
Neonatal Extracellular Superoxide Dismutase Knockout Mice Increase Total Superoxide Dismutase Activity and VEGF Expression after Chronic Hyperoxia


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Model
2.2. Tissue Collection and Processing
2.3. Lung Histology
2.4. Immunofluorescence
2.5. Western Blotting
2.6. SOD Activity Assay
2.7. sGC Activity Assay
2.8. Statistics
3. Results
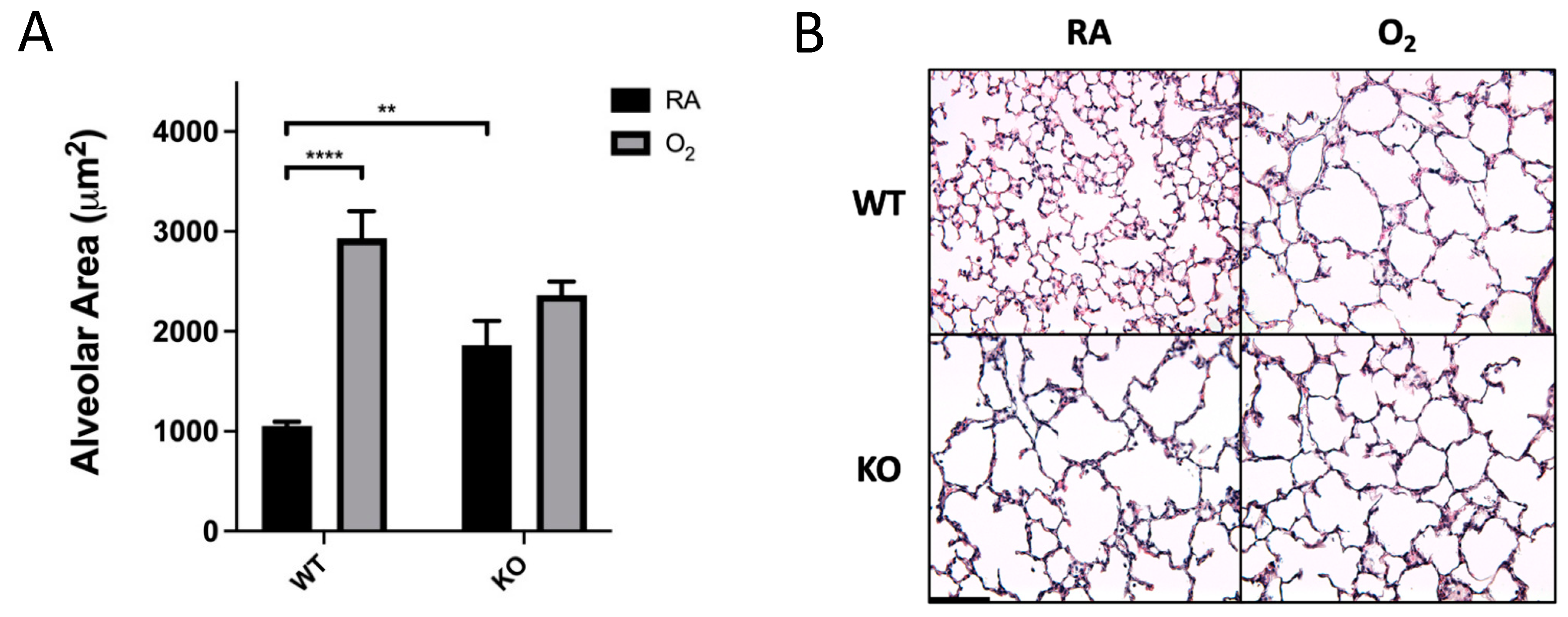
3.1. Combined SOD3 KO and Hyperoxia Exposure Does Not Cause More Alveolar Simplification Than SOD3 KO or Hyperoxia Alone
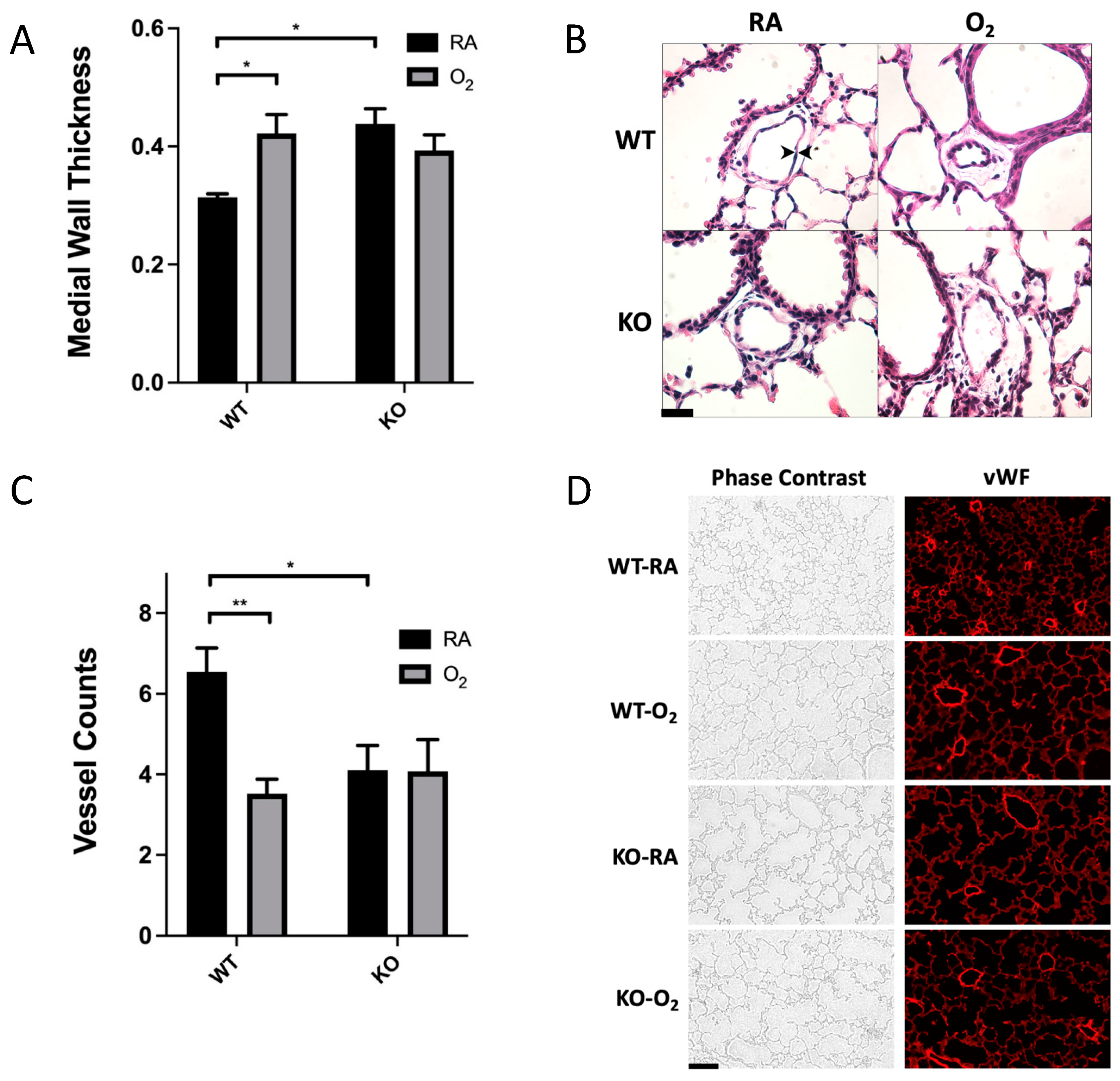
3.2. Combined SOD3 KO and Hyperoxia Exposure Does Not Cause More Microvascular Remodeling Than SOD3 KO or Hyperoxia Alone
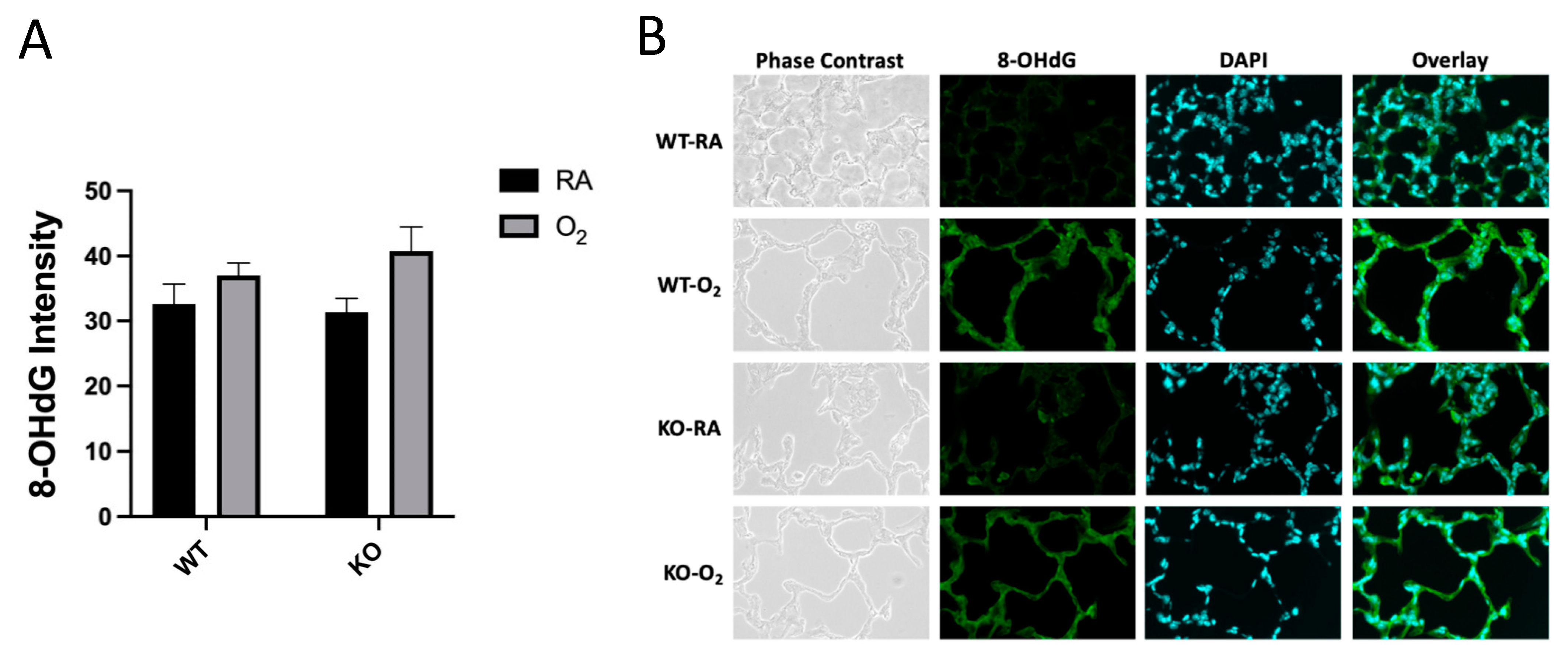
3.3. DNA Oxidation Was Not Affected by Genotype
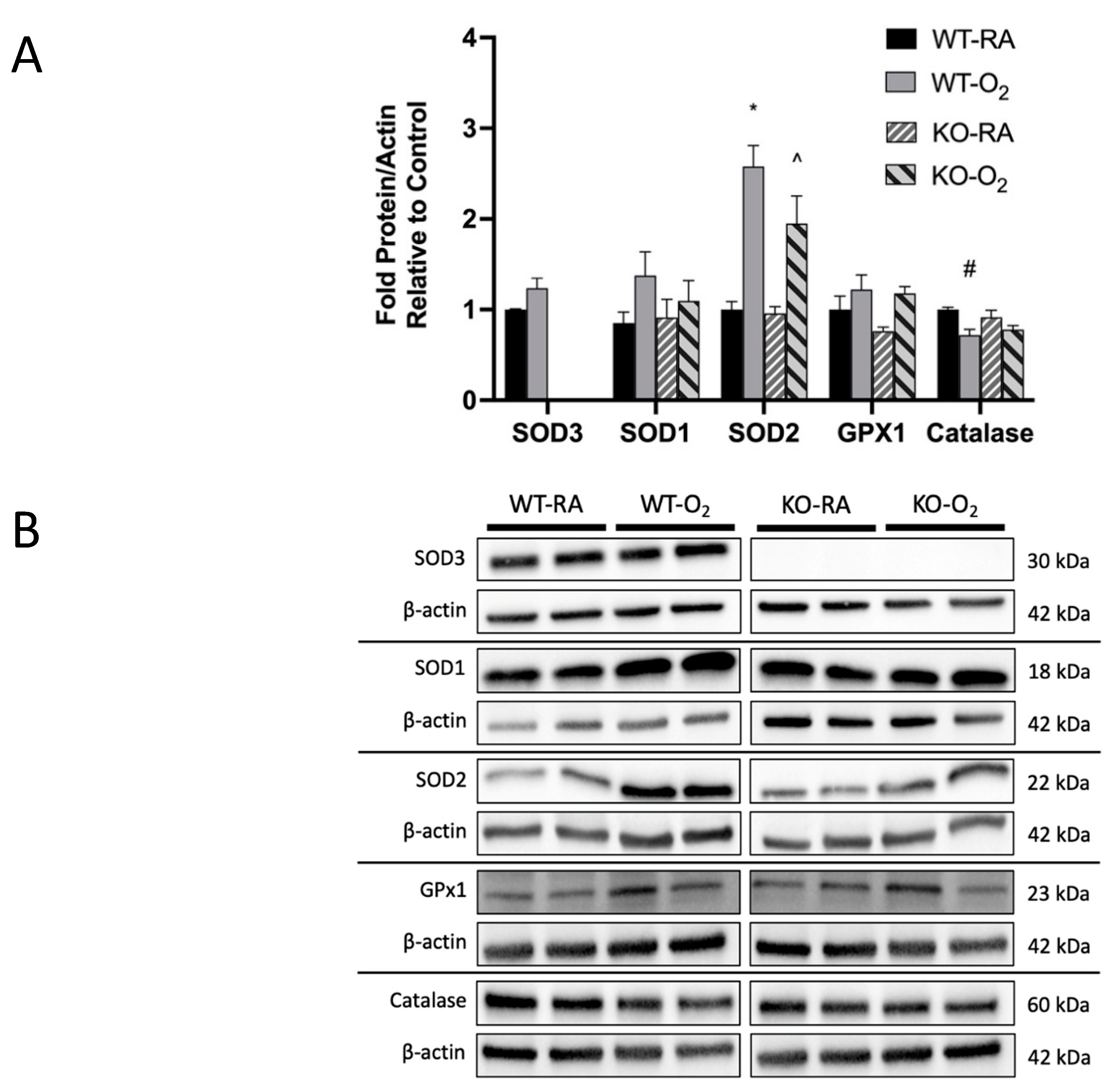
3.4. SOD3 Protein Was Absent in KO Mice, but Genotype Did Not Affect Other Antioxidant Protein Expression
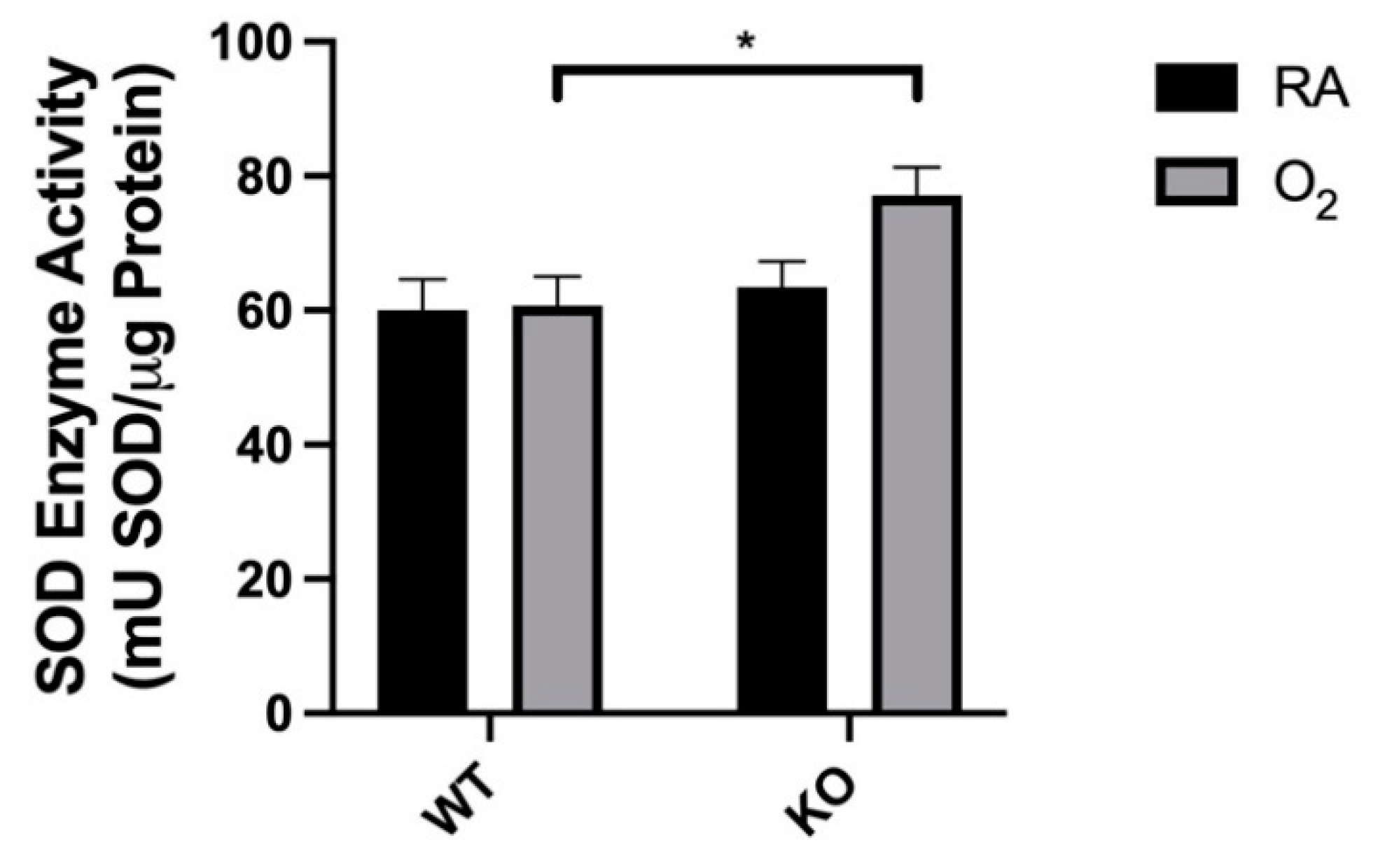
3.5. Combined SOD3 KO and Hyperoxia Increased Total SOD Activity
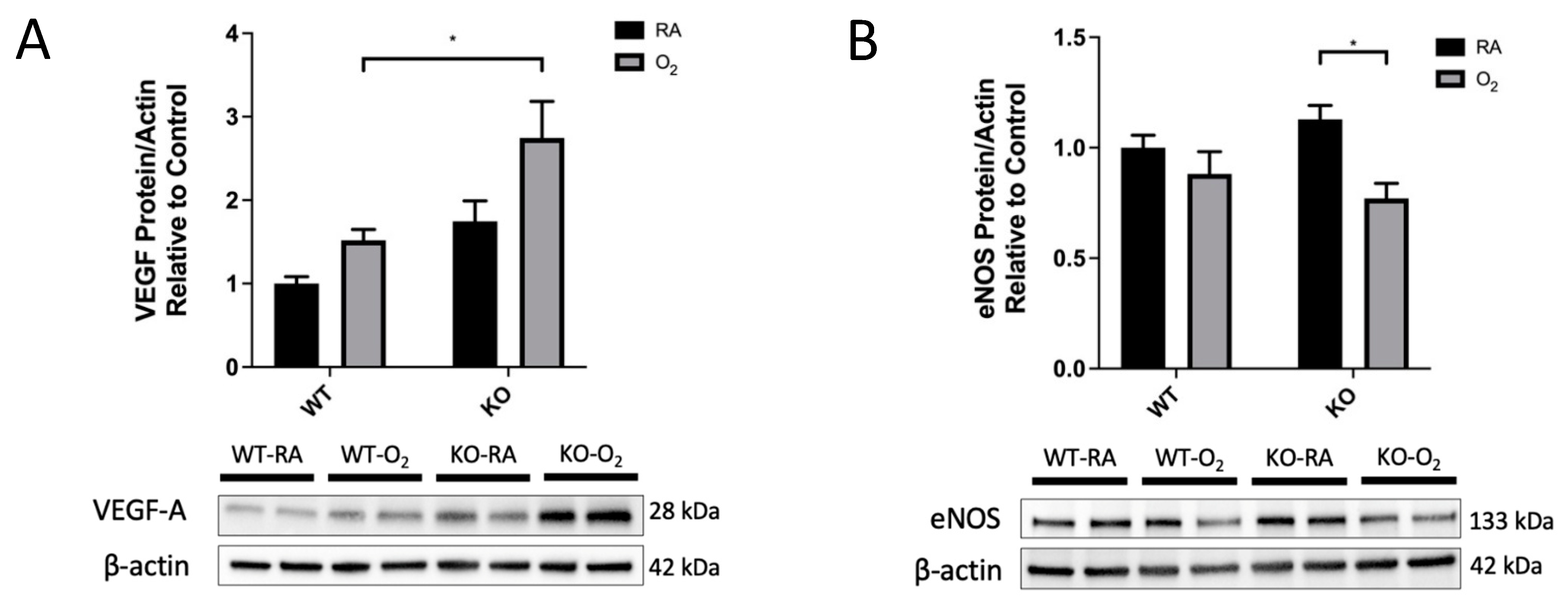
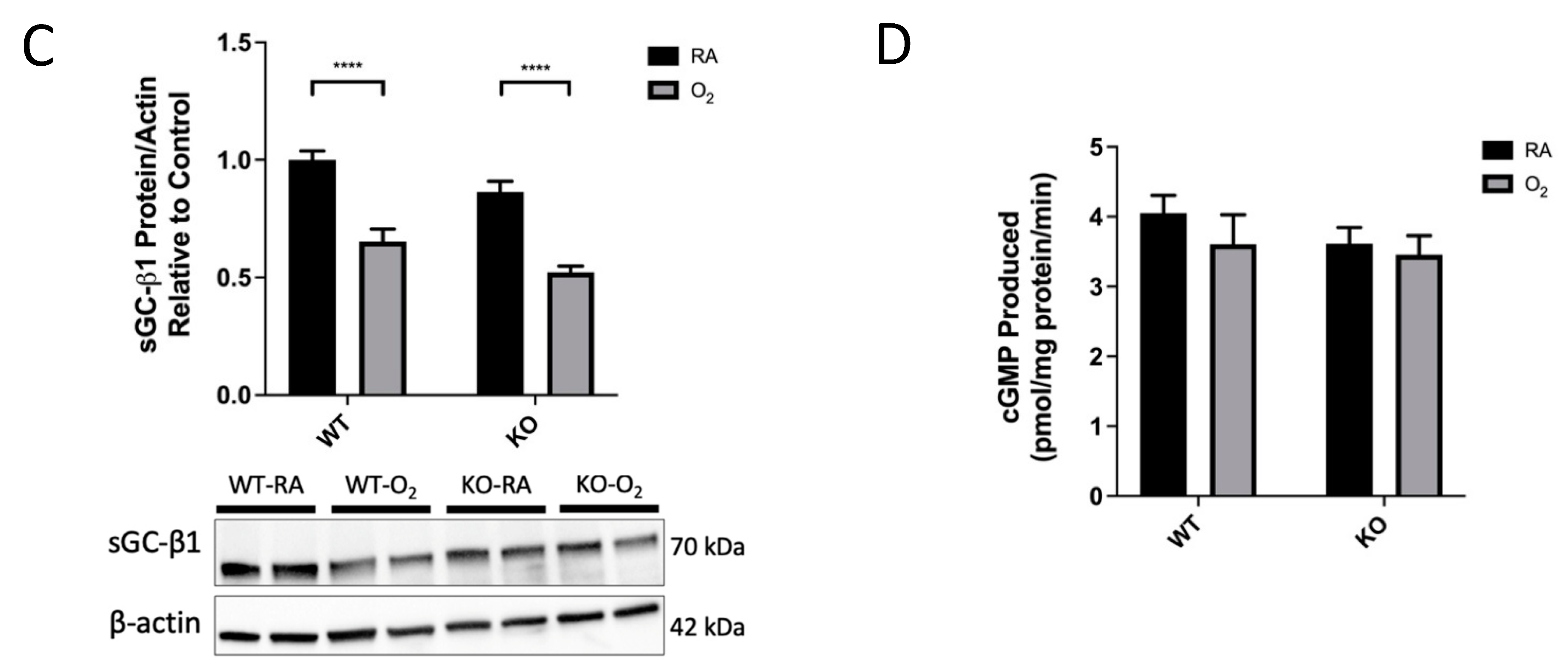
3.6. KO-O2 Mice Increased VEGF and Decreased eNOS in Response to Hyperoxia
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stroustrup, A.; Trasande, L. Epidemiological Characteristics and Resource Use in Neonates With Bronchopulmonary Dysplasia: 1993–2006. Pediatrics 2010, 126, 291–297. [Google Scholar] [CrossRef]
- Poindexter, B.B.; Feng, R.; Schmidt, B.; Aschner, J.L.; Ballard, R.A.; Hamvas, A.; Reynolds, A.M.; Shaw, P.A.; Jobe, A.H. Prematurity and Respiratory Outcomes Program Comparisons and Limitations of Current Definitions of Bronchopulmonary Dysplasia for the Prematurity and Respiratory Outcomes Program. Ann. Am. Thorac. Soc. 2015, 12, 1822–1830. [Google Scholar] [CrossRef] [Green Version]
