
Characterization of the Inducible and Slow-Releasing Hydrogen Sulfide and Persulfide Donor P*: Insights into Hydrogen Sulfide Signaling

 , ,
, , 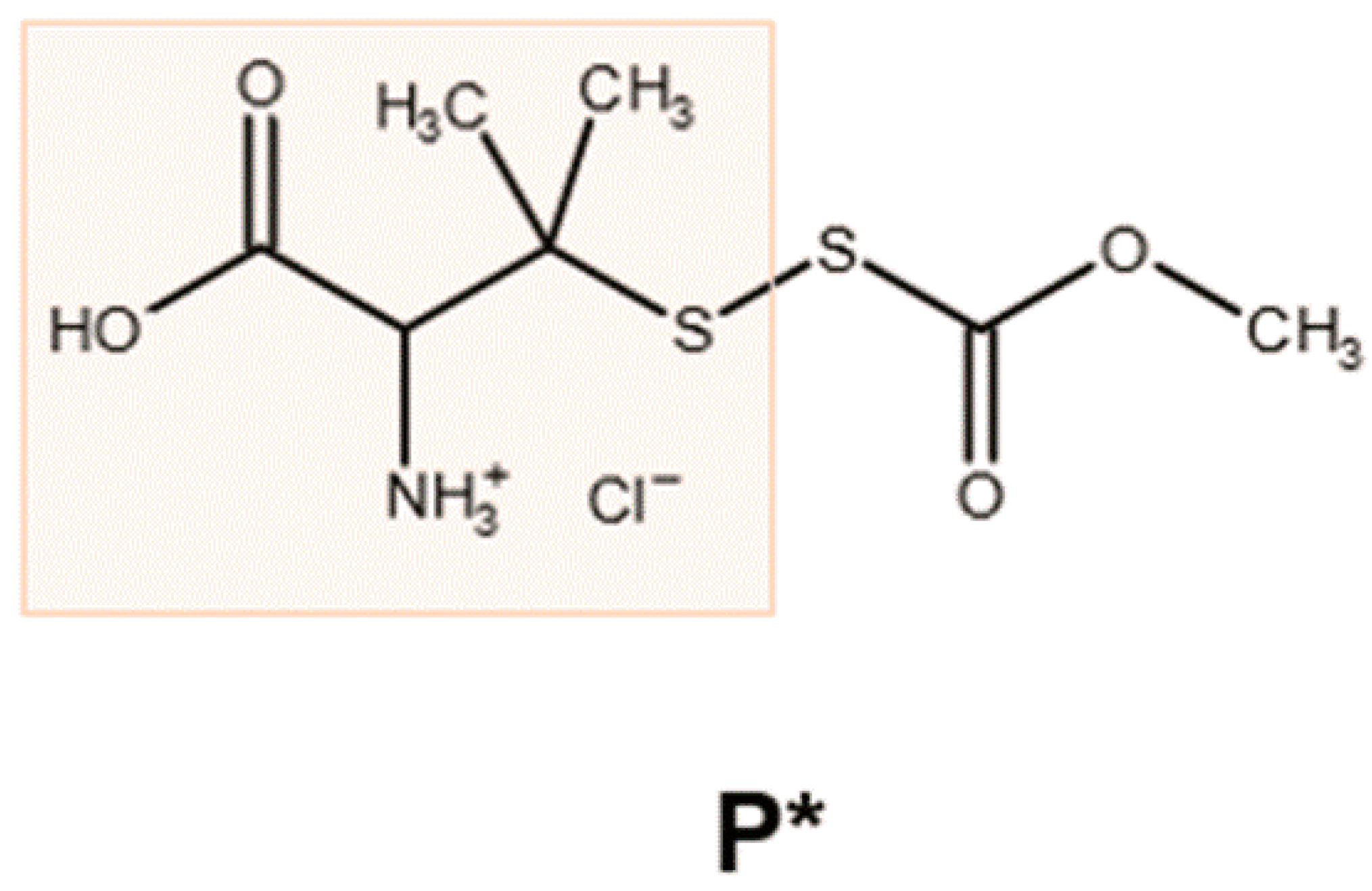{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture
2.3. H2S Fluorescence Measurement
2.4. Trypan Blue Staining
2.5. MTT Measurement
2.6. Western Blot Analysis
2.7. Preparation of Nuclear-Enriched Fractions
2.8. Transfection
2.9. Superoxide Measurement
2.10. Quantification of IL-6
2.11. Quantitative Polymerase Chain Reaction (qPCR)
2.12. Statistical Analysis
3. Results
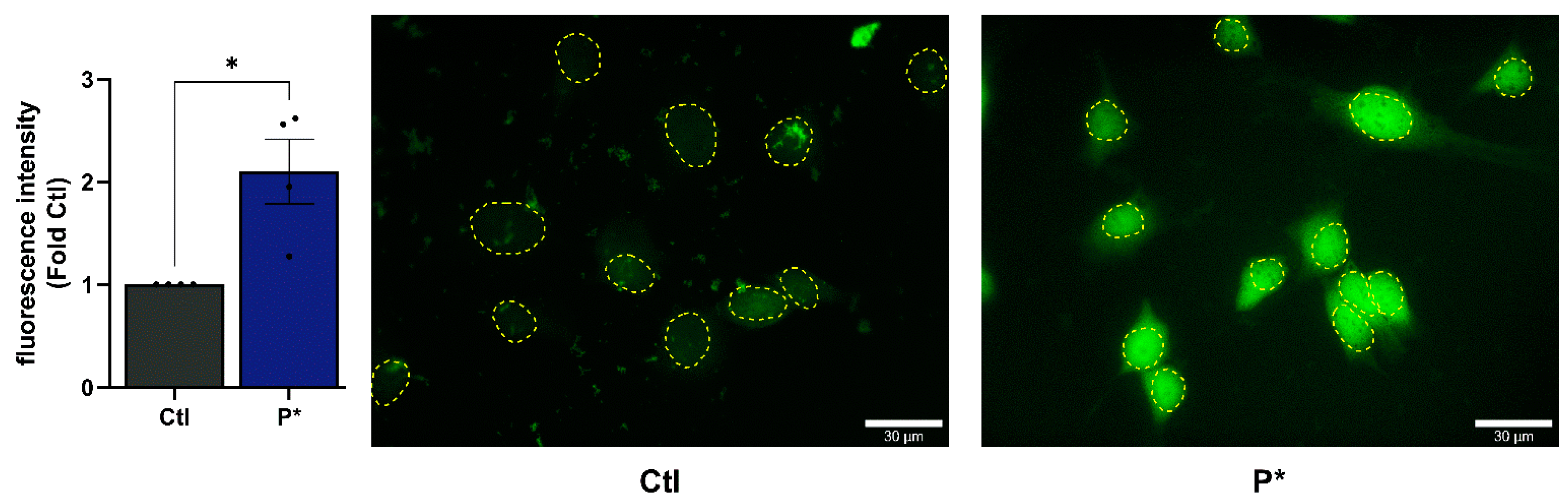
3.1. H2S Release and Cellular Uptake of P*
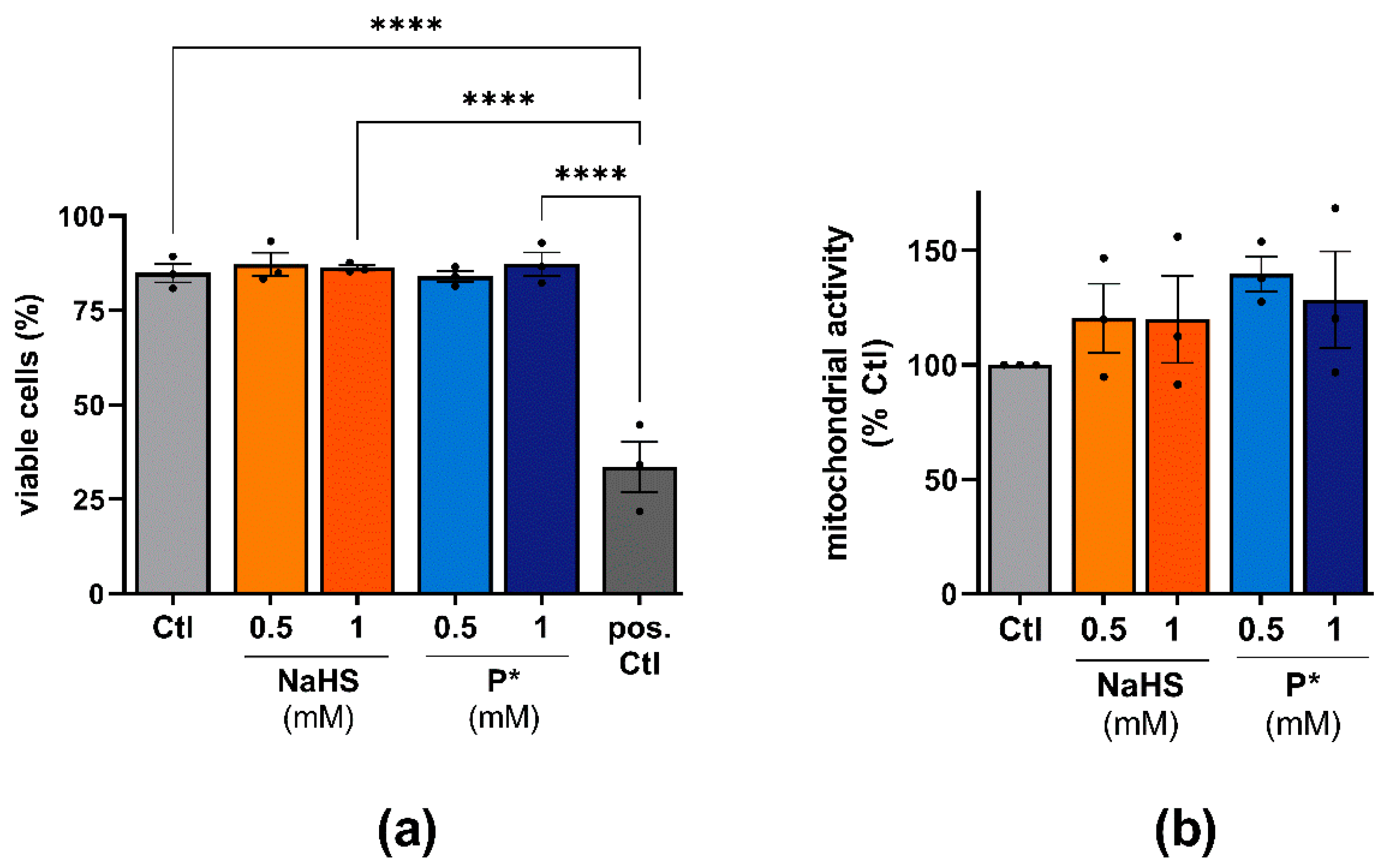
3.2. Cell Viability in Response to P* Administration
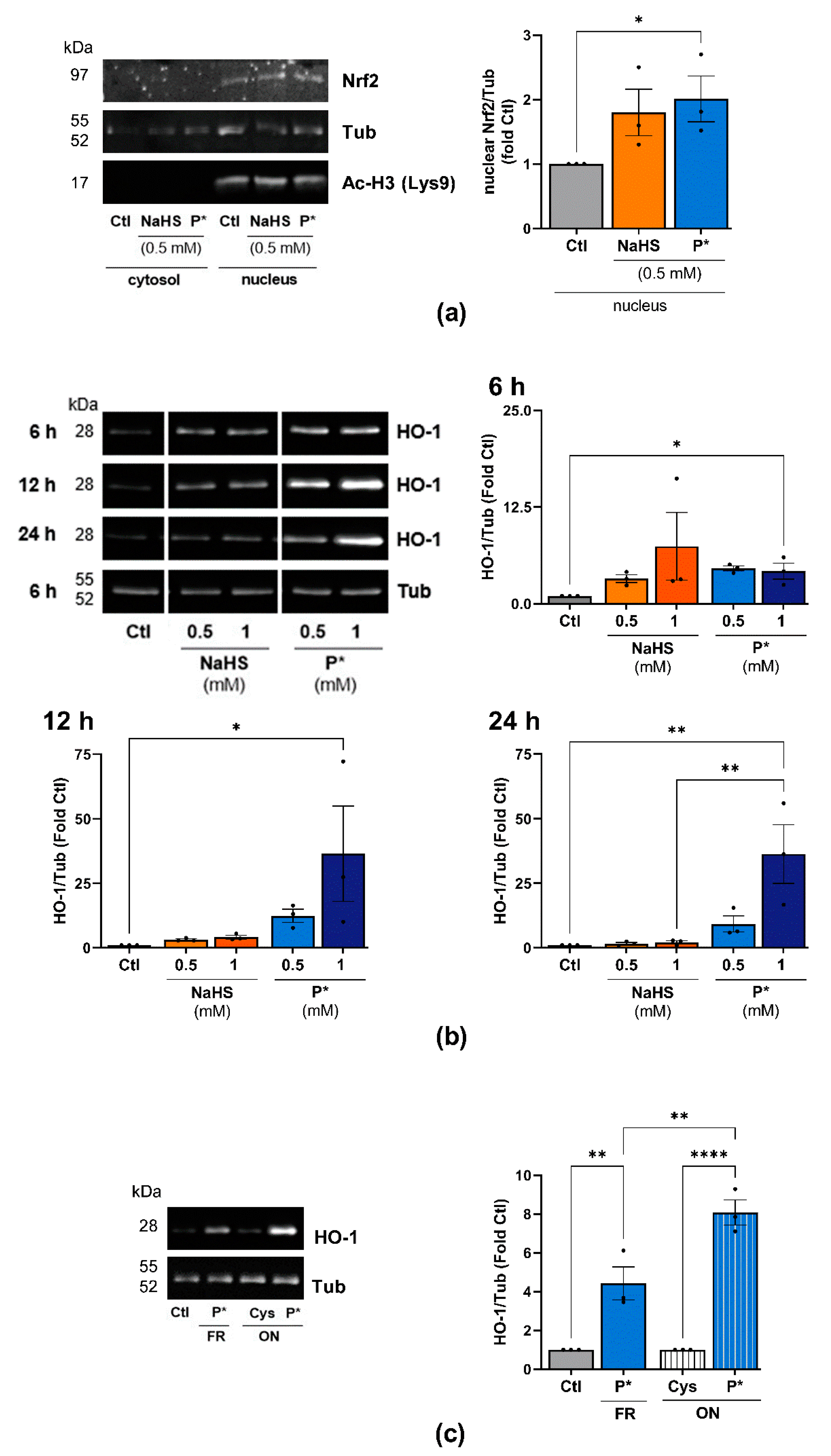
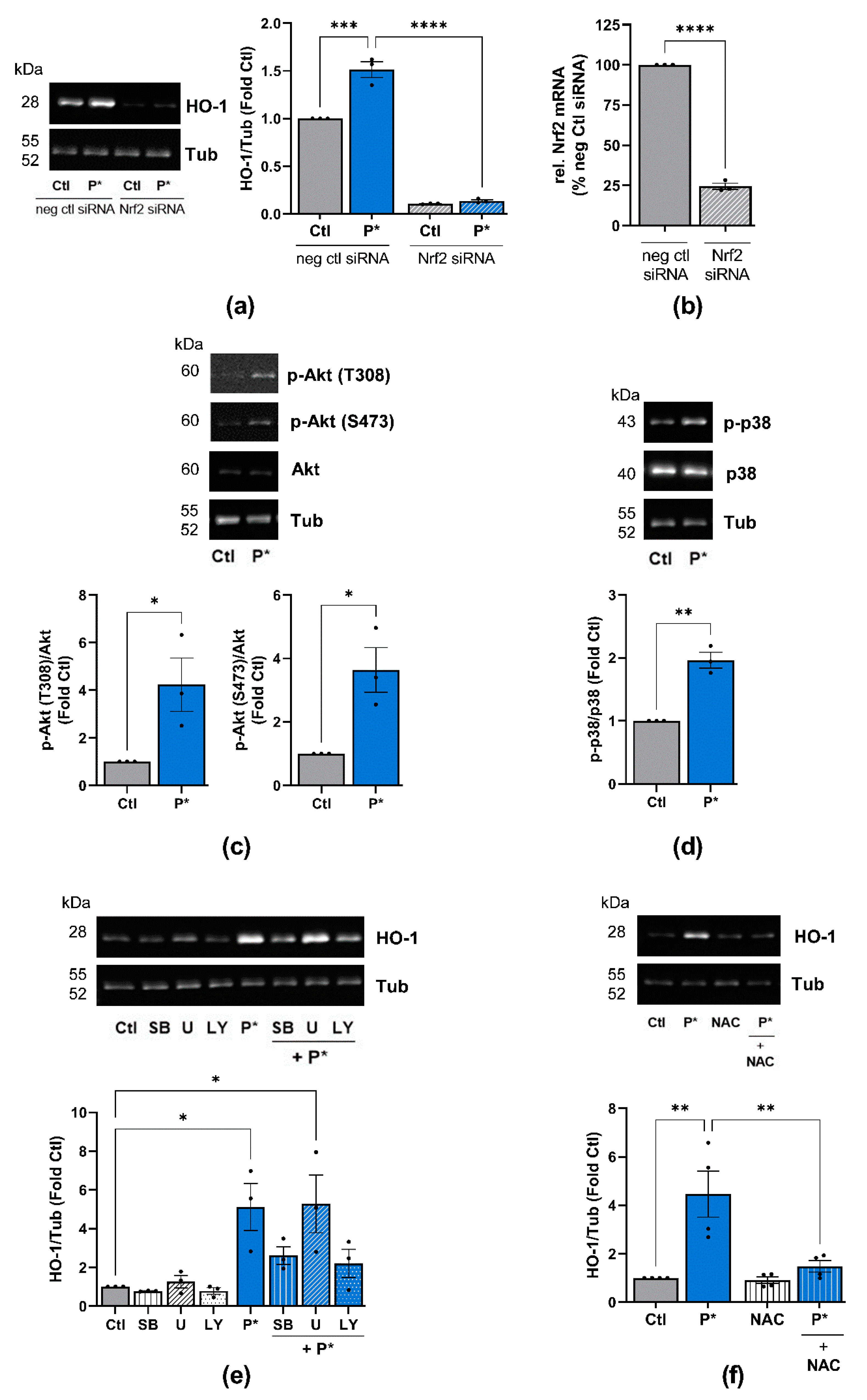
3.3. P* Activates the Nrf2 Pathway
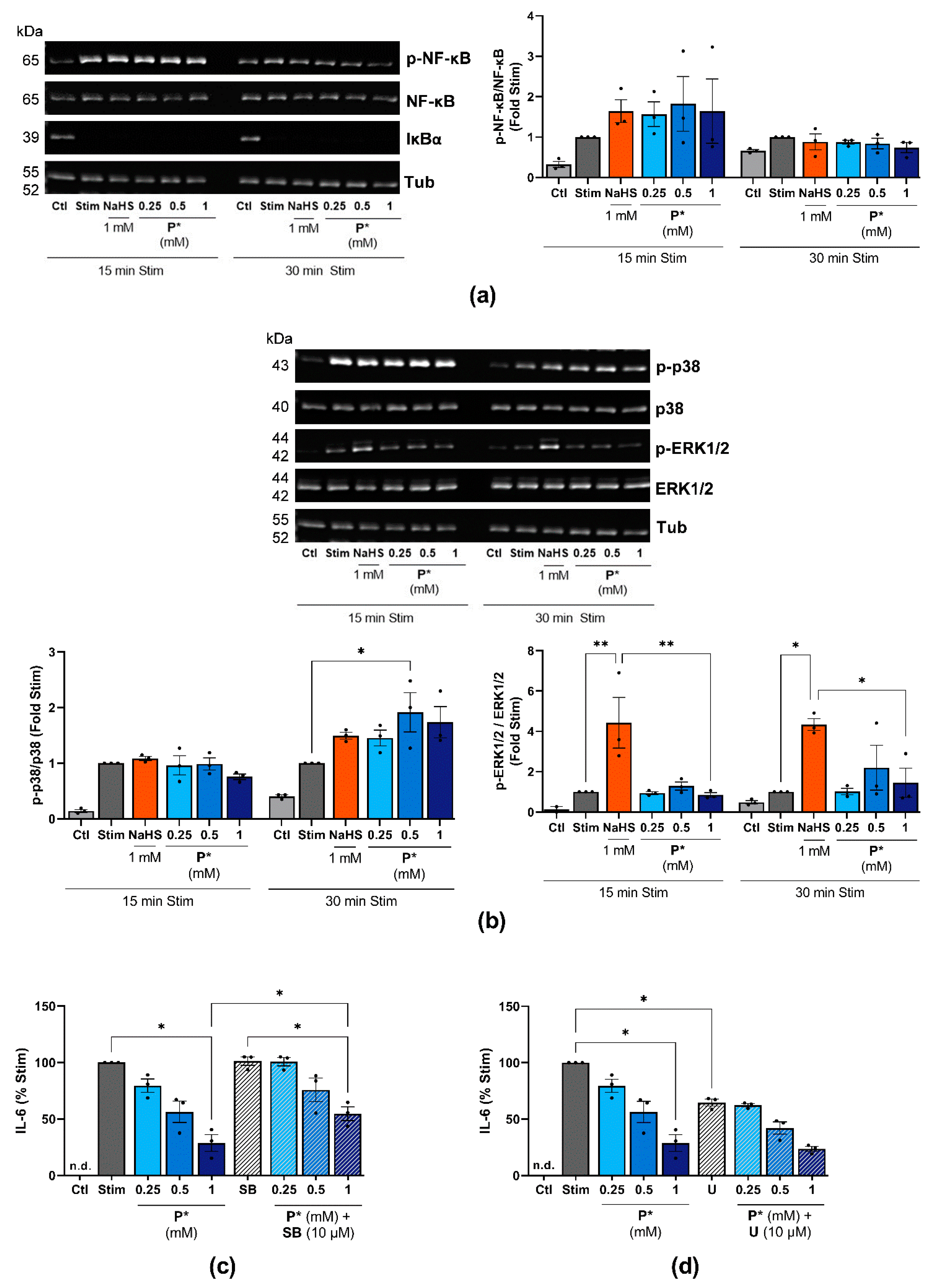
3.4. P* Induces HO-1 via Activation of Nrf2 as Well as PI3K/Akt and p38 MAPK Pathways
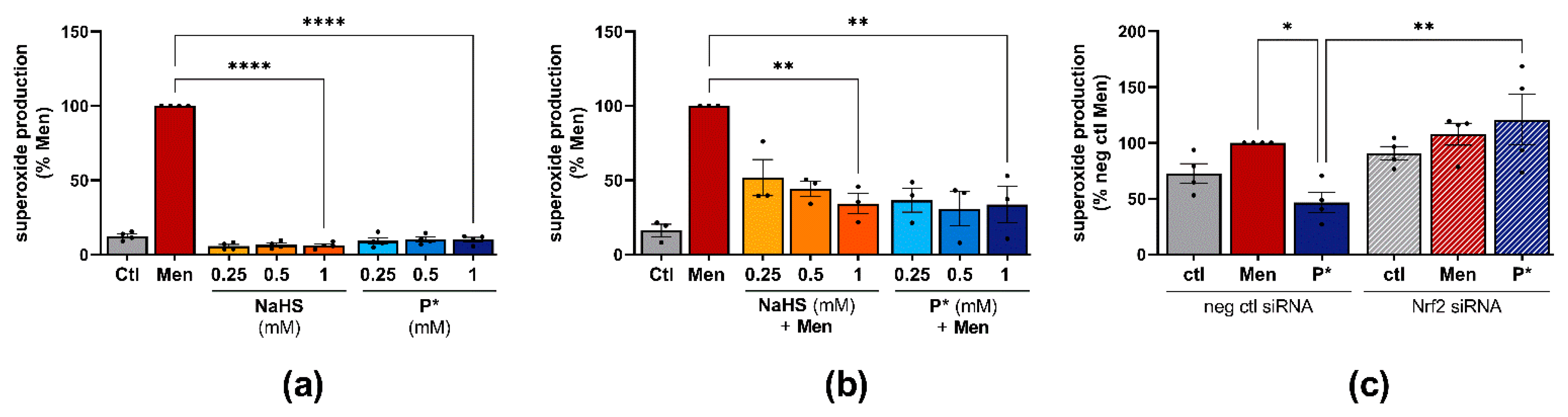
3.5. P* Exerts Antioxidative Effects
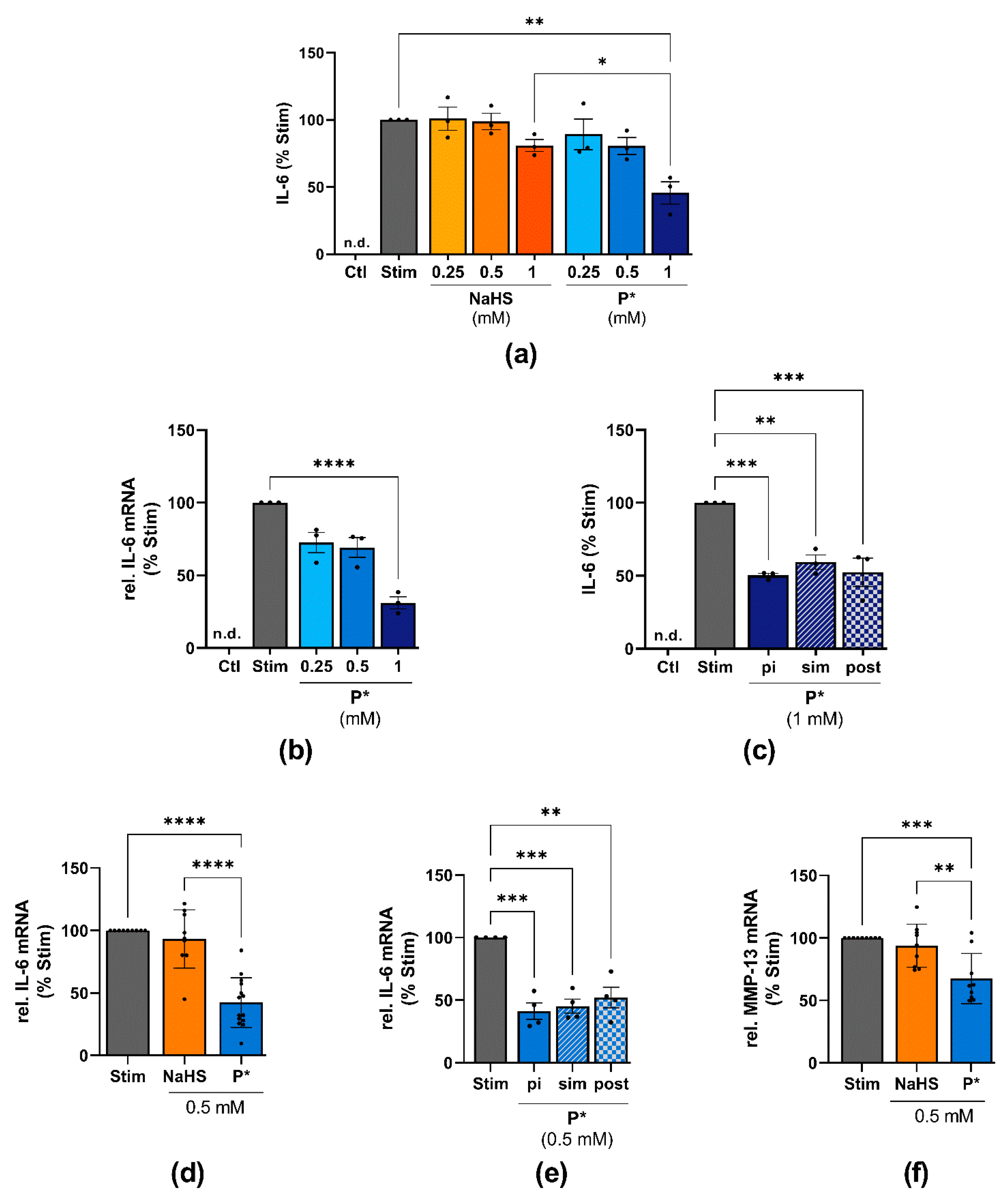
3.6. Effects of P* on IL-6 Expression in ATDC5 Cells and Primary Human Chondrocytes
3.7. P*-Mediated HO-1 Induction and IL-6 Decline
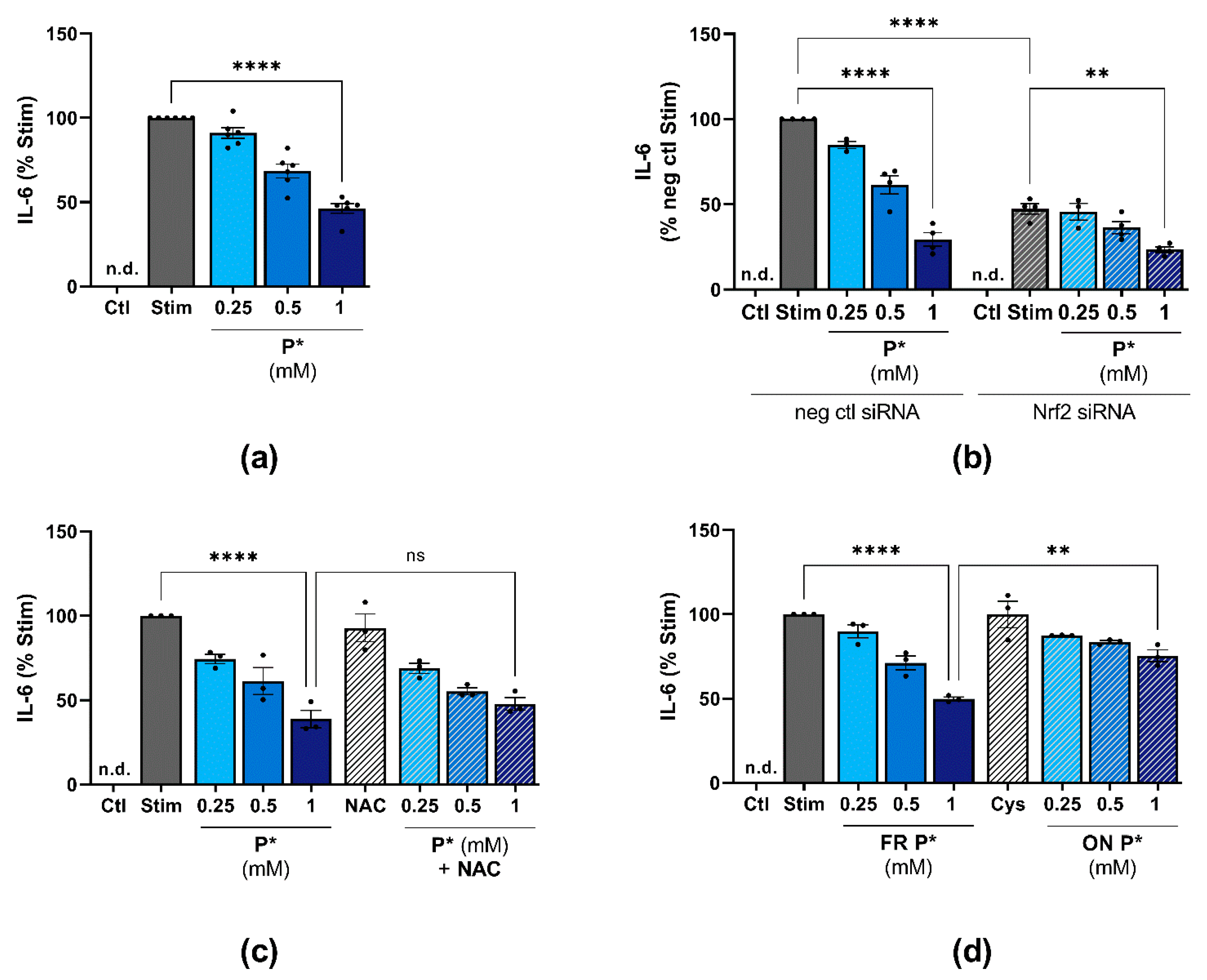
3.8. P* Decreases IL-6 Levels Dependent on H2S Release in ATDC5 Cells
3.9. P* Decreases IL-6 Levels Dependent on p38 MAPK Activation
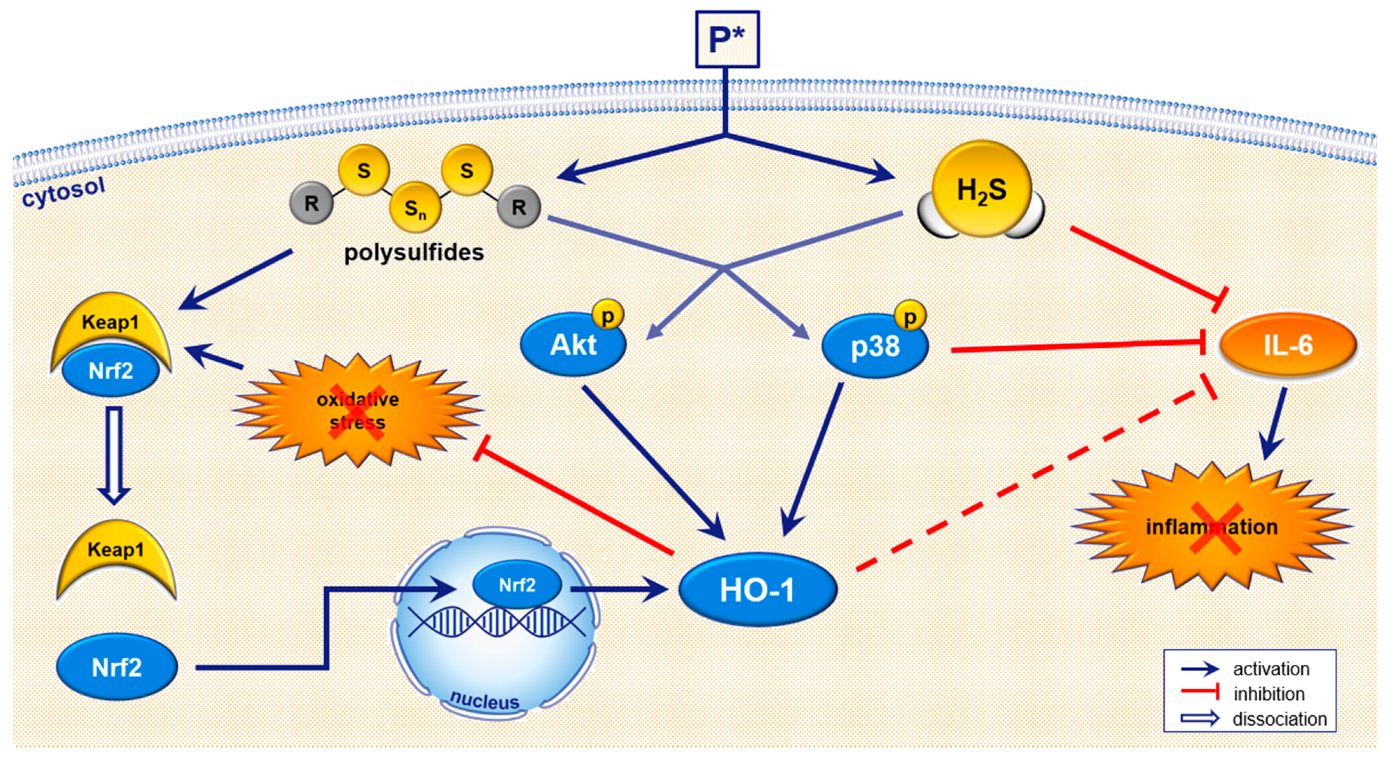
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Glyn-Jones, S.; Palmer, A.J.R.; Agricola, R.; Price, A.J.; Vincent, T.L.; Weinans, H.; Carr, A.J. Osteoarthritis. Lancet 2015, 386, 376–387. [Google Scholar] [CrossRef]
- Hunter, D.J.; Bierma-Zeinstra, S. Osteoarthritis. Lancet 2019, 393, 1745–1759. [Google Scholar] [CrossRef]
- Bannuru, R.R.; Osani, M.C.; Vaysbrot, E.E.; Arden, N.K.; Bennell, K.; Bierma-Zeinstra, S.M.A.; Kraus, V.B.; Lohmander, L.S.; Abbott, J.H.; Bhandari, M.; et al. OARSI guidelines for the non-surgical management of knee, hip, and polyarticular osteoarthritis. Osteoarthr. Cartil. 2019, 27, 1578–1589. [Google Scholar] [CrossRef] [Green Version]
- Sieghart, D.; Liszt, M.; Wanivenhaus, A.; Bröll, H.; Kiener, H.; Klösch, B.; Steiner, G. Hydrogen sulphide decreases IL -1β-induced activation of fibroblast-like synoviocytes from patients with osteoarthritis. J. Cell. Mol. Med. 2014, 19, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Burguera, E.; Vela-Anero, Á.; Magalhães, J.; Meijide-Faílde, R.; Blanco, F. Effect of hydrogen sulfide sources on inflammation and catabolic markers on interleukin 1β-stimulated human articular chondrocytes. Osteoarthr. Cartil. 2014, 22, 1026–1035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, B.; Schantz, J.-T.; Haigh, R.; Wood, M.E.; Moore, P.K.; Viner, N.; Spencer, J.P.E.; Winyard, P.; Whiteman, M. Inducible hydrogen sulfide synthesis in chondrocytes and mesenchymal progenitor cells: Is H2S a novel cytoprotective mediator in the inflamed joint? J. Cell. Mol. Med. 2012, 16, 896–910. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Shao, Z.; Gu, M.; Ni, L.; Shi, Y.; Yan, Y.; Wu, A.; Jin, H.; Chen, J.; Pan, X.; et al. Hydrogen sulfide protects against IL-1β-induced inflammation and mitochondrial dysfunction-related apoptosis in chondrocytes and ameliorates osteoarthritis. J. Cell. Physiol. 2021, 236, 4369–4386. [Google Scholar] [CrossRef] [PubMed]
