
Molecular Mechanisms Related to Oxidative Stress in Retinitis Pigmentosa

 ,
,  ,
, 
Abstract
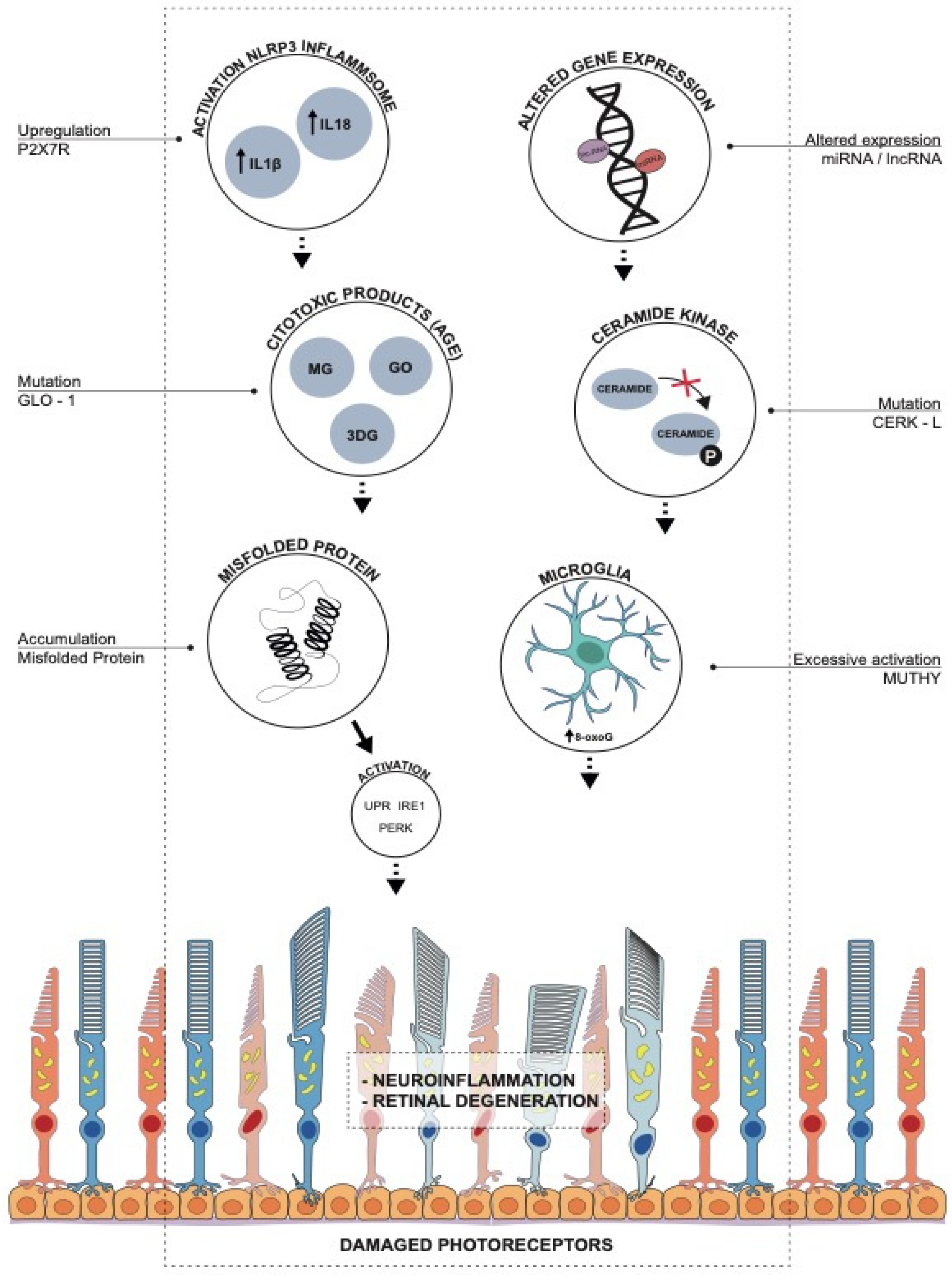
:1. Introduction
Association between RP-Causative Mutations and Activation of UPR, PERK and IRE1 as a Response to Oxidative Stress
2. Mutations in Endogenous Antioxidant Pathways: MUTYH, CERKL and GLO1
2.1. Role of 8-Oxoguanine and MUTYH in RP
2.2. Oxidative Stress in RP: The Role of CERKL
2.3. Glyoxalase 1 (GLO1) Related Genes and Pathways
2.3.1. AUTS2
2.3.2. ARHGAP21 and PTPN13
2.3.3. FMNL2
2.3.4. UBC, MYO18A, EPS15, ANKH
2.3.5. RFFL, FBXW2, CAND1
2.3.6. SIK3
2.3.7. IPO7
2.3.8. MRPS33, MORC4, MCPH1, NFIA, CTIF and LMBRD1
3. miRNA Altered Expression and Oxidative Stress:
4. Role of Long Non-Codingrna
5. P2X7 Receptor and Inflammation in RP
6. The Dual Role of Microglia in RP: Between Neurotoxicity and Neuroprotection
7. Conclusions
Funding
Conflicts of Interest
Abbreviations
| 3-DG | 3-DeoxyGlucosone |
| 8-oxoG | 8oxoGuanine |
| ACACA | Acetyl-CoA Carboxylase Alpha |
| AGEs | Advanced Glycation End products |
| AIPL1 | Aryl Hydrocarbon Receptor Interacting Protein Like 1 |
| AKT1 | AKT Serine/Threonine Kinase 1 |
| ALEs | Advanced lipoxidation End-products |
| ANKH | Progressive ankylosis protein homolog |
| ARF | Alternate Reading Frame |
| ARHGAP21 | Rho GTPase Activating Protein 21 |
| AUTS2 | Cytoplasmatic activator of transcription and developmental regulator |
| BDNF-AS | Brain-derived neurotrophic factor Antisense |
| BER | Base Exicision Repair |
| C3 | Complement Component 3 |
| CAND1 | Cullin-associated NEDD8-dissociated protein 1 |
| CAP1 | Cyclase Associated Actin Cytoskeleton Regulatory Protein 1 |
| CERKL | Ceramide Kinase Like |
| CR3 | Complement Receptor 3 |
| CRD | Cone-Rode Dystrophy |
| CREB | cAMP response element-binding protein |
| CRNDE | Colorectal Neoplasia Differentially Expressed |
| CSF1R | Colony Stimulator Factor 1 Receptor (PLX5622) |
| CTIF | Cap Binding Complex Dependent Translation Initiation Factor |
| CYTOR | Cytoskeleton Regulator RNA |
| EPS15 | Epidermal Growth Factor Receptor Pathway Substrate 15 |
| FBXW2 | F-box/WD repeat-containing protein 2 |
| FMNL2 | Formin Like Protein 2 |
| FMR1 | FMRP Translational Regulator 1 |
| GAPDH | Glyceraldehyde 3-phosphate Dehydrogenase |
| GLO1 | Glyoxalase 1 |
| GO | Glyoxal |
| hMTH1 | Human MutT Homologue |
| HSP | Heat Shock Protein |
| iBRB | Inner Blood-Retinal Barrier |
| IGF1 | insulin-like growth factor-1 |
| IL-18 | Interleukin-18 |
| IL-1β | Interleukin-1β |
| IPO3 | Transportin-3 |
| IPO7 | Importin 7 |
| IRE1 | Inositol-Requiring Enzyme 1 |
| KLHL7 | Kelch Like Family Member 7 |
| LC3-II | Lipidated form of LC3 (Microtubule-associated protein 1A/1B-light chain 3) |
| LMBRD1 | Limb Development Membrane Protein Domain 1 |
| lnc-RNA | long non-coding-RNA |
| MCPH | Microcephalin |
| MG | MethylGlyoxal |
| MIR31HG | MicroRNA 31 Host Gene |
| miRNA | microRNA |
| miRNome | Murine miRNA |
| MNX | Motor Neuron And Pancreas Homeobox 1 |
| MNX-AS1 | antisense transcript of MNX1 |
| MORC 4 | MORC family CW-type zinc finger protein 4 |
| MRPS33 | 28S ribosomal protein S33 |
| mTOR | Mammalian target of Rapamycin |
| MUTYH | mutY DNA glycosylase |
| MYO18A | Myosin XVIIIA |
| NFIA | Nuclear Factor I A |
| NLS | Nuclear Localization Signals |
| NPC | Nuclear Pore Complex |
| ONL | Outer Nuclear Layer |
| OS | Oxidative Stress |
| P2X7R | P2X 7 receptor |
| PARP | Poly(ADP-ribose) polymerase |
| PEDF | Pigment Epithelium-derived Factor |
| PERK | PKR-like Endoplasmic Reticulum Kinase |
| PI3K | Phosphoinositide 3 Kinase |
| PINK1 | PTEN-induced kinase 1 |
| POS | Photoreceptor Outer Segment |
| PPAR | peroxisome proliferator-activated receptor |
| PRC1 | Polycomb Complex 1 |
| PTEN | Phosphatase and tensin homolog |
| PTPN13 | Protein Tyrosine Phosphatase Non-Receptor type 13 |
| RAC1 | Rac Family Small GTPase 1 |
| RDH11 | Retinol Dehydrogenase 11 |
| RE | Rough Endoplasmic reticulum |
| RFFL | E3 ubiquitin-protein ligase rififylin |
| ROS | Reactive Oxygen Species |
| RPE | Retinal Pigment Epithelium |
| SIK3 | SIK Family Kinase 3 |
| SL | SphingoLipid |
| SOD | Superoxide Dismutase |
| SRGAP1 | Slit-Robo Rho GTPase Activating Protein 1 |
| SSB | Single Strand Breaks |
| TUG1 | Taurine Up-Regulated 1 |
| UBC | Ubiquitin C |
| UPR | Unfolded Protein Response |
| USH1G | Usher syndrome type-1G protein |
| VEGF | Vascular Endothelial Growth Factor |
| VIM | Vimentin |
References