- Baraldi, E.; Filippone, M. Chronic Lung Disease after Premature Birth. N. Engl. J. Med. 2007, 357, 1946–1955. [Google Scholar] [CrossRef] [Green Version]
- Saugstad, O.D. Bronchopulmonary Dysplasia-Oxidative Stress and Antioxidants. Semin. Neonatol. 2003, 8, 39–49. [Google Scholar] [CrossRef]
- Berkelhamer, S.K.; Kim, G.A.; Radder, J.E.; Wedgwood, S.; Czech, L.; Steinhorn, R.H.; Schumacker, P.T. Developmental Differences in Hyperoxia-Induced Oxidative Stress and Cellular Responses in the Murine Lung. Free Radic. Biol. Med. 2013, 61, 51–60. [Google Scholar] [CrossRef] [Green Version]
- Heinonen, K.; Mononen, I.; Mononen, T.; Parviainen, M.; Penttilä, I.; Launiala, K. Plasma Vitamin C Levels Are Low in Premature Infants Fed Human Milk. Am. J. Clin. Nutr. 1986, 43, 923–924. [Google Scholar] [CrossRef] [Green Version]
- Ochoa, J.J.; Ramirez-Tortosa, M.C.; Palomino, N.; Robles, R.; Mataix, J.; Huertas, J.R.; Quiles, J.L. Oxidative Stress in Erythrocytes from Premature and Full-Term Infants During Their First 72 h of Life. Free Radic. Res. 2003, 37, 317–322. [Google Scholar] [CrossRef] [Green Version]
- Autor, A.P.; Frank, L.; Roberts, R.J. Developmental Characteristics of Pulmonary Superoxide Dismutase: Relationship to Idiopathic Respiratory Distress Syndrome. Pediatric Res. 1976, 10, 154–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ofman, G.; Tipple, T.E. Antioxidants & Bronchopulmonary Dysplasia: Beating the System or Beating a Dead Horse? Free Radic. Biol. Med. 2019, 142, 138–145. [Google Scholar] [CrossRef]
- Rosenfeld, W.; Evans, H.; Concepcion, L.; Jhaveri, R.; Schaeffer, H.; Friedman, A. Prevention of Bronchopulmonary Dysplasia by Administration of Bovine Superoxide Dismutase in Preterm Infants with Respiratory Distress Syndrome. J. Pediatrics 1984, 105, 781–785. [Google Scholar] [CrossRef]
