327. [Google Scholar] [CrossRef] [Green Version]
- Siletsky, S.A.; Gennis, R.B. Time-resolved electrometric study of the F→O transition in cytochrome c oxidase. The effect of Zn2+ ions on the positive side of the membrane. Biochemistry 2021, 86, 105–122. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.C. Mechanisms of inflammatory neurodegeneration: iNOS and NADPH oxidase. Biochem. Soc. Trans. 2007, 35, 1119–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giuffre, A.; Stubauer, G.; Sarti, P.; Brunori, M.; Zumft, W.G.; Buse, G.; Soulimane, T. The heme-copper oxidases of Thermus thermophilus catalyze the reduction of nitric oxide: Evolutionary implications. Proc. Natl. Acad. Sci. USA 1999, 96, 14718–14723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, T.; Lin, I.J.; Chen, Y.; Fee, J.A.; Moenne-Loccoz, P. Fourier transform infrared characterization of a CuB-nitrosyl complex in cytochrome ba3 from Thermus thermophilus: Relevance to NO reductase activity in heme-copper terminal oxidases. J. Am. Chem. Soc. 2007, 129, 14952–14958. [Google Scholar] [CrossRef] [PubMed] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borisov, V.B.; Siletsky, S.A.; Nastasi, M.R.; Forte, E. ROS Defense Systems and Terminal Oxidases in Bacteria. Antioxidants 2021, 10, 839. https://doi.org/10.3390/antiox10060839
Borisov VB, Siletsky SA, Nastasi MR, Forte E. ROS Defense Systems and Terminal Oxidases in Bacteria. Antioxidants. 2021; 10(6):839. https://doi.org/10.3390/antiox10060839
Chicago/Turabian StyleBorisov, Vitaliy B., Sergey A. Siletsky, Martina R. Nastasi, and Elena Forte. 2021. "ROS Defense Systems and Terminal Oxidases in Bacteria" Antioxidants 10, no. 6: 839. https://doi.org/10.3390/antiox10060839
APA StyleBorisov, V. B., Siletsky, S. A., Nastasi, M. R., & Forte, E. (2021). ROS Defense Systems and Terminal Oxidases in Bacteria. Antioxidants, 10(6), 839. https://doi.org/10.3390/antiox10060839