- Campa, C.; Gallenga, C.E.; Bolletta, E.; Perri, P. The Role of Gene Therapy in the Treatment of Retinal Diseases: A Review. Curr. Gene Ther. 2017, 17, 194–213. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, F.S.; Gallenga, C.E.; Bonifazzi, C.; Perri, P. A challenge to the striking genotypic heterogeneity of retinitis pigmentosa: A better understanding of the pathophysiology using the newest genetic strategies. Eye 2016, 30, 1542–1548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef] [PubMed]
- Gorbatyuk, M.S.; Starra, C.R.; Gorbatyuk, O.S. Endoplasmic reticulum stress: New insights into the pathogenesis and treatment of retinal degenerative diseases. Prog. Retin. Eye Res. 2020, 79, 100860. [Google Scholar] [CrossRef]
- Domènech, E.B.; Marfany, G. The Relevance of Oxidative Stress in the Pathogenesis and Therapy of Retinal Dystrophies. Antioxidants 2020, 9, 347. [Google Scholar] [CrossRef] [Green Version]
- Kroeger, H.; Chiang, W.C.; Felden, J.; Nguyen, A.; Lin, J.H. ER stress and unfolded protein response in ocular health and disease. FEBS J. 2019, 286, 399–412. [Google Scholar] [CrossRef]
- Donato, L.; Scimone, C.; Nicocia, G.; Denaro, L.; Robledo, R.; Sidoti, A.; D’Angelo, R. GLO1 gene polymorphisms and their association with retinitis pigmentosa: A case–control study in a Sicilian population. Mol. Biol. Rep. 2018, 45, 1349–1355. [Google Scholar] [CrossRef]
- Boya, P. Why autophagy is good for retinal ganglion cells? Eye 2017, 31, 185–190. [Google Scholar] [CrossRef] [Green Version]
- Boya, P.; Esteban-Martínez, L.; Serrano-Puebla, A.; Gómez-Sintes, R.; Villarejo-Zori, B. Autophagy in the eye: Development, degeneration, and aging. Prog. Retin. Eye Res. 2016, 55, 206–245. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Muela, N.; Hernández-Pinto, A.M.; Serrano-Puebla, A.; García-Ledo, L.; Latorre, S.H.; de la Rosa, E.J.; Boya, P. Lysosomal membrane permeabilization and autophagy blockade contribute to photoreceptor cell death in a mouse model of retinitis pigmentosa. Cell Death Differ. 2015, 22, 476–487. [Google Scholar] [CrossRef] [Green Version]
- Donato, L.; Bramanti, P.; Scimone, C.; Rinaldi, C.; Giorgianni, F.; Beranova-Giorgianni, S.; Koirala, D.; D’Angelo, R.; Sidoti, A. miRNAexpression profile of retinal pigment epithelial cells under oxidative stress conditions. FEBS Open Bio 2018, 8, 219–233. [Google Scholar] [CrossRef] [Green Version]
- Jingwen, C.; Zhenqian, H.; Xin, W.; Jaqi, K.; Yan, R.; Wei, G.; Xiang, L.; Jingmei, W.; Weidong, D.; Yusaku, N.; et al. Oxidative stress induces different tissue dependent effects on Mutyh-deficient mice. Free Radic. Biol. Med. 2019, 143, 482–493. [Google Scholar]
- Douglas, M.; Nunez, N.N.; Burnside, M.A.; Bradshaw, K.M.; David, S.S. Repair of 8-oxoG: A Mismatches by the MUTYH Glycosylase: Mechanism, Metals and Medicine. Free Radic. Biol. Med. 2017, 107, 202–215. [Google Scholar]
- Foti, J.J.; Devadoss, B.; Winkler, J.A.; Collins, J.J.; Walker, G.C. Oxidation of the guanine nucleotide pool underlies cell death by bactericidal antibiotics. Science 2012, 336, 315–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakatake, S.; Murakami, Y.; Ikeda, Y.; Morioka, N.; Tachibana, T.; Fujiwara, K.; Yoshida, N.; Notomi, S.; Hisatomi, T.; Yoshida, S.; et al. MUTYH promotes oxidative microglial activation and inherited retinal degeneration. JCI Insight 2016, 1, e87781. [Google Scholar] [CrossRef] [Green Version]
- Oka, S.; Nakabeppu, Y. DNA glycosylase encoded byMUTYH functions as a molecular switch for programmed cell death under oxidative stress to suppress tumorigenesis. Cancer Sci. 2011, 102, 677–682. [Google Scholar] [CrossRef] [PubMed]
- Oka, S.; Ohno, M.; Tsuchimoto, D.; Sakumi, K.; Furuichi, M.; Nakabeppu, Y. Two distinct pathways of cell death triggered by oxidative damage to nuclear and mitochondrial DNAs. EMBO J. 2008, 27, 421–432. [Google Scholar] [CrossRef] [Green Version]
- Murakami, Y.; Ikeda, Y.; Yoshida, N.; Notomi, S.; Hisatomi, T.; Oka, S.; De Luca, G.; Yonemitsu, Y.; Bignami, M.; Nakabeppu, Y.; et al. MutT homolog-1 attenuates oxidative DNA damage and delays photoreceptor cell death in inherited retinal degeneration. Am. J. Pathol. 2012, 181, 1378–1386. [Google Scholar] [CrossRef] [PubMed]
- Usui, S.; Oveson, B.C.; Lee, S.Y.; Jo, Y.J.; Yoshida, T.; Miki, A.; Miki, K.; Iwase, T.; Lu, L.; Campochiaro, P.A. NADPH oxidase plays a central role in cone cell death in retinitis pigmentosa. J. Neurochem. 2009, 110, 1028–1037. [Google Scholar] [CrossRef] [Green Version]