- Davis, J.M.; Parad, R.B.; Michele, T.; Allred, E.; Price, A.; Rosenfeld, W. Pulmonary Outcome at 1 Year Corrected Age in Premature Infants Treated at Birth With Recombinant Human CuZn Superoxide Dismutase. Pediatrics 2003, 111, 469–476. [Google Scholar] [CrossRef]
- Suresh, G.; Davis, J.M.; Soll, R. Superoxide Dismutase for Preventing Chronic Lung Disease in Mechanically Ventilated Preterm Infants. Cochrane Database Syst. Rev. 2001. [Google Scholar] [CrossRef]
- Buonocore, G.; Groenendaal, F. Anti-Oxidant Strategies. Semin. Fetal Neonatal Med. 2007, 12, 287–295. [Google Scholar] [CrossRef]
- Jr, C.A.S.; McEvoy, C.T.; Aschner, J.L.; Kirk, A.; Rosas-Salazar, C.; Cook-Mills, J.M.; Moore, P.E.; Walsh, W.F.; Hartert, T.V. Update on Vitamin E and Its Potential Role in Preventing or Treating Bronchopulmonary Dysplasia. NEO 2018, 113, 366–378. [Google Scholar] [CrossRef] [Green Version]
- Finkel, T. Signal Transduction by Reactive Oxygen Species. J. Cell Biol. 2011, 194, 7–15. [Google Scholar] [CrossRef] [Green Version]
- Marklund, S.L. Human Copper-Containing Superoxide Dismutase of High Molecular Weight. Proc. Natl. Acad. Sci. USA 1982, 79, 7634–7638. [Google Scholar] [CrossRef] [Green Version]
- Oury, T.D.; Day, B.J.; Crapo, J.D. Extracellular Superoxide Dismutase in Vessels and Airways of Humans and Baboons. Free Radic. Biol. Med. 1996, 20, 957–965. [Google Scholar] [CrossRef]
- Woo, H.A.; Yim, S.H.; Shin, D.H.; Kang, D.; Yu, D.-Y.; Rhee, S.G. Inactivation of Peroxiredoxin I by Phosphorylation Allows Localized H2O2 Accumulation for Cell Signaling. Cell 2010, 140, 517–528. [Google Scholar] [CrossRef] [Green Version]
- Delaney, C.; Wright, R.H.; Tang, J.-R.; Woods, C.; Villegas, L.; Sherlock, L.; Savani, R.C.; Abman, S.H.; Nozik-Grayck, E. Lack of EC-SOD Worsens Alveolar and Vascular Development in a Neonatal Mouse Model of Bleomycin-Induced Bronchopulmonary Dysplasia and Pulmonary Hypertension. Pediatr. Res. 2015, 78, 634–640. [Google Scholar] [CrossRef] [Green Version]