- Nasi, S.; Ehirchiou, D.; Chatzianastasiou, A.; Nagahara, N.; Papapetropoulos, A.; Bertrand, J.; Cirino, G.; So, A.; Busso, N. The protective role of the 3-mercaptopyruvate sulfurtransferase (3-MST)-hydrogen sulfide (H2S) pathway against experimental osteoarthritis. Arthritis Res. 2020, 22, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meijide-Failde, R.; Blanco, F.J. Hydrogen Sulfide and Inflammatory Joint Diseases. Curr. Drug Targets 2017, 18, 1641–1652. [Google Scholar] [CrossRef]
- Wang, R. Two’s company, three’s a crowd: Can H2S be the third endogenous gaseous transmitter? FASEB J. 2002, 16, 1792–1798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuto, J.M.; Carrington, S.J.; Tantillo, D.J.; Harrison, J.G.; Ignarro, L.J.; Freeman, B.A.; Chen, A.; Wink, D.A. Small Molecule Signaling Agents: The Integrated Chemistry and Biochemistry of Nitrogen Oxides, Oxides of Carbon, Dioxygen, Hydrogen Sulfide, and Their Derived Species. Chem. Res. Toxicol. 2012, 25, 769–793. [Google Scholar] [CrossRef]
- Szabó, C. Hydrogen sulphide and its therapeutic potential. Nat. Rev. Drug Discov. 2007, 6, 917–935. [Google Scholar] [CrossRef]
- Whiteman, M.; Winyard, P. Hydrogen sulfide and inflammation: The good, the bad, the ugly and the promising. Expert Rev. Clin. Pharmacol. 2011, 4, 13–32. [Google Scholar] [CrossRef] [PubMed]
- Mustafa, A.K.; Gadalla, M.M.; Sen, N.; Kim, S.; Mu, W.; Gazi, S.K.; Barrow, R.K.; Yang, G.; Wang, R.; Snyder, S.H. H2S Signals Through Protein S-Sulfhydration. Sci. Signal. 2009, 2, ra72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, B.D.; Snyder, S.H. H2S signalling through protein sulfhydration and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Calvert, J.; Jha, S.; Gundewar, S.; Elrod, J.; Ramachandran, A.; Pattillo, C.B.; Kevil, C.; Lefer, D.J. Hydrogen Sulfide Mediates Cardioprotection Through Nrf2 Signaling. Circ. Res. 2009, 105, 365–374. [Google Scholar] [CrossRef] [Green Version]
- Xie, Z.-Z.; Liu, Y.; Bian, J.-S. Hydrogen Sulfide and Cellular Redox Homeostasis. Oxidative Med. Cell. Longev. 2016, 2016, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, G.; Zhao, K.; Ju, Y.; Mani, S.; Cao, Q.; Puukila, S.; Khaper, N.; Wu, L.; Wang, R. Hydrogen Sulfide Protects Against Cellular Senescence via S-Sulfhydration of Keap1 and Activation of Nrf2. Antioxid. Redox Signal. 2013, 18, 1906–1919. [Google Scholar] [CrossRef] [PubMed]
- Hourihan, J.M.; Kenna, J.G.; Hayes, J.D. The Gasotransmitter Hydrogen Sulfide Induces Nrf2-Target Genes by Inactivating the Keap1 Ubiquitin Ligase Substrate Adaptor Through Formation of a Disulfide Bond Between Cys-226 and Cys-613. Antioxid. Redox Signal. 2013, 19, 465–481. [Google Scholar] [CrossRef] [PubMed]
