
Oxidative Stress as a Therapeutic Target of Cardiac Remodeling

 , , , , , ,
, , , , , , 
Abstract
:
{kind=link}
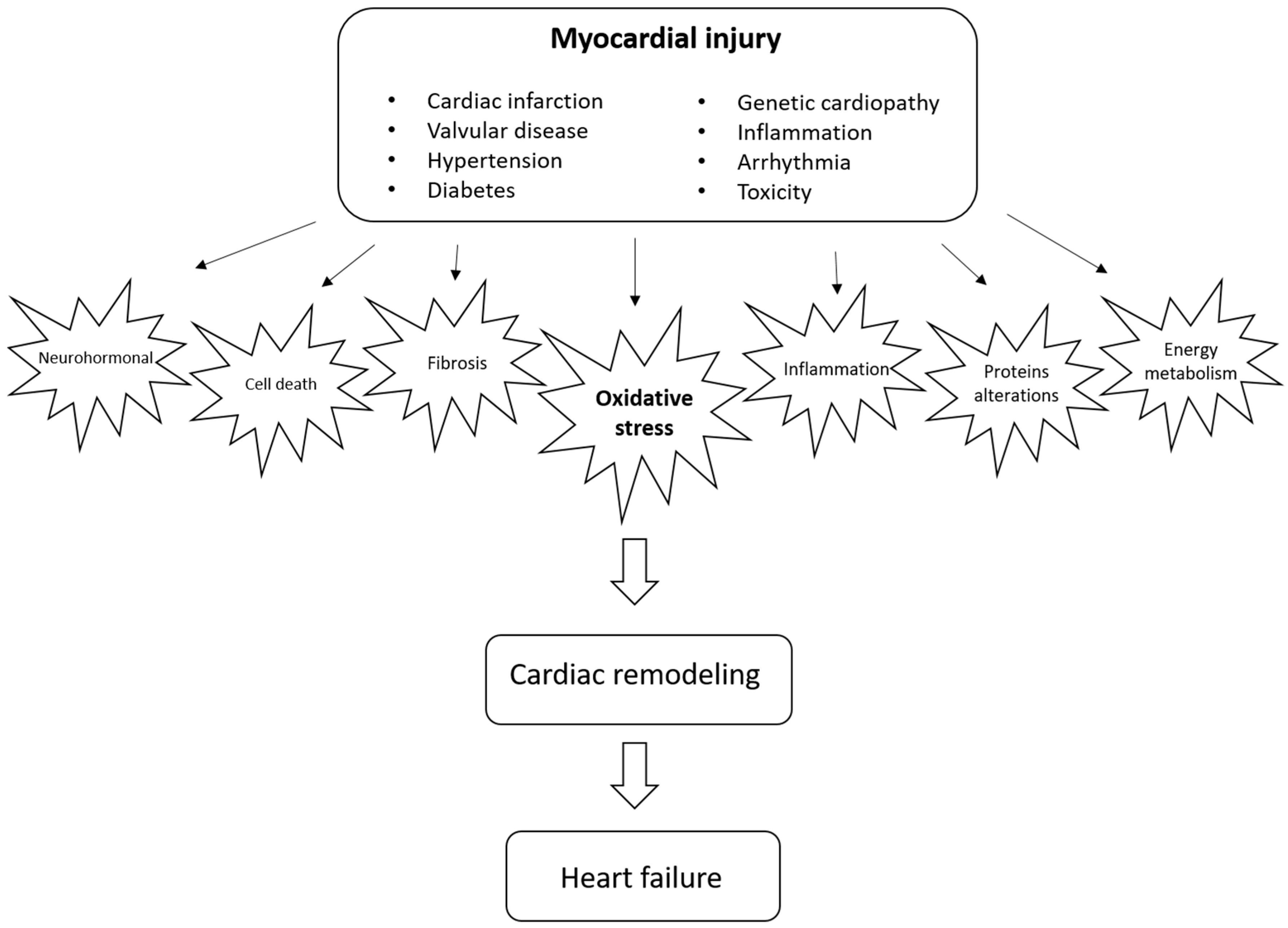{kind=link}
1. Introduction
2. Cardiac Remodeling
3. Oxidative Stress as a Potential Modulator of Cardiac Remodeling
4. Evidence of Oxidative Stress as a Therapeutic Target against Cardiac Remodeling
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Azevedo, P.S.; Polegato, B.F.; Minicucci, M.F.; Paiva, S.A.R.; Zornoff, L.A.M. Cardiac remodeling: Concepts, clinical impact, pathophysiological mechanisms and pharmacologic treatment. Arq. Bras. Cardiol. 2016, 106, 62–69. [Google Scholar] [CrossRef]
- Yang, D.; Liu, H.Q.; Liu, F.Y.; Tang, N.; Guo, Z.; Ma, S.Q.; An, P.; Wang, M.Y.; Wu, H.M.; Yang, Z.; et al. The roles of noncardiomyocytes in cardiac remodeling. Int. J. Biol. Sci. 2020, 16, 2414–2429. [Google Scholar] [CrossRef]
- Leancă, S.A.; Crișu, D.; Petriș, A.O.; Afrăsânie, I.; Genes, A.; Costache, A.D.; Tesloianu, D.N.; Costache, I.I. Left ventricular remodeling after myocardial infarction: From physiopathology to treatment. Life 2022, 12, 1111. [Google Scholar] [CrossRef]
- Shah, A.K.; Bhullar, S.K.; Elimban, V.; Dhalla, N.S. Oxidative stress as a mechanism for functional alterations in cardiac hypertrophy and heart failure. Antioxidants 2021, 10, 931. [Google Scholar] [CrossRef]
- Ramachandra, C.J.A.; Cong, S.; Chan, X.; Yap, E.P.; Yu, F.; Hausenloy, D.J. Oxidative stress in cardiac hypertrophy: From molecular mechanisms to novel therapeutic targets. Free Radic. Biol. Med. 2021, 166, 297–312. [Google Scholar] [CrossRef]
- Tsutsui, H.; Kinugawa, S.; Matsushima, S. Oxidative stress and heart failure. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H2181–H2190. [Google Scholar] [CrossRef] [Green Version]
- Pisoschi, A.M.; Pop, A. The role of antioxidants in the chemistry of oxidative stress: A review. Eur. J. Med. Chem. 2015, 97, 55–74. [Google Scholar] [CrossRef]
- Halliwell, B.; Gutteridge, J.M.; Cross, C.E. Free radicals, antioxidants, and human disease: Where are we now? J. Lab. Clin. Med. 1992, 119, 598–620. [Google Scholar]
- Riley, P.A. Free radicals in biology: Oxidative stress and the effects of ionizing radiation. Int. J. Radiat. Biol. 1994, 65, 27–33. [Google Scholar] [CrossRef]
- López-Alarcón, C.; Denicola, A. Evaluating the antioxidant capacity of natural products: A review on chemical and cellular-based assays. Anal. Chim. Acta 2013, 763, 1–10. [Google Scholar] [CrossRef]
- Evans, J.L.; Goldfine, I.D.; Maddux, B.A.; Grodsky, G.M. Are oxidative stress–activated signaling pathways mediators of insulin resistance and β-Cell dysfunction? Diabetes 2003, 52, 1–8. [Google Scholar] [CrossRef]
- Gutteridge, J.M.C. Biological origin of free radicals, and mechanisms of antioxidant protection. Chem. Biol. Interact. 1994, 91, 133–140. [Google Scholar] [CrossRef]