- Carlsson, L.M.; Jonsson, J.; Edlund, T.; Marklund, S.L. Mice Lacking Extracellular Superoxide Dismutase Are More Sensitive to Hyperoxia. Proc. Natl. Acad. Sci. USA 1995, 92, 6264–6268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, D.; Guo, H.; Xu, X.; Lu, Z.; Fassett, J.; Hu, X.; Xu, Y.; Tang, Q.; Hu, D.; Somani, A.; et al. Exacerbated Pulmonary Arterial Hypertension and Right Ventricular Hypertrophy in Animals with Loss of Function of Extracellular Superoxide Dismutase. Hypertension 2011, 58, 303–309. [Google Scholar] [CrossRef] [Green Version]
- Folz, R.J.; Abushamaa, A.M.; Suliman, H.B. Extracellular Superoxide Dismutase in the Airways of Transgenic Mice Reduces Inflammation and Attenuates Lung Toxicity Following Hyperoxia. J. Clin. Investig. 1999, 103, 1055–1066. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, M.N.; Suliman, H.B.; Folz, R.J.; Nozik-Grayck, E.; Golson, M.L.; Mason, S.N.; Auten, R.L. Extracellular Superoxide Dismutase Protects Lung Development in Hyperoxia-Exposed Newborn Mice. Am. J. Respir. Crit. Care Med. 2003, 167, 400–405. [Google Scholar] [CrossRef] [Green Version]
- Perveen, S.; Patel, H.; Arif, A.; Younis, S.; Codipilly, C.N.; Ahmed, M. Role of EC-SOD Overexpression in Preserving Pulmonary Angiogenesis Inhibited by Oxidative Stress. PLoS ONE 2012, 7, e51945. [Google Scholar] [CrossRef] [Green Version]
- Oury, T.D.; Day, B.J.; Crapo, J.D. Extracellular Superoxide Dismutase: A Regulator of Nitric Oxide Bioavailability. Lab. Invest. 1996, 75, 617–636. [Google Scholar]
- Brady, T.C.; Chang, L.Y.; Day, B.J.; Crapo, J.D. Extracellular Superoxide Dismutase Is Upregulated with Inducible Nitric Oxide Synthase after NF-Kappa B Activation. Am. J. Physiol. 1997, 273, L1002–L1006. [Google Scholar] [CrossRef]