- Ndisang, J.F.; Jadhav, A.; Mishra, M. The Heme Oxygenase System Suppresses Perirenal Visceral Adiposity, Abates Renal Inflammation and Ameliorates Diabetic Nephropathy in Zucker Diabetic Fatty Rats. PLoS ONE 2014, 9, e87936. [Google Scholar] [CrossRef] [PubMed]
- Minamino, T.; Christou, H.; Hsieh, C.-M.; Liu, Y.; Dhawan, V.; Abraham, N.G.; Perrella, M.A.; Mitsialis, S.A.; Kourembanas, S. Targeted expression of heme oxygenase-1 prevents the pulmonary inflammatory and vascular responses to hypoxia. Proc. Natl. Acad. Sci. USA 2001, 98, 8798–8803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishikawa, K.; Sugawara, D.; Wang, X.-P.; Suzuki, K.; Itabe, H.; Maruyama, Y.; Lusis, A.J. Heme Oxygenase-1 Inhibits Atherosclerotic Lesion Formation in LDL-Receptor Knockout Mice. Circ. Res. 2001, 88, 506–512. [Google Scholar] [CrossRef] [PubMed]
- Hung, S.-Y.; Liou, H.-C.; Kang, K.-H.; Wu, R.-M.; Wen, C.-C.; Fu, W.-M. Overexpression of Heme Oxygenase-1 Protects Dopaminergic Neurons against 1-Methyl-4-Phenylpyridinium-Induced Neurotoxicity. Mol. Pharmacol. 2008, 74, 1564–1575. [Google Scholar] [CrossRef] [Green Version]
- Vulapalli, S.R.; Chen, Z.; Chua, B.H.L.; Wang, T.; Liang, C.-S. Cardioselective overexpression of HO-1 prevents I/R-induced cardiac dysfunction and apoptosis. Am. J. Physiol. Circ. Physiol. 2002, 283, H688–H694. [Google Scholar] [CrossRef] [Green Version]
- Takada, T.; Miyaki, S.; Ishitobi, H.; Hirai, Y.; Nakasa, T.; Igarashi, K.; Lotz, M.K.; Ochi, M. Bach1 deficiency reduces severity of osteoarthritis through upregulation of heme oxygenase-1. Arthritis Res. Ther. 2015, 17, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Kapturczak, M.H.; Wasserfall, C.; Brusko, T.; Campbell-Thompson, M.; Ellis, T.M.; Atkinson, M.A.; Agarwal, A. Heme Oxygenase-1 Modulates Early Inflammatory Responses: Evidence from the Heme Oxygenase-1-Deficient Mouse. Am. J. Pathol. 2004, 165, 1045–1053. [Google Scholar] [CrossRef]
- Abraham, N.G.; Kappas, A. Pharmacological and Clinical Aspects of Heme Oxygenase. Pharmacol. Rev. 2008, 60, 79–127. [Google Scholar] [CrossRef] [Green Version]
- Greiner, R.; Pálinkás, Z.; Bäsell, K.; Becher, D.; Antelmann, H.; Nagy, P.; Dick, T.P. Polysulfides Link H2S to Protein Thiol Oxidation. Antioxid. Redox Signal. 2013, 19, 1749–1765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ida, T.; Sawa, T.; Ihara, H.; Tsuchiya, Y.; Watanabe, Y.; Kumagai, Y.; Suematsu, M.; Motohashi, H.; Fujii, S.; Matsunaga, T.; et al. Reactive cysteine persulfides and S-polythiolation regulate oxidative stress and redox signaling. Proc. Natl. Acad. Sci. USA 2014, 111, 7606–7611. [Google Scholar] [CrossRef] [Green Version]
- Kimura, Y.; Mikami, Y.; Osumi, K.; Tsugane, M.; Oka, J.; Kimura, H. Polysulfides are possible H 2 S-derived signaling molecules in rat brain. FASEB J. 2013, 27, 2451–2457. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Macinkovic, I.; Devarie-Baez, N.O.; Pan, J.; Park, C.-M.; Carroll, K.S.; Filipovic, M.R.; Xian, M. Detection of Protein S-Sulfhydration by a Tag-Switch Technique. Angew. Chem. Int. Ed. 2013, 53, 575–581. [Google Scholar] [CrossRef] [Green Version]