- Conti, P.; Lauritano, D.; Caraffa, A.; Gallenga, C.E.; Kritas, S.K.; Ronconi, G.; Martinotti, S. Microglia and mast cells generate proinflammatory cytokines in the brain and worsen inflammatory state: Suppressor effect of IL-37. Eur. J. Pharmacol. 2020, 15, 875. [Google Scholar] [CrossRef]
- Tuson, M.; Garanto, A.; Gonzàlez-Duarte, R.; Marfany, G. Overexpression of CERKL, a gene responsible for retinitis pigmentosa in humans, protects cells from apoptosis induced by oxidative stress. Mol. Vis. 2009, 15, 168–180. [Google Scholar]
- Fathinajafabadi, A.; Pérez-Jiménez, E.; Riera, M.; Knecht, E.; Gonzàez-Duarte, R. CERKL, a retinal disease gene, encodes an mRNA-binding protein that localizes in compact and untranslated mRNPs associated with microtubules. PLoS ONE 2014, 9, e87898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Wang, L.; Zhang, J.; Huang, M.; Wong, F.; Liu, X.; Liu, F.; Cui, X.; Yang, G.; Chen, J.; et al. CERKL interacts with mitochondrial TRX2 and protects retinal cells from oxidative stress-induced apoptosis. Biochim. Biophys. Acta Mol. Basis Dis. 2014, 1842, 1121–1129. [Google Scholar] [CrossRef] [Green Version]
- Hannun, Y.A.; Obeid, L.M. The ceramide-centric universe of lipid- mediated cell regulation: Stress encounters of the lipid kind. J. Biol. Chem. 2002, 277, 25847–25850. [Google Scholar] [CrossRef] [Green Version]
- Hannun, Y.A.; Obeid, L.M. Principles of bioactive lipid signalling: Lessons from sphingolipids. Nat. Rev. Mol. Cell Biol. 2008, 9, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Tuson, M.; Marfany, G.; Gonzalez-Duarte, R. Mutation of CERKL, a novel human ceramide kinase gene, causes autosomal recessive retinitis pigmentosa (RP26). Am. J. Hum Genet. 2004, 74, 128–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auslender, N.; Sharon, D.; Abbasi, A.H.; Garzozi, H.J.; Banin, E.; Ben- Yosef, T. A common founder mutation of CERKL underlies autosomal recessive retinal degeneration with early macular involvement among Yemenite Jews. Investig. Ophthalmol. Vis. Sci. 2007, 48, 5431–5438. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.A.; Anwar, S.; Aljarbou, A.N.; Al-Orainy, M.; Aldebasi, Y.H.; Islam, S.; Younus, H. Protective effect of thymoquinone on glucose or methylglyoxal-induced glycation of superoxide dismutase. Int. J. Biol. Macromol. 2014, 65, 16–20. [Google Scholar] [CrossRef]
- Groener, J.B.; Oikonomou, D.; Cheko, R.; Kender, Z.; Zemva, J.; Kihm, L.; Muckenthaler, M.; Peters, V.; Fleming, T.; Kopf, S.; et al. Methylglyoxal and Advanced Glycation End Products in Patients with Diabetes—What We Know so Far and the Missing Links. Exp. Clin. Endocrinol. Diabetes 2019, 127, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Nedic, O.; Rattan, S.I.; Grune, T.; Trougakos, I.P. Molecular effects of advanced glycation end products on cell signalling pathways, ageing and pathophysiology. Free Radic. Res. 2013, 47, 28–38. [Google Scholar] [CrossRef]
- Lee, H.J.; Howell, S.K.; Sanford, R.J.; Beisswenger, P.J. Methylglyoxal can modify GAPDH activity and structure. Ann. N. Y. Acad. Sci. 2005, 1043, 135–145. [Google Scholar] [CrossRef]
- Wautier, M.P.; Guillausseau, P.J.; Wautier, J.L. Activation of the receptor for advanced glycation end products and consequences on health. Diabetes Metab. Syndr. 2017, 11, 305–309. [Google Scholar] [CrossRef]
- Xu, J.; Chen, L.J.; Yu, J.; Wang, H.J.; Zhang, F.; Liu, Q.; Wu, J. Involvement of Advanced Glycation End Products in the Pathogenesis of Diabetic Retinopathy. Cell Physiol. Biochem. 2018, 48, 705–717. [Google Scholar] [CrossRef]
- Grigsby, J.G.; Allen, D.M.; Ferrigno, A.S.; Vellanki, S.; Pouw, C.E.; Hejny, W.A.; Tsin, A.T.C. Autocrine and Paracrine Secretion of Vascular Endothelial Growth Factor in the Pre-Hypoxic Diabetic Retina. Curr. Diabetes Rev. 2017, 13, 161–174. [Google Scholar] [CrossRef]
- Chen, M.; Curtis, T.M.; Stitt, A.W. Advanced glycation end products and diabetic retinopathy. Curr. Med. Chem. 2013, 20, 3234–3240. [Google Scholar] [CrossRef]
- Datta, S.; Cano, M.; Ebrahimi, K.; Wang, L.; Handa, J.T. The impact of oxidative stress and inflammation on RPE degeneration in non-neovascular AMD. Prog. Retin. Eye Res. 2017, 60, 201–218. [Google Scholar] [CrossRef] [PubMed]