- Friebe, A.; Schultz, G.; Koesling, D. Stimulation of Soluble Guanylate Cyclase by Superoxide Dismutase Is Mediated by NO. Biochem. J. 1998, 335 Pt 3, 527–531. [Google Scholar] [CrossRef] [Green Version]
- Farrow, K.N.; Lakshminrusimha, S.; Reda, W.J.; Wedgwood, S.; Czech, L.; Gugino, S.F.; Davis, J.M.; Russell, J.A.; Steinhorn, R.H. Superoxide Dismutase Restores ENOS Expression and Function in Resistance Pulmonary Arteries from Neonatal Lambs with Persistent Pulmonary Hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 295, L979–L987. [Google Scholar] [CrossRef] [Green Version]
- Young, S.L.; Evans, K.; Eu, J.P. Nitric Oxide Modulates Branching Morphogenesis in Fetal Rat Lung Explants. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002, 282, L379–L385. [Google Scholar] [CrossRef] [Green Version]
- Han, R.N.N.; Stewart, D.J. Defective Lung Vascular Development in Endothelial Nitric Oxide Synthase-Deficient Mice. Trends Cardiovasc. Med. 2006, 16, 29–34. [Google Scholar] [CrossRef]
- Bachiller, P.R.; Cornog, K.H.; Kato, R.; Buys, E.S.; Roberts, J.D. Soluble Guanylate Cyclase Modulates Alveolarization in the Newborn Lung. Am. J. Physiol. Lung Cell Mol. Physiol. 2013, 305, L569–L581. [Google Scholar] [CrossRef] [Green Version]
- Parton, R.G.; del Pozo, M.A. Caveolae as Plasma Membrane Sensors, Protectors and Organizers. Nat. Rev. Mol. Cell Biol. 2013, 14, 98–112. [Google Scholar] [CrossRef]
- Ahmed, M.N.; Codipilly, C.; Hogg, N.; Auten, R.L. The Protective Effect of Overexpression of Extracellular Superoxide Dismutase on Nitric Oxide Bioavailability in the Lung after Exposure to Hyperoxia Stress. Exp. Lung Res. 2011, 37, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Oshikawa, J.; Urao, N.; Kim, H.W.; Kaplan, N.; Razvi, M.; McKinney, R.; Poole, L.B.; Fukai, T.; Ushio-Fukai, M. Extracellular SOD-Derived H2O2 Promotes VEGF Signaling in Caveolae/Lipid Rafts and Post-Ischemic Angiogenesis in Mice. PLoS ONE 2010, 5, e10189. [Google Scholar] [CrossRef] [Green Version]