- Koike, S.; Nishimoto, S.; Ogasawara, Y. Cysteine persulfides and polysulfides produced by exchange reactions with H2S protect SH-SY5Y cells from methylglyoxal-induced toxicity through Nrf2 activation. Redox Biol. 2017, 12, 530–539. [Google Scholar] [CrossRef]
- Kim, S.; Lee, H.-G.; Park, S.-A.; Kundu, J.K.; Keum, Y.-S.; Cha, Y.-N.; Na, H.-K.; Surh, Y.-J. Keap1 Cysteine 288 as a Potential Target for Diallyl Trisulfide-Induced Nrf2 Activation. PLoS ONE 2014, 9, e85984. [Google Scholar] [CrossRef] [Green Version]
- Koike, S.; Ogasawara, Y.; Shibuya, N.; Kimura, H.; Ishii, K. Polysulfide exerts a protective effect against cytotoxicity caused byt-buthylhydroperoxide through Nrf2 signaling in neuroblastoma cells. FEBS Lett. 2013, 587, 3548–3555. [Google Scholar] [CrossRef] [Green Version]
- Olson, K.R. H2S and polysulfide metabolism: Conventional and unconventional pathways. Biochem. Pharmacol. 2018, 149, 77–90. [Google Scholar] [CrossRef]
- Xu, S.; Hamsath, A.; Neill, D.L.; Wang, Y.; Yang, C.-T.; Xian, M. Strategies for the Design of Donors and Precursors of Reactive Sulfur Species. Chem. A Eur. J. 2019, 25, 4005–4016. [Google Scholar] [CrossRef]
- Szabo, C.; Papapetropoulos, A. International Union of Basic and Clinical Pharmacology. CII: Pharmacological Modulation of H2S Levels: H2S Donors and H2S Biosynthesis Inhibitors. Pharmacol. Rev. 2017, 69, 497–564. [Google Scholar] [CrossRef] [Green Version]
- Artaud, I.; Galardon, E. A Persulfide Analogue of the Nitrosothiol SNAP: Formation, Characterization and Reactivity. ChemBioChem 2014, 15, 2361–2364. [Google Scholar] [CrossRef] [PubMed]
- Braunstein, I.; Engelman, R.; Yitzhaki, O.; Ziv, T.; Galardon, E.; Benhar, M. Opposing effects of polysulfides and thioredoxin on apoptosis through caspase persulfidation. J. Biol. Chem. 2020, 295, 3590–3600. [Google Scholar] [CrossRef] [PubMed]
- Toegel, S.; Pabst, M.; Wu, S.; Grass, J.; Goldring, M.; Chiari, C.; Kolb, A.; Altmann, F.; Viernstein, H.; Unger, F. Phenotype-related differential α-2,6- or α-2,3-sialylation of glycoprotein N-glycans in human chondrocytes. Osteoarthr. Cartil. 2010, 18, 240–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, K.; Kolesnik, B.; Gorren, A.C.F.; Werner, E.R.; Mayer, B. Cell type-specific recycling of tetrahydrobiopterin by dihydrofolate reductase explains differential effects of 7,8-dihydrobiopterin on endothelial nitric oxide synthase uncoupling. Biochem. Pharmacol. 2014, 90, 246–253. [Google Scholar] [CrossRef] [Green Version]
- Paul, B.D.; Snyder, S.H. H 2 S: A Novel Gasotransmitter that Signals by Sulfhydration. Trends Biochem. Sci. 2015, 40, 687–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, H. Signaling Molecules: Hydrogen Sulfide and Polysulfide. Antioxid. Redox Signal. 2015, 22, 362–376. [Google Scholar] [CrossRef] [Green Version]