- Hou, Y.C.; Janczuk, A.; Wang, P.G. Current trends in the development of nitric oxide donors. Curr. Pharm. Des. 1999, 5, 417–441. [Google Scholar]
- Goldblum, R.R.; McClellan, M.; White, K.; Gonzalez, S.J.; Thompson, B.R.; Vang, H.X.; Cohen, H.; Higgins, L.; Markowski, T.W.; Yang, T.Y.; et al. Oxidative stress pathogenically remodels the cardiac myocyte cytoskeleton via structural alterations to the microtubule lattice. Dev. Cell 2021, 56, 2252–2266. [Google Scholar] [CrossRef]
- Senoner, T.; Dichtl, W. Oxidative stress in cardiovascular diseases: Still a therapeutic target? Nutrients 2019, 11, 2090. [Google Scholar] [CrossRef] [Green Version]
- Sabri, A.; Hughie, H.H.; Lucchesi, P.A. Regulation of hypertrophic and apoptotic signaling pathways by reactive oxygen species in cardiac myocytes. Antioxid. Redox Signal. 2003, 5, 731–740. [Google Scholar] [CrossRef]
- Karamanos, N.K.; Theocharis, A.D.; Piperigkou, Z.; Manou, D.; Passi, A.; Skandalis, S.S.; Vynios, D.H.; Orian-Rousseau, V.; Ricard-Blum, S.; Schmelzer, C.E.H.; et al. A guide to the composition and functions of the extracellular matrix. FEBS J. 2021, 288, 6850–6912. [Google Scholar] [CrossRef]
- Hayashidani, S.; Tsutsui, H.; Ikeuchi, M.; Shiomi, T.; Matsusaka, H.; Kubota, T.; Iamanaka-Yoshida, K.; Itoh, T.; Takeshita, A. Targeted deletion of MMP-2 attenuates early LV rupture and late remodeling after experimental myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H1229–H1235. [Google Scholar] [CrossRef] [Green Version]
- Stones, R.; Benoist, D.; Peckham, M.; White, E. Microtubule proliferation in right ventricular myocytes of rats with monocrotaline-induced pulmonary hypertension. J. Mol. Cell Cardiol. 2013, 56, 91–96. [Google Scholar] [CrossRef] [Green Version]
- Ohi, R.; Zanic, M. Ahead of the curve: New insights into microtubule dynamics. F1000Research 2016, 5, 314. [Google Scholar] [CrossRef]
- van der Pol, A.; van Gilst, W.H.; Voors, A.A.; van der Meer, P. Treating oxidative stress in heart failure: Past, present and future. Eur. J. Heart Fail. 2019, 21, 425–435. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, H.; Kurebayashi, N.; Yamazawa, T.; Murayama, T. Regulatory mechanisms of ryanodine receptor/Ca2+ release channel revealed by recent advancements in structural studies. J. Muscle Res. Cell Motil. 2021, 42, 291–304. [Google Scholar] [CrossRef]
- Kourie, J.I. Interaction of reactive oxygen species with ion transport mechanisms. Am. J. Physiol. 1998, 275, C1–C24. [Google Scholar] [CrossRef]
- Kawakami, M.; Okabe, E. Superoxide anion radical-triggered Ca2+ release from cardiac sarcoplasmic reticulum through ryanodine receptor Ca2+ channel. Mol. Pharmacol. 1998, 53, 497–503. [Google Scholar] [CrossRef] [Green Version]
- Spinale, F.G. Bioactive peptide signaling within the myocardial interstitium and the matrix metalloproteinases. Circ. Res. 2002, 91, 1082–1084. [Google Scholar] [CrossRef] [Green Version]