- Donato, L.; Scimone, C.; Alibrandi, S.; Rinaldi, C.; Sidoti, A.; D’Angelo, R. Transcriptome Analyses of lncRNAs in A2E-Stressed Retinal Epithelial Cells Unveil Advanced Links between Metabolic Impairments Related to Oxidative Stress and Retinitis Pigmentosa. Antioxidants 2020, 9, 318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaarniranta, K.; Koskela, A.; Felszeghy, S.; Kivinen, N.; Salminen, A.; Kauppinen, A. Fatty acids and oxidized lipoproteins contribute to autophagy and innate immunity responses upon the degeneration of retinal pigment epithelium and development of age-related macular degeneration. Biochimie 2019, 159, 49–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peculis, R.; Konrade, I.; Skapare, E.; Fridmanis, D.; NikitinaZake, L.; Lejnieks, A.; Pirags, V.; Dambrova, M.; Klovins, J. Identification of glyoxalase 1 polymorphisms associatedwith enzyme activity. Gene 2013, 515, 140–143. [Google Scholar] [CrossRef] [PubMed]
- Donato, L.; Scimone, C.; Alibrandi, S.; Nicocia, G.; Rinaldi, C.; Sidoti, A.; D’Angelo, R. Discovery of GLO1 New Related Genes and Pathways by RNA-Seq on A2E-Stressed Retinal Epithelial Cells Could Improve Knowledge on Retinitis Pigmentosa. Antioxidants 2020, 9, 416. [Google Scholar] [CrossRef] [PubMed]
- Hori, K.; Hoshino, M. Neuronal Migration and AUTS2 Syndrome. Brain Sci. 2017, 7, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosa, L.R.O.; Soares, G.M.; Silveira, L.R.; Boschero, A.C.; Barbosa-Sampaio, H.C.L. ARHGAP21 as a master regulator of multiple cellular processes. J. Cell Physiol. 2018, 233, 8477–8481. [Google Scholar] [CrossRef] [PubMed]
- Kuchay, S.; Duan, S.; Schenkein, E.; Peschiaroli, A.; Saraf, A.; Florens, L.; Washburn, M.P.; Pagano, M. FBXL2- and PTPL1-mediated degradation of p110-free p85beta regulatory subunit controls the PI(3)K signalling cascade. Nat. Cell Biol. 2013, 15, 472–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villa, F.; Deak, M.; Bloomberg, G.B.; Alessi, D.R.; van Aalten, D.M. Crystal structure of the PTPL1/FAP-1 human tyrosine phosphatase mutated in colorectal cancer: Evidence for a second phosphotyrosine substrate recognition pocket. J. Biol. Chem. 2005, 280, 8180–8187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, H.; Xu, N.; Doycheva, D.M.; Malaguit, J.; Tang, J.; Zhang, J.H. Recombinant Slit2 attenuates neuronal apoptosis via the Robo1-srGAP1 pathway in a rat model of neonatal HIE. Neuropharmacology 2019, 158, 107727. [Google Scholar] [CrossRef]
- Kage, F.; Steffen, A.; Ellinger, A.; Ranftler, C.; Gehre, C.; Brakebusch, C.; Pavelka, M.; Stradal, T.; Rottner, K. FMNL2 and -3 regulate Golgi architecture and anterograde transport downstream of Cdc42. Sci. Rep. 2017, 7, 9791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, X.; Kiru, S.; Gomez, G.A.; Yap, A.S. Regulated recruitment of SRGAP1 modulates RhoA signaling for contractility during epithelial junction maturation. Cytoskeleton 2018, 75, 61–69. [Google Scholar] [CrossRef]
- Buschman, M.D.; Field, S.J. MYO18A: An unusual myosin. Adv. Biol. Regul. 2018, 67, 84–92. [Google Scholar] [CrossRef]
- Van Gils, M.; Nollet, L.; Verly, E.; Deianova, N.; Vanakker, O.M. Cellular signaling in pseudoxanthoma elasticum: An update. Cell Signal. 2019, 55, 119–129. [Google Scholar] [CrossRef]
- Khanobdee, K.; Kolberg, J.B.; Dunlevy, J.R. Nuclear and plasma membrane localization of SH3BP4 in retinal pigment epithelial cells. Mol. Vis. 2004, 10, 933–942. [Google Scholar]
- Majumdar, A.; Ramagiri, S.; Rikhy, R. Drosophila homologue of Eps15 is essential for synaptic vesicle recycling. Exp. Cell Res. 2006, 312, 2288–2298. [Google Scholar] [CrossRef]
- Gan, X.; Wang, C.; Patel, M.; Kreutz, B.; Zhou, M.; Kozasa, T.; Wu, D. Different Raf protein kinases mediate different signaling pathways to stimulate E3 ligase RFFL gene expression in cell migration regulation. J. Biol. Chem. 2013, 288, 33978–33984. [Google Scholar] [CrossRef] [Green Version]
- Sakai, R.; Fukuda, R.; Unida, S.; Aki, M.; Ono, Y.; Endo, A.; Kusumi, S.; Koga, D.; Fukushima, T.; Komada, M.; et al. The integral function of the endocytic recycling compartment is regulated by RFFL-mediated ubiquitylation of Rab11 effectors. J. Cell Sci. 2019, 132. [Google Scholar] [CrossRef] [Green Version]
- Kajiho, H.; Yamamoto, Y.; Sakisaka, T. CAND1 regulates lunapark for the proper tubular network of the endoplasmic reticulum. Sci. Rep. 2019, 9, 13152. [Google Scholar] [CrossRef] [Green Version]
- Uebi, T.; Itoh, Y.; Hatano, O.; Kumagai, A.; Sanosaka, M.; Sasaki, T.; Sasagawa, S.; Doi, J.; Tatsumi, K.; Mitamura, K.; et al. Involvement of SIK3 in glucose and lipid homeostasis in mice. PLoS ONE 2012, 7, e37803. [Google Scholar] [CrossRef]