- Fukuto, J.M.; Hobbs, A.J. A comparison of the chemical biology of hydropersulfides (RSSH) with other protective biological antioxidants and nucleophiles. Nitric Oxide 2021, 107, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Murphy, B.; Bhattacharya, R.; Mukherjee, P. Hydrogen sulfide signaling in mitochondria and disease. FASEB J. 2019, 33, 13098–13125. [Google Scholar] [CrossRef] [Green Version]
- Ferrandiz, M.; Devesa, M.L.F.A.I. Inducers of Heme Oxygenase-1. Curr. Pharm. Des. 2008, 14, 473–486. [Google Scholar] [CrossRef] [PubMed]
- Paine, A.; Eiz-Vesper, B.; Blasczyk, R.; Immenschuh, S. Signaling to heme oxygenase-1 and its anti-inflammatory therapeutic potential. Biochem. Pharmacol. 2010, 80, 1895–1903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prawan, A.; Kundu, J.K.; Surh, Y.-J. Molecular Basis of Heme Oxygenase-1 Induction: Implications for Chemoprevention and Chemoprotection. Antioxid. Redox Signal. 2005, 7, 1688–1703. [Google Scholar] [CrossRef] [PubMed]
- Ryter, S.W.; Alam, J.; Choi, A.M.K. Heme Oxygenase-1/Carbon Monoxide: From Basic Science to Therapeutic Applications. Physiol. Rev. 2006, 86, 583–650. [Google Scholar] [CrossRef] [PubMed]
- Goldring, M.B.; Otero, M. Inflammation in osteoarthritis. Curr. Opin. Rheumatol. 2011, 23, 471–478. [Google Scholar] [CrossRef]
- Weinmann, D.; Kenn, M.; Schmidt, S.; Schmidt, K.; Walzer, S.M.; Kubista, B.; Windhager, R.; Schreiner, W.; Toegel, S.; Gabius, H.-J. Galectin-8 induces functional disease markers in human osteoarthritis and cooperates with galectins-1 and -3. Cell. Mol. Life Sci. 2018, 75, 4187–4205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toegel, S.; Bieder, D.; André, S.; Altmann, F.; Walzer, S.M.; Kaltner, H.; Hofstaetter, J.G.; Windhager, R.; Gabius, H.-J. Glycophenotyping of osteoarthritic cartilage and chondrocytes by RT-qPCR, mass spectrometry, histochemistry with plant/human lectins and lectin localization with a glycoprotein. Arthritis Res. Ther. 2013, 15, R147. [Google Scholar] [CrossRef] [Green Version]
- Hughes, M.N.; Centelles, M.N.; Moore, K.P. Making and working with hydrogen sulfide: The chemistry and generation of hydrogen sulfide in vitro and its measurement in vivo: A review. Free. Radic. Biol. Med. 2009, 47, 1346–1353. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H. Signalling by hydrogen sulfide and polysulfides via protein S -sulfuration. Br. J. Pharmacol. 2020, 177, 720–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuto, J.M.; Ignarro, L.J.; Nagy, P.; Wink, D.A.; Kevil, C.; Feelisch, M.; Cortese-Krott, M.M.; Bianco, C.L.; Kumagai, Y.; Hobbs, A.J.; et al. Biological hydropersulfides and related polysulfides—A new concept and perspective in redox biology. FEBS Lett. 2018, 592, 2140–2152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lepetsos, P.; Papavassiliou, A.G. ROS/oxidative stress signaling in osteoarthritis. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2016, 1862, 576–591. [Google Scholar] [CrossRef] [PubMed]
- Henrotin, Y.; Kurz, B.; Aigner, T. Oxygen and reactive oxygen species in cartilage degradation: Friends or foes? Osteoarthr. Cartil. 2005, 13, 643–654. [Google Scholar] [CrossRef] [Green Version]