- Mitra, A.; Datta, R.; Rana, S.; Sarkar, S. Modulation of NFKB1/p50 by ROS leads to impaired ATP production during MI compared to cardiac hypertrophy. J. Cell Biochem. 2018, 119, 1575–1590. [Google Scholar] [CrossRef]
- Kang, D.; Hamasaki, N. Mitochondrial transcription factor A in the maintenance of mitochondrial DNA: Overview of its multiple roles. Ann. N. Y. Acad. Sci. 2005, 1042, 101–108. [Google Scholar] [CrossRef]
- Ikeuchi, M.; Matsusaka, H.; Kang, D.; Matsushima, S.; Ide, T.; Kubota, T.; Fujiwara, T.; Hamasaki, N.; Takeshita, A.; Sunagawa, K.; et al. Overexpression of mitochondrial transcription factor a ameliorates mitochondrial deficiencies and cardiac failure after myocardial infarction. Circulation 2005, 112, 683–690. [Google Scholar] [CrossRef] [Green Version]
- Li, J.M.; Gall, N.P.; Grieve, D.J.; Chen, M.; Shah, A.M. Activation of NADPH oxidase during progression of cardiac hypertrophy to failure. Hypertension 2002, 40, 477–484. [Google Scholar] [CrossRef] [Green Version]
- Fiorillo, C.; Nediani, C.; Ponziani, V.; Giannini, L.; Celli, A.; Nassi, N.; Formigli, L.; Perna, A.M.; Nassi, P. Cardiac volume overload rapidly induces oxidative stress-mediated myocyte apoptosis and hypertrophy. Biochim. Biophys. Acta 2005, 1741, 173–182. [Google Scholar] [CrossRef] [Green Version]
- Pagan, L.U.; Gomes, M.J.; Gatto, M.; Mota, G.A.F.; Okoshi, K.; Okoshi, M.P. The role of oxidative stress in the aging heart. Antioxidants 2022, 11, 336. [Google Scholar] [CrossRef]
- Mayyas, F.; Aldawod, H.; Alzoubi, K.H.; Khabour, O.; Shihadeh, A.; Eissenberg, T. Comparison of the cardiac effects of electronic cigarette aerosol exposure with waterpipe and combustible cigarette smoke exposure in rats. Life Sci. 2020, 251, 117644. [Google Scholar] [CrossRef]
- Tsutsui, H.; Ide, T.; Hayashidani, S.; Kinugawa, S.; Suematsu, N.; Utsumi, H.; Takeshita, A. Effects of ACE inhibition on left ventricular failure and oxidative stress in dahl salt-sensitive rats. J. Cardiovasc. Pharmacol. 2001, 37, 725–733. [Google Scholar] [CrossRef]
- Savic-Radojevic, A.; Pljesa-Ercegovac, M.; Matic, M.; Simic, D.; Radovanovic, S.; Simic, T. Novel biomarkers of heart failure. Adv. Clin. Chem. 2017, 79, 93–152. [Google Scholar]
- Tang, W.H.W.; Tong, W.; Troughton, R.W.; Martin, M.G.; Shrestha, K.; Borowski, A.; Jasper, S.; Hazen, S.L.; Klein, A.L. Prognostic value and echocardiographic determinants of plasma myeloperoxidase levels in chronic heart failure. J. Am. Coll. Cardiol. 2007, 49, 2364–2370. [Google Scholar] [CrossRef] [Green Version]
- Tromp, J.; Khan, M.A.F.; Mentz, R.J.; O’Connor, C.M.; Metra, M.; Dittrich, H.C.; Ponikowski, P.; Teeerlink, J.R.; Cotteer, G.; Davison, B.; et al. Biomarker profiles of acute heart failure patients with a mid-range ejection fraction. JACC Heart Fail. 2017, 5, 507–517. [Google Scholar] [CrossRef]