- Chong, C.M.; Zheng, W. Artemisinin protects human retinal pigment epithelial cells from hydrogen peroxide-induced oxidative damage through activation of ERK/CREB signaling. Redox Biol. 2016, 9, 50–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kriete, A.; Bosl, W.J.; Booker, G. Rule-based cell systems model of aging using feedback loop motifs mediated by stress responses. PLoS Comput. Biol. 2010, 6, e1000820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golomb, L.; Bublik, D.R.; Wilder, S.; Nevo, R.; Kiss, V.; Grabusic, K.; Volarevic, S.; Oren, M. Importin 7 and exportin 1 link c-Myc and p53 to regulation of ribosomal biogenesis. Mol. Cell. 2012, 45, 222–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavdar Koc, E.; Burkhart, W.; Blackburn, K.; Moseley, A.; Spremulli, L.L. The small subunit of the mammalian mitochondrial ribosome. Identification of the full complement of ribosomal proteins present. J. Biol. Chem. 2001, 276, 19363–19374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Zhuang, Q.; Hu, G.; Geng, S. MORC4 is a novel breast cancer oncogene regulated by miR-193b-3p. J. Cell Biochem. 2019, 120, 4634–4643. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zong, W.; Li, T.; Wang, Y.; Xu, X.; Zhou, Z.W.; Wang, Z.Q. The E3 ubiquitin ligase APC/C(C)(dh1) degrades MCPH1 after MCPH1-betaTrCP2-Cdc25A-mediated mitotic entry to ensure neurogenesis. EMBO J. 2017, 36, 3666–3681. [Google Scholar] [CrossRef] [PubMed]
- Clark, B.S.; Stein-O’Brien, G.L.; Shiau, F.; Cannon, G.H.; Davis-Marcisak, E.; Sherman, T.; Santiago, C.P.; Hoang, T.V.; Rajaii, F.; James-Esposito, R.E.; et al. Single-Cell RNA-Seq Analysis of Retinal Development Identifies NFI Factors as Regulating Mitotic Exit and Late-Born Cell Specification. Neuron 2019, 102, 1111–1126. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.; Park, J.; Kim, Y.K. Crosstalk between translation and the aggresome-autophagy pathway. Autophagy 2018, 14, 1079–1081. [Google Scholar] [CrossRef]
- Kawaguchi, K.; Okamoto, T.; Morita, M.; Imanaka, T. Translocation of the ABC transporter ABCD4 from the endoplasmic reticulum to lysosomes requires the escort protein LMBD1. Sci. Rep. 2016, 6, 30183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinoshita, Y.; Nogami, K.; Jomura, R.; Akanuma, S.I.; Abe, H.; Inouye, M.; Kubo, Y.; Hosoya, K.I. Investigation of Receptor-Mediated Cyanocobalamin (Vitamin B12) Transport across the Inner Blood-Retinal Barrier Using Fluorescence-Labeled Cyanocobalamin. Mol. Pharm. 2018, 15, 3583–3594. [Google Scholar] [CrossRef]
- He, L.; Hannon, G.J. MicroRNAs: Small RNAs with a big role in gene regulation. Nat. Rev. Genet. 2004, 5, 522–531. [Google Scholar] [CrossRef]
- Zamore, P.D.; Haley, B. Ribo-gnome: The big world of small RNAs. Science 2005, 309, 1519–1524. [Google Scholar] [CrossRef] [Green Version]
- Ohana, R.; Weiman-Kelman, B.; Raviv, S.; Tamm, E.R.; Pasmanik-Chor, M.; Rinon, A.; Netanely, D.; Shamir, R.; Solomon, A.S.; Ashery-Padan, R. MicroRNAs are essential for differentiation of the retinal pigmented epithelium and maturation of adjacent photoreceptors. Development 2015, 142, 2487–2498. [Google Scholar]
- Romano, G.L.; Platania, C.B.M.; Drago, F.; Salomone, S.; Ragusa, M.; Barbagallo, C.; Di Pietro, C.; Purrello, M.; Reibaldi, M.; Avitabile, T.; et al. Retinal and circulating miRNAs in age-related macular degeneration: An in vivo animal and human study. Front. Pharmacol. 2017, 8, 168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, B.; Maidana, D.E.; Dib, B.; Miller, J.B.; Bouzika, P.; Miller, J.W.; Vavvas, D.G.; Lin, H. MiR-17-3p Exacerbates Oxidative Damage in Human Retinal Pigment Epithelial Cells. PLoS ONE 2016, 11, e0160887. [Google Scholar] [CrossRef] [Green Version]
- Kruk, J.; Kubasik-Kladna, K.; Aboul-Enein, H.Y. The role oxidative stress in the pathogenesis of eye diseases: Current status and a dual role of physical activity. Mini Rev. Med. Chem. 2015, 16, 241–257. [Google Scholar] [CrossRef] [PubMed]
- Mao, K.; Shu, W.; Qiu, Q.; Gu, Q.; Wu, X. Salvianolic acid A protects retinal pigment epithelium from OX-LDL-induced inflammation in an age-related macular degeneration model. Discov. Med. 2017, 23, 129–147. [Google Scholar] [PubMed]
- Kim, J.H.; Lee, S.J.; Kim, K.W.; Yu, Y.S.; Kim, J.H. Oxidized low density lipoprotein-induced senescence of retinal pigment epithelial cells is followed by outer blood-retinal barrier dysfunction. Int. J. Biochem. Cell Biol. 2012, 44, 808–814. [Google Scholar] [CrossRef] [PubMed]