- Sarady, J.K.; Otterbein, S.L.; Liu, F.; Otterbein, L.E.; Choi, A.M.K. Carbon Monoxide Modulates Endotoxin-Induced Production of Granulocyte Macrophage Colony-Stimulating Factor in Macrophages. Am. J. Respir. Cell Mol. Biol. 2002, 27, 739–745. [Google Scholar] [CrossRef]
- Stocker, R.; Yamamoto, Y.; McDonagh, A.F.; Glazer, A.N.; Ames, B.N. Bilirubin is an antioxidant of possible physiological importance. Science 1987, 235, 1043–1046. [Google Scholar] [CrossRef]
- Otterbein, L.E.; Soares, M.P.; Yamashita, K.; Bach, F.H. Heme oxygenase-1: Unleashing the protective properties of heme. Trends Immunol. 2003, 24, 449–455. [Google Scholar] [CrossRef]
- Nitti, M.; Piras, S.; Marinari, U.M.; Moretta, L.; Pronzato, M.A.; Furfaro, A.L. HO-1 Induction in Cancer Progression: A Matter of Cell Adaptation. Antioxidants 2017, 6, 29. [Google Scholar] [CrossRef] [PubMed]
- Deng, R.; Wang, S.-M.; Yin, T.; Ye, T.-H.; Shen, G.-B.; Li, L.; Zhao, J.-Y.; Sang, Y.-X.; Duan, X.-G.; Wei, Y.-Q. Inhibition of Tumor Growth and Alteration of Associated Macrophage Cell Type by an HO-1 Inhibitor in Breast Carcinoma-Bearing Mice. Oncol. Res. Featur. Preclin. Clin. Cancer Ther. 2012, 20, 473–482. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.-Y.; Yeh, W.-L.; Huang, S.-M.; Tang, C.-H.; Lin, H.-Y.; Chou, S.-J. Osteopontin increases heme oxygenase–1 expression and subsequently induces cell migration and invasion in glioma cells. Neuro Oncol. 2012, 14, 1367–1378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sunamura, M.; Duda, D.G.; Ghattas, M.H.; Lozonschi, L.; Motoi, F.; Yamauchi, J.; Matsuno, S.; Shibahara, S.; Abraham, N.G. Heme oxygenase-1 accelerates tumor angiogenesis of human pancreatic cancer. Angiogenesis 2003, 6, 15–24. [Google Scholar] [CrossRef]
- Was, H.; Cichoń, T.; Smolarczyk, R.; Rudnicka, D.; Stopa, M.; Chevalier, C.; Leger, J.J.; Lackowska, B.; Grochot-Przeczek, A.; Bojkowska, K.; et al. Overexpression of Heme Oxygenase-1 in Murine Melanoma: Increased Proliferation and Viability of Tumor Cells, Decreased Survival of Mice. Am. J. Pathol. 2006, 169, 2181–2198. [Google Scholar] [CrossRef] [Green Version]
- Na, H.-K.; Surh, Y.-J. Oncogenic potential of Nrf2 and its principal target protein heme oxygenase-1. Free. Radic. Biol. Med. 2014, 67, 353–365. [Google Scholar] [CrossRef]
- Fu, L.; Liu, K.; He, J.; Tian, C.; Yu, X.; Yang, J. Direct Proteomic Mapping of Cysteine Persulfidation. Antioxid. Redox Signal. 2020, 33, 1061–1076. [Google Scholar] [CrossRef]
- Li, L.; Rose, P.; Moore, P.K. Hydrogen Sulfide and Cell Signaling. Annu. Rev. Pharmacol. Toxicol. 2011, 51, 169–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Zhao, L.; Mao, J.; Huang, J.; Chen, J. Antioxidant Effects of Hydrogen Sulfide on Left Ventricular Remodeling in Smoking Rats Are Mediated via PI3K/Akt-Dependent Activation of Nrf2. Toxicol. Sci. 2014, 144, 197–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, M.; Zhuo, C.; Jiang, R.; Chen, G.; Shan, J.; Ping, J.; Tian, H.; Wang, L.; Lin, C.; Hu, L. Protective effect of hydrogen sulphide against myocardial hypertrophy in mice. Oncotarget 2017, 8, 22344–22352. [Google Scholar] [CrossRef]