- Wang, H.; Chen, Q.; Li, Y.; Jing, X.; Yang, J. Prognostic value of growth differentiation factor-15 in Chinese patients with heart failure: A prospective observational study. Cardiol. J. 2018, 25, 245–253. [Google Scholar] [CrossRef] [Green Version]
- Yamagami, F.; Tajiri, K.; Doki, K.; Hattori, M.; Honda, J.; Aita, S.; Harunari, T.; Yamasaki, H.; Murakoshi, N.; Sekiguchi, Y.; et al. Indoxyl sulphate is associated with atrial fibrillation recurrence after catheter ablation. Sci. Rep. 2018, 8, 17276. [Google Scholar] [CrossRef] [Green Version]
- Wollert, K.C.; Kempf, T. Growth differentiation factor 15 in heart failure: An update. Curr. Heart Fail. Rep. 2012, 9, 337–345. [Google Scholar] [CrossRef]
- Dou, L.; Jourde-Chiche, N.; Faure, V.; Cerini, C.; Berland, Y.; Dignat-George, F.; Brunet, P. The uremic solute indoxyl sulfate induces oxidative stress in endothelial cells. J. Thromb. Haemost. 2007, 5, 1302–1308. [Google Scholar] [CrossRef]
- Yang, P.S.; Kim, T.; Uhm, J.S.; Park, S.; Joung, B.; Lee, M.H.; Pak, H.N. High plasma level of soluble RAGE is independently associated with a low recurrence of atrial fibrillation after catheter ablation in diabetic patient. Europace 2016, 18, 1711–1718. [Google Scholar] [CrossRef]
- Goidescu, C.M.; Chiorescu, R.M.; Diana, M.H.L.; Mocan, M.; Stoia, M.A.; Anton, F.P.; Farcas, A.D. ACE2 and Apelin-13: Biomarkers with a prognostic value in congestive heart failure. Dis. Markers 2021, 2021, 5569410. [Google Scholar] [CrossRef]
- Vilhena, J.C.; Cunha, L.L.M.; Jorge, T.M.; Machado, M.L.; Soares, R.A.; Santos, I.B.; Bem, G.F.; Fernandes-Santos, C.; Ognibene, D.T.; Moura, R.S.; et al. Açaí reverses adverse cardiovascular remodeling in renovascular hypertension: A comparative effect with Enalapril. J. Cardiovasc. Pharmacol. 2021, 77, 673–684. [Google Scholar] [CrossRef]
- Arnoso, B.J.M.; Magliaccio, F.M.; Araújo, C.A.; Soares, R.A.; Santos, I.B.; Bem, G.F.; Fernandes-Santos, C.; Ognibene, D.T.; Moura, R.S.; Resende, A.C.; et al. Açaí seed extract (ASE) rich in proanthocyanidins improves cardiovascular remodeling by increasing antioxidant response in obese high-fat diet-fed mice. Chem. Biol. Interact. 2022, 351, 109721. [Google Scholar]
- Figueiredo, A.M.; Cardoso, A.C.; Pereira, B.L.B.; Silva, R.A.C.; Ripa, A.F.G.D.; Pinelli, T.F.B.; Oliveira, B.C.; Rafacho, B.P.M.; Ishikawa, L.I.W.; Azevedo, P.S.; et al. Açai supplementation (Euterpe oleracea Mart.) attenuates cardiac remodeling after myocardial infarction in rats through different mechanistic pathways. PLoS ONE 2022, 17, e0264854. [Google Scholar] [CrossRef]
- Oliveira, B.C.; Santos, P.P.; Figueiredo, A.M.; Rafacho, B.P.M.; Ishikawa, L.; Zanati, S.G.; Fernandes, A.A.H.; Azevedo, P.S.; Polegato, B.F.; Zornoff, L.A.M.; et al. Influência do consumo de suco de laranja (Citrus sinensis) na remodelação cardíaca de ratos submetidos a infarto do miocárdio. Arq. Bras. Cardiol. 2021, 116, 1127–1136. [Google Scholar] [CrossRef]