- Hoppe, G.; Marmorstein, A.D.; Pennock, E.A.; Hoff, H.F. Oxidized low density lipoprotein-induced inhibition of processing of photoreceptor outer segments by RPE. Investig. Ophthalmol. Vis. Sci. 2001, 42, 2714–2720. [Google Scholar]
- Yu, A.L.; Lorenz, R.L.; Haritoglou, C.; Kampik, A.; Welge-Lussen, U. Biological effects of native and oxidized low-density lipoproteins in cultured human retinal pigment epithelial cells. Exp. Eye Res. 2009, 88, 495–503. [Google Scholar] [CrossRef] [PubMed]
- Saneipour, M.; Ghatreh-Samani, K.; Heydarian, E.; Farrokhi, E.; Abdian, N. Adiponectin inhibits oxidized low density lipoprotein-induced increase in matrix metalloproteinase 9 expression in vascular smooth muscle cells. ARYA Atheroscler. 2015, 11, 191–195. [Google Scholar]
- Yating, Q.; Yuan, Y.; Wei, Z.; Qing, G.; Xingwei, W.; Qiu, Q.; Lili, Y. Oxidized LDL induces apoptosis of human retinal pigment epithelium through activation of ERK-Bax/Bcl-2 signaling pathways. Curr. Eye Res. 2015, 40, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.A.; Kanuga, N.; Romero, I.A.; Greenwood, J.; Luthert, P.J.; Cheetham, M.E. Oxidative stress affects the junctional integrity of retinal pigment epithelial cells. Investig. Ophthalmol. Vis. Sci. 2004, 45, 675–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopp, F.; Mendell, J.T. Functional Classification and Experimental Dissection of Long Noncoding RNAs. Cell 2018, 172, 393–407. [Google Scholar] [CrossRef] [Green Version]
- Kondo, Y.; Shinjo, K.; Katsushima, K. Long non-coding RNAs as an epigenetic regulator in human cancers. Cancer Sci. 2017, 108, 1927–1933. [Google Scholar] [CrossRef] [Green Version]
- Wawrzyniak, O.; Zarebska, Z.; Rolle, K.; Gotz-Wieckowska, A. Circular and long non-coding RNAs and their role in ophthalmologic diseases. Acta Biochim. Pol. 2018, 65, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Donato, L.; Scimone, C.; Rinaldi, C.; D’Angelo, R.; Sidoti, A. Non-coding RNAome of RPE cells under oxidative stress suggests unknown regulative aspects of Retinitis pigmentosa etiopathogenesis. Sci. Rep. 2018, 8, 16638. [Google Scholar] [CrossRef]
- Manelyte, L.; Strohner, R.; Gross, T.; Langst, G. Chromatin targeting signals, nucleosome positioning mechanism and non-coding RNA-mediated regulation of the chromatin remodeling complex NoRC. PLoS Genet. 2014, 10, e1004157. [Google Scholar] [CrossRef] [Green Version]
- Thapar, R. Regulation of DNA Double-Strand Break Repair by Non-Coding RNAs. Molecules 2018, 23, 2789. [Google Scholar] [CrossRef] [Green Version]
- Han, X.; Yang, Y.; Sun, Y.; Qin, L.; Yang, Y. LncRNA TUG1 affects cell viability by regulating glycolysis in osteosarcoma cells. Gene 2018, 674, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Wang, M.; Yang, H.; Mao, L.; He, Q.; Jin, H.; Ye, Z.M.; Luo, X.Y.; Xia, Y.P.; Hu, B. LncRNA TUG1 sponges microRNA-9 to promote neurons apoptosis by up-regulated Bcl2l11 under ischemia. Biochem. Biophys. Res. Commun. 2017, 485, 167–173. [Google Scholar] [CrossRef]
- Li, Y.; Xu, F.; Xiao, H.; Han, F. Long noncoding RNA BDNF-AS inversely regulated BDNF and modulated high-glucose induced apoptosis in human retinal pigment epithelial cells. J. Cell Biochem. 2018, 119, 817–823. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.K.; Lee, E.J.; Kim, Y.H.; Kim, D.Y.; Oh, H.; Kim, S.I.; Kang, Y.H. Chrysin Ameliorates Malfunction of Retinoid Visual Cycle through Blocking Activation of AGE-RAGE-ER Stress in Glucose-Stimulated Retinal Pigment Epithelial Cells and Diabetic Eyes. Nutrients 2018, 10, 1046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Millar, C.A.; Powell, K.A.; Hickson, G.R.; Bader, M.F.; Gould, G.W. Evidence for a role for ADP-ribosylation factor 6 in insulin-stimulated glucose transporter-4 (GLUT4) trafficking in 3T3-L1 adipocytes. J. Biol. Chem. 1999, 274, 17619–17625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellis, B.C.; Graham, L.D.; Molloy, P.L. CRNDE, a long non-coding RNA responsive to insulin/IGF signaling, regulates genes involved in central metabolism. Biochim. Biophys. Acta 2014, 1843, 372–386. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Xi, X.; Mei, Y.; Zhao, X.; Zhou, L.; Ma, M.; Liu, S.; Zha, X.; Yang, Y. High-glucose induces retinal pigment epithelium mitochondrial pathways of apoptosis and inhibits mitophagy by regulating ROS/PINK1/Parkin signal pathway. Biomed. Pharmacother. 2019, 111, 1315–1325. [Google Scholar] [CrossRef] [PubMed]