- Lohninger, L.; Tomasova, L.; Praschberger, M.; Hintersteininger, M.; Erker, T.; Gmeiner, B.M.; Laggner, H. Hydrogen sulphide induces HIF-1α and Nrf2 in THP-1 macrophages. Biochimie 2015, 112, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Ono, K.; Tsutsuki, H.; Ihara, H.; Islam, W.; Akaike, T.; Sawa, T. Enhanced Cellular Polysulfides Negatively Regulate TLR4 Signaling and Mitigate Lethal Endotoxin Shock. Cell Chem. Biol. 2019, 26, 686–698.e4. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, M.; Gaddam, R.R. Hydrogen Sulfide in Inflammation: A Novel Mediator and Therapeutic Target. Antioxid. Redox Signal. 2021, 34, 1368–1377. [Google Scholar] [CrossRef]
- Zanardo, R.C.O.; Brancaleone, V.; Distrutti, E.; Fiorucci, S.; Cirino, G.; Wallace, J.L. Hydrogen sulfide is an endogenous modulator of leukocyte-mediated inflammation. FASEB J. 2006, 20, 2118–2120. [Google Scholar] [CrossRef] [PubMed]
- Vaamonde-García, C.; Burguera, E.F.; Vela-Anero, Á.; Hermida-Gómez, T.; Filgueira-Fernández, P.; Fernández-Rodríguez, J.A.; Meijide-Faílde, R.; Blanco, F.J. Intraarticular Administration Effect of Hydrogen Sulfide on an In Vivo Rat Model of Osteoarthritis. Int. J. Mol. Sci. 2020, 21, 7421. [Google Scholar] [CrossRef]
- Hosseinzadeh, A.; Jafari, D.; Kamarul, T.; Bagheri, A.; Sharifi, A.M. Evaluating the Protective Effects and Mechanisms of Diallyl Disulfide on Interlukin-1β-Induced Oxidative Stress and Mitochondrial Apoptotic Signaling Pathways in Cultured Chondrocytes. J. Cell. Biochem. 2017, 118, 1879–1888. [Google Scholar] [CrossRef] [PubMed]
- Batallé, G.; Cabarga, L.; Pol, O. The Inhibitory Effects of Slow-Releasing Hydrogen Sulfide Donors in the Mechanical Allodynia, Grip Strength Deficits, and Depressive-Like Behaviors Associated with Chronic Osteoarthritis Pain. Antioxidants 2019, 9, 31. [Google Scholar] [CrossRef] [Green Version]
- Wruck, C.J.; Streetz, K.; Pavic, G.; Götz, M.E.; Tohidnezhad, M.; Brandenburg, L.-O.; Varoga, D.; Eickelberg, O.; Herdegen, T.; Trautwein, C.; et al. Nrf2 Induces Interleukin-6 (IL-6) Expression via an Antioxidant Response Element within the IL-6 Promoter. J. Biol. Chem. 2011, 286, 4493–4499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgener, A.-V.; Bantug, G.R.; Meyer, B.J.; Higgins, R.; Ghosh, A.; Bignucolo, O.; Ma, E.H.; Loeliger, J.; Unterstab, G.; Geigges, M.; et al. SDHA gain-of-function engages inflammatory mitochondrial retrograde signaling via KEAP1–Nrf2. Nat. Immunol. 2019, 20, 1311–1321. [Google Scholar] [CrossRef]
- Oh, G.-S.; Pae, H.-O.; Lee, B.-S.; Kim, B.-N.; Kim, J.-M.; Kim, H.-R.; Jeon, S.B.; Jeon, W.K.; Chae, H.-J.; Chung, H.-T. Hydrogen sulfide inhibits nitric oxide production and nuclear factor-κB via heme oxygenase-1 expression in RAW264.7 macrophages stimulated with lipopolysaccharide. Free. Radic. Biol. Med. 2006, 41, 106–119. [Google Scholar] [CrossRef] [PubMed]
- Kloesch, B.; Liszt, M.; Steiner, G.; Bröll, J. Inhibitors of p38 and ERK1/2 MAPkinase and hydrogen sulphide block constitutive and IL-1β-induced IL-6 and IL-8 expression in the human chondrocyte cell line C-28/I2. Rheumatol. Int. 2012, 32, 729–736. [Google Scholar] [CrossRef]