- Ruan, Y.; Jin, Q.; Zeng, J.; Ren, F.; Xie, Z.; Ji, K.; Wu, L.; Wu, J.; Li, L. Grape seed proanthocyanidin extract ameliorates cardiac remodelling after myocardial infarction through PI3K/AKT pathway in Mice. Front. Pharmacol. 2020, 11, 585984. [Google Scholar] [CrossRef]
- Silva, R.C.; Polegato, B.F.; Azevedo, P.S.; Fernandes, A.A.; Okoshi, K.; Paiva, S.A.R.; Minicucci, M.F.; Zornoff, L.A.M. Jaboticaba (Myrciaria jaboticaba) attenuates ventricular remodeling after myocardial infarction in rats. Antioxidants 2022, 11, 249. [Google Scholar] [CrossRef]
- Zivarpour, P.; Reiner, Ž.; Hallajzadeh, J.; Mirsafaei, L. Resveratrol and cardiac fibrosis prevention and treatment. Curr. Pharm. Biotechnol. 2022, 23, 190–200. [Google Scholar] [CrossRef]
- Morris, G.; Anderson, G.; Dean, O.; Berk, M.; Galecki, P.; Martin-Subero, M.; Maes, M. The glutathione system: A new drug arget in neuroimmune disorders. Mol. Neurobiol. 2014, 50, 1059–1084. [Google Scholar] [CrossRef]
- Tanzilli, G.; Arrivi, A.; Placanica, A.; Viceconte, N.; Cammisotto, V.; Nocella, C.; Barillà, F.; Torromeo, C.; Pucci, G.; Acconcia, M.C.; et al. Glutathione infusion before and 3 days after primary angioplasty blunts ongoing NOX2-mediated inflammatory response. J. Am. Heart Assoc. 2021, 10, e020560. [Google Scholar] [CrossRef] [PubMed]
- Gal, R.; Deres, L.; Toth, K.; Halmosi, R.; Habon, T. The effect of resveratrol on the cardiovascular system from molecular mechanisms to clinical results. Int. J. Mol. Sci. 2021, 22, 10152. [Google Scholar] [CrossRef] [PubMed]
- Raj, P.; Sayfee, K.; Parikh, M.; Yu, L.; Wigle, J.; Netticadan, T.; Zieroth, S. Comparative and combinatorial effects of resveratrol and sacubitril/valsartan alongside valsartan on cardiac remodeling and dysfunction in MI-induced rats. Molecules 2021, 26, 5006. [Google Scholar] [CrossRef] [PubMed]
- Magyar, K.; Halmosi, R.; Palfi, A.; Feher, G.; Czopf, L.; Fulop, A.; Battyany, i.; Sumego, B.; Toth, K.; Szabados, E. Cardioprotection by resveratrol: A human clinical trial in patients with stable coronary artery disease. Clin. Hemorheol. Microcirc. 2012, 50, 179–187. [Google Scholar] [CrossRef]
- Gal, R.; Deres, L.; Horvath, O.; Eros, K.; Sandor, B.; Urban, P.; Soos, S.; Marton, Z.; Sumegi, B.; Toth, K.; et al. Resveratrol improves heart function by moderating inflammatory processes in patients with systolic heart failure. Antioxidants 2020, 9, 1108. [Google Scholar] [CrossRef]
- Guimarães, S.S.; Cruz, W.S.; Silva, L.; Maciel, G.; Huguenin, A.B.; Carvalho, M.; Costa, B.; Silva, G.; Costa, C.; D’Ippolito, J.A.; et al. Effect of L-carnitine supplementation on reverse remodeling in patients with ischemic heart disease undergoing coronary artery bypass grafting: A randomized, placebo-controlled trial. Ann. Nutr. Metab. 2017, 70, 106–110. [Google Scholar] [CrossRef]
- van Straaten, H.M.; Man, A.M.S.; Waard, M.C. Vitamin C revisited. Crit. Care 2014, 18, 460. [Google Scholar]