- Shen, B.-Q.; Lee, D.Y.; Zioncheck, T.F. Vascular Endothelial Growth Factor Governs Endothelial Nitric-Oxide Synthase Expression via a KDR/Flk-1 Receptor and a Protein Kinase C Signaling Pathway*. J. Biol. Chem. 1999, 274, 33057–33063. [Google Scholar] [CrossRef] [Green Version]
- Simons, M.; Gordon, E.; Claesson-Welsh, L. Mechanisms and Regulation of Endothelial VEGF Receptor Signalling. Nat. Rev. Mol. Cell Biol. 2016, 17, 611–625. [Google Scholar] [CrossRef]
- Archer, S.L.; Johnson, G.J.; Gebhard, R.L.; Castleman, W.L.; Levine, A.S.; Westcott, J.Y.; Voelkel, N.F.; Nelson, D.P.; Weir, E.K. Effect of Dietary Fish Oil on Lung Lipid Profile and Hypoxic Pulmonary Hypertension. J. Appl. Physiol. 1989, 66, 1662–1673. [Google Scholar] [CrossRef]
- Perez, M.; Lee, K.J.; Cardona, H.J.; Taylor, J.M.; Robbins, M.E.; Waypa, G.B.; Berkelhamer, S.K.; Farrow, K.N. Aberrant CGMP Signaling Persists during Recovery in Mice with Oxygen-Induced Pulmonary Hypertension. PLoS ONE 2017, 12, e0180957. [Google Scholar] [CrossRef] [Green Version]
- Bradford, M.M. A Rapid and Sensitive Method for the Quantitation of Microgram Quantities of Protein Utilizing the Principle of Protein-Dye Binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Gupta, A.; Perez, M.; Lee, K.J.; Taylor, J.M.; Farrow, K.N. SOD2 Activity Is Not Impacted by Hyperoxia in Murine Neonatal Pulmonary Artery Smooth Muscle Cells and Mice. Int. J. Mol. Sci. 2015, 16, 6373–6390. [Google Scholar] [CrossRef] [Green Version]
- Datta, A.; Kim, G.A.; Taylor, J.M.; Gugino, S.F.; Farrow, K.N.; Schumacker, P.T.; Berkelhamer, S.K. Mouse Lung Development and NOX1 Induction during Hyperoxia Are Developmentally Regulated and Mitochondrial ROS Dependent. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 309, L369–L377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherlock, L.G.; Trumpie, A.; Hernandez-Lagunas, L.; McKenna, S.; Fisher, S.; Bowler, R.; Wright, C.J.; Delaney, C.; Nozik-Grayck, E. Redistribution of Extracellular Superoxide Dismutase Causes Neonatal Pulmonary Vascular Remodeling and PH but Protects Against Experimental Bronchopulmonary Dysplasia. Antioxidants 2018, 7, 42. [Google Scholar] [CrossRef] [Green Version]
- Yamakura, F.; Kawasaki, H. Post-Translational Modifications of Superoxide Dismutase. Biochim. Biophys. Acta (BBA)–Proteins Proteom. 2010, 1804, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q. Role of Nrf2 in Oxidative Stress and Toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [Green Version]
- Ma, D.; Gao, W.; Liu, J.; Kong, D.; Zhang, Y.; Qian, M. Mechanism of Oxidative Stress and Keap-1/Nrf2 Signaling Pathway in Bronchopulmonary Dysplasia. Medicine (Baltimore) 2020, 99. [Google Scholar] [CrossRef]
- Gerber, H.-P.; McMurtrey, A.; Kowalski, J.; Yan, M.; Keyt, B.A.; Dixit, V.; Ferrara, N. Vascular Endothelial Growth Factor Regulates Endothelial Cell Survival through the Phosphatidylinositol 3′-Kinase/Akt Signal Transduction Pathway: REQUIREMENT FOR Flk-1/KDR ACTIVATION*. J. Biol. Chem. 1998, 273, 30336–30343. [Google Scholar] [CrossRef] [Green Version]
- Brown, K.R.S.; England, K.M.; Goss, K.L.; Snyder, J.M.; Acarregui, M.J. VEGF Induces Airway Epithelial Cell Proliferation in Human Fetal Lung in Vitro. Am. J. Physiol. Lung Cell. Mol. Physiol. 2001, 281, L1001–L1010. [Google Scholar] [CrossRef]
- Hong, Y.A.; Lim, J.H.; Kim, M.Y.; Kim, Y.; Park, H.S.; Kim, H.W.; Choi, B.S.; Chang, Y.S.; Kim, H.W.; Kim, T.-Y.; et al. Extracellular Superoxide Dismutase Attenuates Renal Oxidative Stress Through the Activation of Adenosine Monophosphate-Activated Protein Kinase in Diabetic Nephropathy. Antioxid. Redox Signal. 2017, 28, 1543–1561. [Google Scholar] [CrossRef]