- Ha, C.; Tian, S.; Sun, K.; Wang, D.; Lv, J.; Wang, Y. Hydrogen sulfide attenuates IL-1β-induced inflammatory signaling and dysfunction of osteoarthritic chondrocytes. Int. J. Mol. Med. 2015, 35, 1657–1666. [Google Scholar] [CrossRef] [Green Version]
- Brown, K.K.; Heitmeyer, S.A.; Hookfin, E.B.; Hsieh, L.; Buchalova, M.; Taiwo, Y.O.; Janusz, M.J. P38 MAP kinase inhibitors as potential therapeutics for the treatment of joint degeneration and pain associated with osteoarthritis. J. Inflamm. 2008, 5, 22. [Google Scholar] [CrossRef] [Green Version]
- Loeser, R.F.; Erickson, E.A.; Long, D.L. Mitogen-activated protein kinases as therapeutic targets in osteoarthritis. Curr. Opin. Rheumatol. 2008, 20, 581–586. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Shirai, T.; Morishita, S.; Uchida, S.; Saeki-Miura, K.; Makishima, F. p38 Mitogen-Activated Protein Kinase Functionally Contributes to Chondrogenesis Induced by Growth/Differentiation Factor-5 in ATDC5 Cells. Exp. Cell Res. 1999, 250, 351–363. [Google Scholar] [CrossRef]
- Stanton, L.-A.; Sabari, S.; Sampaio, A.V.; Underhill, T.M.; Beier, F. p38 MAP kinase signalling is required for hypertrophic chondrocyte differentiation. Biochem. J. 2004, 378, 53–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thouverey, C.; Caverzasio, J. Focus on the p38 MAPK signaling pathway in bone development and maintenance. BoneKEy Rep. 2015, 4, 711. [Google Scholar] [CrossRef] [Green Version]
- Giampaoli, S.; Valeriani, F.; Gianfranceschi, G.; Vitali, M.; Delfini, M.; Festa, M.; Bottari, E.; Spica, V.R. Hydrogen sulfide in thermal spring waters and its action on bacteria of human origin. Microchem. J. 2013, 108, 210–214. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trummer, M.; Galardon, E.; Fischer, A.; Toegel, S.; Mayer, B.; Steiner, G.; Kloesch, B. Characterization of the Inducible and Slow-Releasing Hydrogen Sulfide and Persulfide Donor P*: Insights into Hydrogen Sulfide Signaling. Antioxidants 2021, 10, 1049. https://doi.org/10.3390/antiox10071049
Trummer M, Galardon E, Fischer A, Toegel S, Mayer B, Steiner G, Kloesch B. Characterization of the Inducible and Slow-Releasing Hydrogen Sulfide and Persulfide Donor P*: Insights into Hydrogen Sulfide Signaling. Antioxidants. 2021; 10(7):1049. https://doi.org/10.3390/antiox10071049
Chicago/Turabian StyleTrummer, Modesta, Erwan Galardon, Anita Fischer, Stefan Toegel, Bernd Mayer, Guenter Steiner, and Burkhard Kloesch. 2021. "Characterization of the Inducible and Slow-Releasing Hydrogen Sulfide and Persulfide Donor P*: Insights into Hydrogen Sulfide Signaling" Antioxidants 10, no. 7: 1049. https://doi.org/10.3390/antiox10071049
APA StyleTrummer, M., Galardon, E., Fischer, A., Toegel, S., Mayer, B., Steiner, G., & Kloesch, B. (2021). Characterization of the Inducible and Slow-Releasing Hydrogen Sulfide and Persulfide Donor P*: Insights into Hydrogen Sulfide Signaling. Antioxidants, 10(7), 1049. https://doi.org/10.3390/antiox10071049