- Malik, A.; Bagchi, A.K.; Vinayak, K.; Akolkar, G.; Slezak, J.; Belló-Klein, A.; Jassal, D.S.; Singal, P.K. Vitamin C: Historical perspectives and heart failure. Heart Fail. Rev. 2021, 26, 699–709. [Google Scholar] [CrossRef]
- Akolkar, G.; Dias, D.S.; Ayyappan, P.; Bagchi, A.K.; Jassal, D.S.; Salemi, V.M.C.; Irigoyen, M.C.; Angelis, K.; Singal, P.K. Vitamin C mitigates oxidative/nitrosative stress and inflammation in doxorubicin-induced cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H795–H809. [Google Scholar] [CrossRef]
- Novaes, R.D.; Santos, E.C.; Fialho, M.D.C.Q.; Gonçalves, W.G.; Sequetto, P.L.; Talvani, A.; Gonçalves, R.V. Nonsteroidal anti-inflammatory is more effective than anti-oxidant therapy in counteracting oxidative/nitrosative stress and heart disease in T. cruzi -infected mice. Parasitology 2017, 144, 904–916. [Google Scholar] [CrossRef]
- Antonic, M.; Lipovec, R.; Gregorcic, F.; Juric, P.; Kosir, G. Perioperative ascorbic acid supplementation does not reduce the incidence of postoperative atrial fibrillation in on-pump coronary artery bypass graft patients. J. Cardiol. 2017, 69, 98–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Ding, Z.; Yang, F.; Dai, Y.; Chen, P.; Theus, S.; Singh, S.; Budhiraja, M.; Mehta, J.L. Modulation of myocardial injury and collagen deposition following ischaemia–reperfusion by linagliptin and liraglutide, and both together. Clin. Sci. 2016, 130, 1353–1362. [Google Scholar] [CrossRef] [PubMed]
- Koniari, I.; Velissaris, D.; Kounis, N.G.; Koufou, E.; Artopoulou, E.; Gregorio, C.; Mplani, V.; Paraskevas, T.; Tsigkas, G.; Hung, M.Y.; et al. Anti-diabetic therapy, heart failure and oxidative stress: An update. J. Clin. Med. 2022, 11, 4660. [Google Scholar] [CrossRef] [PubMed]
- Bigagli, E.; Luceri, C.; Dicembrini, I.; Tatti, L.; Scavone, F.; Giovannelli, L.; Mannucci, E.; Lodovici, M. Effect of dipeptidyl-peptidase 4 inhibitors on circulating oxidative stress biomarkers in patients with type 2 diabetes mellitus. Antioxidants 2020, 9, 233. [Google Scholar] [CrossRef] [Green Version]
- Heidenreich, P.A.; Bozkurt, B.; Aguilar, D.; Allen, L.A.; Byun, J.J.; Colvin, M.M.; Deswal, A.; Drazner, M.H.; Dunlay, S.M.; Evers, L.R.; et al. 2022 AHA/ACC/HFSA guideline for the management of heart failure: Executive summary: A report of the american college of cardiology/american heart association joint committee on clinical practice guidelines. Circulation 2022, 145, e876–e894. [Google Scholar] [CrossRef]
- Braunwald, E. Gliflozins in the management of cardiovascular disease. N. Engl. J. Med. 2022, 386, 2024–2034. [Google Scholar] [CrossRef]
- Gager, G.M.; von Lewinski, D.; Sourij, H.; Jilma, B.; Eyileten, C.; Filipiak, K.; Hulsmann, M.; Kubica, J.; Postula, M.; Siller-Matula, J.M. Effects of SGLT2 inhibitors on ion homeostasis and oxidative stress associated mechanisms in heart failure. Biomed. Pharmacother. 2021, 143, 112169. [Google Scholar] [CrossRef]