- Yanagi, Y. Role of Peoxisome Proliferator Activator Receptor gamma on Blood Retinal Barrier Breakdown. PPAR Res. 2008, 2008, doi. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, J.H.; Lavail, M.M. Misfolded proteins and retinal dystrophies. Adv. Exp. Med. Biol. 2010, 664, 115–121. [Google Scholar] [PubMed] [Green Version]
- Lundkvist, A.; Reichenbach, A.; Betsholtz, C.; Carmeliet, P.; Wolburg, H.; Pekny, M. Under stress, the absence of intermediate filaments from Muller cells in the retina has structural and functional consequences. J. Cell Sci. 2004, 117, 3481–3488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossignol, R.; Ranchon-Cole, I.; Paris, A.; Herzine, A.; Perche, A.; Laurenceau, D.; Bertrand, P.; Cercy, C.; Pichon, J.; Mortaud, S.; et al. Visual sensorial impairments in neurodevelopmental disorders: Evidence for a retinal phenotype in Fragile X Syndrome. PLoS ONE 2014, 9, e105996. [Google Scholar] [CrossRef] [Green Version]
- Adinolfi, E.; Giuliani, A.L.; De Marchi, E.; Pegoraro, A.; Orioli, E.; Di Virgilio, F. The P2X7 receptor: A main player in inflammation. Biochem. Pharmacol. 2018, 151, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Di Virgilio, F.; Tang, Y.; Sarti, A.C.; Rossato, M. A rationale for targeting the P2X7 receptor in Coronavirus disease 19. Br. J. Pharmacol. 2020, 177, 4990–4994. [Google Scholar] [CrossRef] [PubMed]
- Di Virgilio, F.; Dal Ben, D.; Sarti, A.C.; Giuliani, A.L.; Falzoni, S. The P2X7 Receptor in Infection and Inflammation. Immunity 2017, 47, 15–31. [Google Scholar] [CrossRef] [Green Version]
- Savio, L.E.B.; De Andrade Mello, P.; Da Silva, C.G.; Coutinho-Silva, R. The P2X7 Receptor in Inflammatory Diseases: Angel or Demon? Front. Pharmacol. 2018, 9, 52. [Google Scholar] [PubMed] [Green Version]
- Reichenbach, A.; Bringmann, A. Purinergic signaling in retinal degeneration and regeneration. Neuropharmacology 2016, 104, 194–211. [Google Scholar] [CrossRef] [PubMed]
- Calzaferri, F.; Ruiz-Ruiz, C.; Diego, A.M.G.; Pascual, R.; Méndez-López, I.; Cano-Abad, M.F.; García, A.G. The purinergic P2X7 receptor as a potential drug target to combat neuroinflammation in neurodegenerative diseases. Med. Res. Rev. 2020, 40, 2427–2465. [Google Scholar] [CrossRef] [PubMed]
- Appelbaum, T.; Santana, E.; Aguirre, G.D. Strong Upregulation of Inflammatory Genes Accompanies Photoreceptor demise in Canine Models of Retinal Degeneration. PLoS ONE 2017, 12, e0177224. [Google Scholar]
- Block, M.L.; Zecca, L.; Hong, J.S. Microglia-mediated neurotoxicity: Uncovering the molecular mechanism. Nat. Rev. Neurosci. 2007, 8, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Subhramanyam, C.S.; Wang, C.; Hu, Q.; Dheen, S.T. Microglia-mediated neuroinflammation in neurodegenerative diseases. Semin. Cell Dev. Biol. 2019, 94, 112–120. [Google Scholar] [CrossRef]
- Peng, B.; Xiao, J.; Wang, K.; So, K.F.; Tipoe, G.L.; Lin, B. Suppression of microglial activation is neuroprotective in a mouse model of human retinitis pigmentosa. J. Neurosci. 2014, 34, 8139–8150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.K.; Xue, Y.; Cepko, C.L. Microglia modulation by TGF-Beta1 protects cones in mouse models of retinal degeneration. J. Clin. Investig. 2020, 130, 4360–4369. [Google Scholar] [PubMed]
- Smith, J.A.; Das, A.; Ray, S.K.; Banik, N.L. Role of pro-inflammatory cytokines released from microglia in neurodegenerative diseases. Brain Res. Bull. 2012, 87, 10–20. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Chitnis, T.; Weiner, H.L. CNS inflammation and neurodegeneration. J. Clin. Investig. 2017, 127, 3577–3587. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Ma, W.; Zhao, L.; Fariss, R.N.; Wong, W.T. Adaptive Muller Cell Responses to Microglial Activation Mediate Neuroprotection and Coordinates Inflammation in The Retina. J. Neuroinflamm. 2011, 8, 173. [Google Scholar] [CrossRef] [Green Version]
- Taylor, R.A.; Chang, C.F.; Goods, B.A.; Hammond, M.D.; Mac Grory, B.; Ai, Y.; Steinschneider, A.F.; Renfroe, S.C.; Askenase, M.H.; McCullough, L.D.; et al. TGF-beta1 modulates microglial phenotype and promotes recovery after intracerebral hemorrhage. J. Clin. Investig. 2017, 127, 280–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Koren, E.G.; Yu, C.; Klingeborn, M.; Wong, A.Y.; Prigge, C.L.; Mathew, R.; Kalnitsky, J.; Msallam, R.A.; Silvin, A.; Kay, J.N.; et al. Microglial function is distinct in different anatomical locations during retinal homeostasis and degenration. Immunity 2019, 50, 723–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rutar, M.; Valter, K.; Natoli, R.; Provis, J.M. Synthesis and propagation of complement C3by microglia/monocytes in the aging retina. PLoS ONE. 2014, 9, e93343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silverman, S.M.; Ma, W.; Wang, X.; Zhao, L.; Wong, W.T. C3- and CR3- dependent microglial clearence protects photoreceptors in retinitis pigmentosa. J. Exp. Med. 2019, 216, 1925–1943. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Pathway Involved | Effects | Association with RP | References | |