- Wert, K.J.; Velez, G.; Cross, M.R.; Wagner, B.A.; Teoh-Fitzgerald, M.L.; Buettner, G.R.; McAnany, J.J.; Olivier, A.; Tsang, S.H.; Harper, M.M.; et al. Extracellular Superoxide Dismutase (SOD3) Regulates Oxidative Stress at the Vitreoretinal Interface. Free Radic Biol. Med. 2018, 124, 408–419. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.W.; Lin, A.; Guldberg, R.E.; Ushio-Fukai, M.; Fukai, T. Essential Role of Extracellular SOD in Reparative Neovascularization Induced by Hindlimb Ischemia. Circ. Res. 2007, 101, 409–419. [Google Scholar] [CrossRef] [Green Version]
- Förstermann, U.; Sessa, W.C. Nitric Oxide Synthases: Regulation and Function. Eur Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mathias, M.; Taylor, J.; Mendralla, E.; Perez, M. Neonatal Extracellular Superoxide Dismutase Knockout Mice Increase Total Superoxide Dismutase Activity and VEGF Expression after Chronic Hyperoxia. Antioxidants 2021, 10, 1236. https://doi.org/10.3390/antiox10081236
Mathias M, Taylor J, Mendralla E, Perez M. Neonatal Extracellular Superoxide Dismutase Knockout Mice Increase Total Superoxide Dismutase Activity and VEGF Expression after Chronic Hyperoxia. Antioxidants. 2021; 10(8):1236. https://doi.org/10.3390/antiox10081236
Chicago/Turabian StyleMathias, Maxwell, Joann Taylor, Elizabeth Mendralla, and Marta Perez. 2021. "Neonatal Extracellular Superoxide Dismutase Knockout Mice Increase Total Superoxide Dismutase Activity and VEGF Expression after Chronic Hyperoxia" Antioxidants 10, no. 8: 1236. https://doi.org/10.3390/antiox10081236
APA StyleMathias, M., Taylor, J., Mendralla, E., & Perez, M. (2021). Neonatal Extracellular Superoxide Dismutase Knockout Mice Increase Total Superoxide Dismutase Activity and VEGF Expression after Chronic Hyperoxia. Antioxidants, 10(8), 1236. https://doi.org/10.3390/antiox10081236