- Cowie, M.R.; Fisher, M. SGLT2 inhibitors: Mechanisms of cardiovascular benefit beyond glycaemic control. Nat. Rev. Cardiol. 2020, 17, 761–772. [Google Scholar] [CrossRef]
- Li, C.; Zhang, J.; Xue, M.; Li, X.; Han, F.; Liu, X.; Xu, L.; Lu, Y.; Cheng, Y.; Li, T.; et al. SGLT2 inhibition with empagliflozin attenuates myocardial oxidative stress and fibrosis in diabetic mice heart. Cardiovasc. Diabetol. 2019, 18, 15. [Google Scholar] [CrossRef]
- Kondo, H.; Akoumianakis, I.; Badi, I.; Akawi, N.; Kotanidis, C.P.; Polkinghorne, M.; Stadiotti, I.; Sommariva, E.; Antonopoulos, A.S.; Carena, M.C.; et al. Effects of canagliflozin on human myocardial redox signalling: Clinical implications. Eur. Heart J. 2021, 42, 4947–4960. [Google Scholar] [CrossRef]
- Mohan, M.; Al-Talabany, S.; McKinnie, A.; Mordi, I.R.; Singh, J.S.S.; Gandy, S.J.; Baig, F.; Hussain, M.S.; Bhalraam, U.; Khan, F.; et al. A randomized controlled trial of metformin on left ventricular hypertrophy in patients with coronary artery disease without diabetes: The MET-REMODEL trial. Eur. Heart J. 2019, 40, 3409–3417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vannuccini, F.; Campora, A.; Barilli, A.; Palazzuol, A. Vericiguat in heart failure: Characteristics, scientific evidence and potential clinical applications. Biomedicines 2022, 10, 2471. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, P.W.; Pieske, B.; Anstrom, K.J.; Ezekowitz, J.; Hernandez, A.F.; Butler, J.; Lam, C.S.; Ponikowski, P.; Voors, A.A.; Jia, G.; et al. Vericiguat in patients with heart failure and reduced ejection fraction. N. Engl. J. Med. 2020, 382, 1883–1893. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martins, D.; Garcia, L.R.; Queiroz, D.A.R.; Lazzarin, T.; Tonon, C.R.; Balin, P.d.S.; Polegato, B.F.; de Paiva, S.A.R.; Azevedo, P.S.; Minicucci, M.F.; et al. Oxidative Stress as a Therapeutic Target of Cardiac Remodeling. Antioxidants 2022, 11, 2371. https://doi.org/10.3390/antiox11122371
Martins D, Garcia LR, Queiroz DAR, Lazzarin T, Tonon CR, Balin PdS, Polegato BF, de Paiva SAR, Azevedo PS, Minicucci MF, et al. Oxidative Stress as a Therapeutic Target of Cardiac Remodeling. Antioxidants. 2022; 11(12):2371. https://doi.org/10.3390/antiox11122371
Chicago/Turabian StyleMartins, Danilo, Leonardo Rufino Garcia, Diego Aparecido Rios Queiroz, Taline Lazzarin, Carolina Rodrigues Tonon, Paola da Silva Balin, Bertha Furlan Polegato, Sergio Alberto Rupp de Paiva, Paula Schmidt Azevedo, Marcos Ferreira Minicucci, and et al. 2022. "Oxidative Stress as a Therapeutic Target of Cardiac Remodeling" Antioxidants 11, no. 12: 2371. https://doi.org/10.3390/antiox11122371
APA StyleMartins, D., Garcia, L. R., Queiroz, D. A. R., Lazzarin, T., Tonon, C. R., Balin, P. d. S., Polegato, B. F., de Paiva, S. A. R., Azevedo, P. S., Minicucci, M. F., & Zornoff, L. (2022). Oxidative Stress as a Therapeutic Target of Cardiac Remodeling. Antioxidants, 11(12), 2371. https://doi.org/10.3390/antiox11122371