|---|---|---|---|---|
| CHRONIC ACTIVATION OF PERK AND IRE1 | Misfolded proteins: UPR activation | Activation of pro-apoptotic programs Pro-inflammatory signalling Dysfunctional autophagy, free cytosolic Ca2+ | Altered protein synthesis rate in the retina and retinal degeneration | [4,5] |
| MUTYH MUTATION | DNA repair | Formation of single-strand breaks (SSBs) of DNA Disturbed homeostasis and cell death Oxidative microglial activation | Retinal degeneration and neuroinflammation in RP | [16,18,20] |
| CERKL MUTATION | Oxidative stress protection | Activation of pro-apoptotic programs | Accelerated progression of retinal neurodegeneration. | [21,26,27] |
| GLO1 MUTATION | Detoxification of cytotoxic products of glycolysis | Inactivation of antioxidant enzymes (glutathione peroxidase and SOD enzymes) | Hyperinflammation and permanent tissue damage Vascular dysfunction Altered transduction pathways and genetic expression in EPR | [28,30,33,38] |
| Gene | Effects of Mutation | Consequences | References |
|---|---|---|---|
| AUTS2, ANKH | Alteration of actin filament structure and activity | RPE apoptosis | [41] |
| SIK3, IPO3, MRPS33 | Dysregulation of energy metabolism and translation machinery | Cell death | [56,58,59] |
| ARHGAP2, PTPN13 | Alteration of cell migration and proliferation Modification of cell polarity, cell adhesion, Golgi regulation Impairment of intracellular trafficking and glucose homeostasis | RPE apoptosis | [42,43,44,45] |
| FMNL2 | Alteration of actin polymerization and organization of the cytoskeleton | Alteration of vesicular trafficking of RPE cells | [46,47] |
| UBC, MYO18A, EPS15, ANKH | Impairment of intracellular transport processes | Influence of vesicular trafficking of RPE cells (essential for POS renewal and visual cycle intermediate regeneration) AGE accumulation | [48,49,50,51] |
| RFFL, FBXW2, CAND1 | ER stress Accumulation of misfolded proteins AGEs and ROS production | Cell death Alteration of cellular respiration process Impairment of glycolytic metabolism in RPE cells. | [52,53,54] |
| SIK3 | Defect in energy metabolism Decrease of mitochondrial respiration Up-regulation of autophagy | Cellular antioxidant mechanisms impairment Alteration of cellular respiration process | [56,57] |
| IPO7 | Ribosomal biogenesis stress Nucleolar morphology changes | p53-dependent growth arrest | [58] |
| MRPS33, MORC4, MCPH1, NFIA, CTIF, LMBRD1 | Damage of mitochondrial protein synthesis Arrests in DNA damage repair Impairment of mitotic exit and cell differentiation Impairment of mRNA and protein quality control Alteration of transport and metabolism of cobalamin | RPE apoptosis and retinal degeneration Decreased distribution of cyanocobalamin (vitamin B12) from blood to retina | [59,62,63,64] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gallenga, C.E.; Lonardi, M.; Pacetti, S.; Violanti, S.S.; Tassinari, P.; Di Virgilio, F.; Tognon, M.; Perri, P. Molecular Mechanisms Related to Oxidative Stress in Retinitis Pigmentosa. Antioxidants 2021, 10, 848. https://doi.org/10.3390/antiox10060848
Gallenga CE, Lonardi M, Pacetti S, Violanti SS, Tassinari P, Di Virgilio F, Tognon M, Perri P. Molecular Mechanisms Related to Oxidative Stress in Retinitis Pigmentosa. Antioxidants. 2021; 10(6):848. https://doi.org/10.3390/antiox10060848
Chicago/Turabian StyleGallenga, Carla Enrica, Maria Lonardi, Sofia Pacetti, Sara Silvia Violanti, Paolo Tassinari, Francesco Di Virgilio, Mauro Tognon, and Paolo Perri. 2021. "Molecular Mechanisms Related to Oxidative Stress in Retinitis Pigmentosa" Antioxidants 10, no. 6: 848. https://doi.org/10.3390/antiox10060848
APA StyleGallenga, C. E., Lonardi, M., Pacetti, S., Violanti, S. S., Tassinari, P., Di Virgilio, F., Tognon, M., & Perri, P. (2021). Molecular Mechanisms Related to Oxidative Stress in Retinitis Pigmentosa. Antioxidants, 10(6), 848. https://doi.org/10.3390/antiox10060848