
HNO Protects the Myocardium against Reperfusion Injury, Inhibiting the mPTP Opening via PKCε Activation

 ,
,  ,
, 
 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
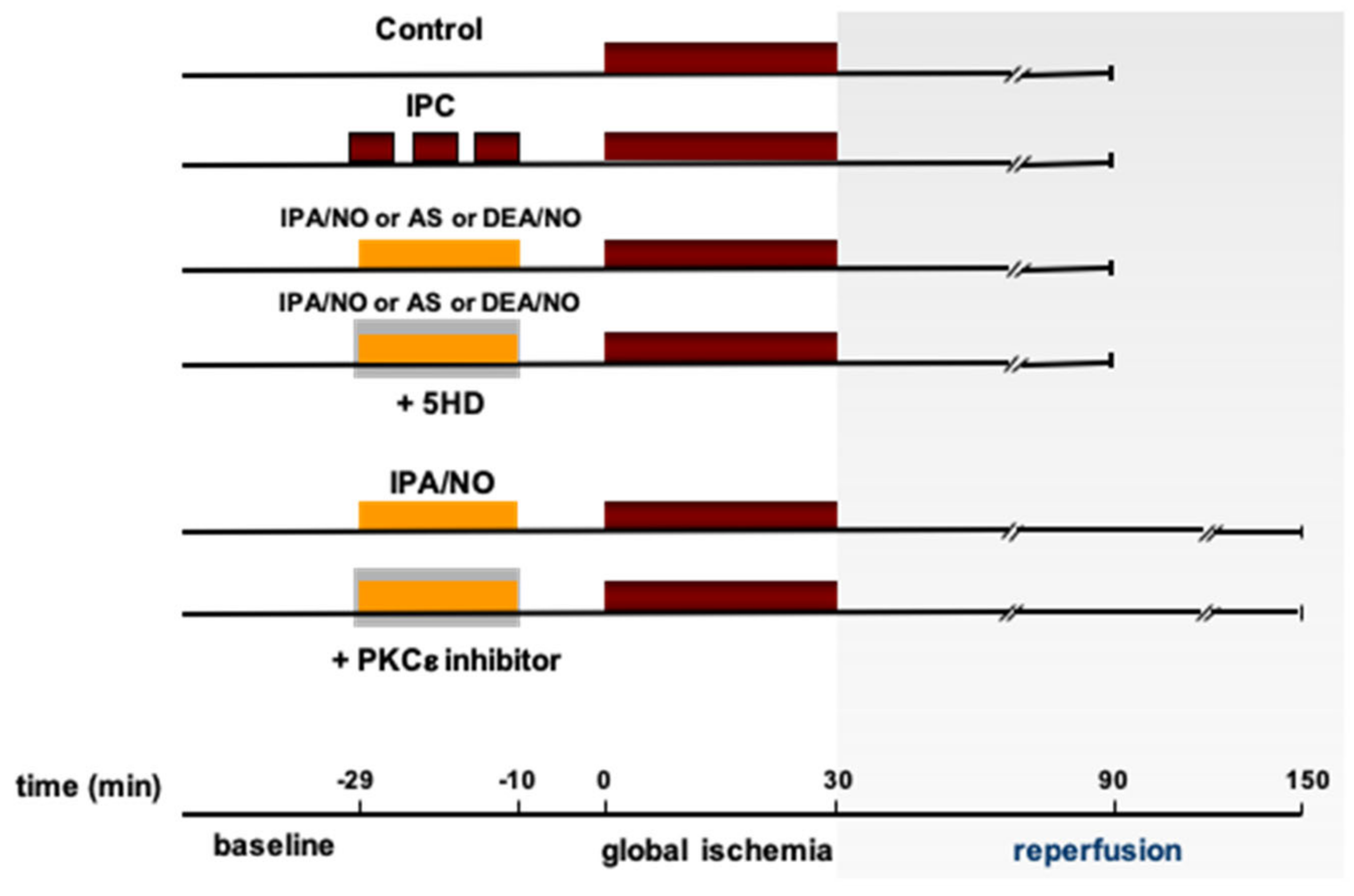
2.1. Experimental Protocols
2.2. Assessment of Myocardial Injury
2.3. Western Blotting
2.4. Immunohistochemistry Preparation
2.5. Cells
2.6. mPTP ROS Threshold (tmPTP) Measurements
2.7. Chemicals
2.8. Statistical Analysis
3. Results
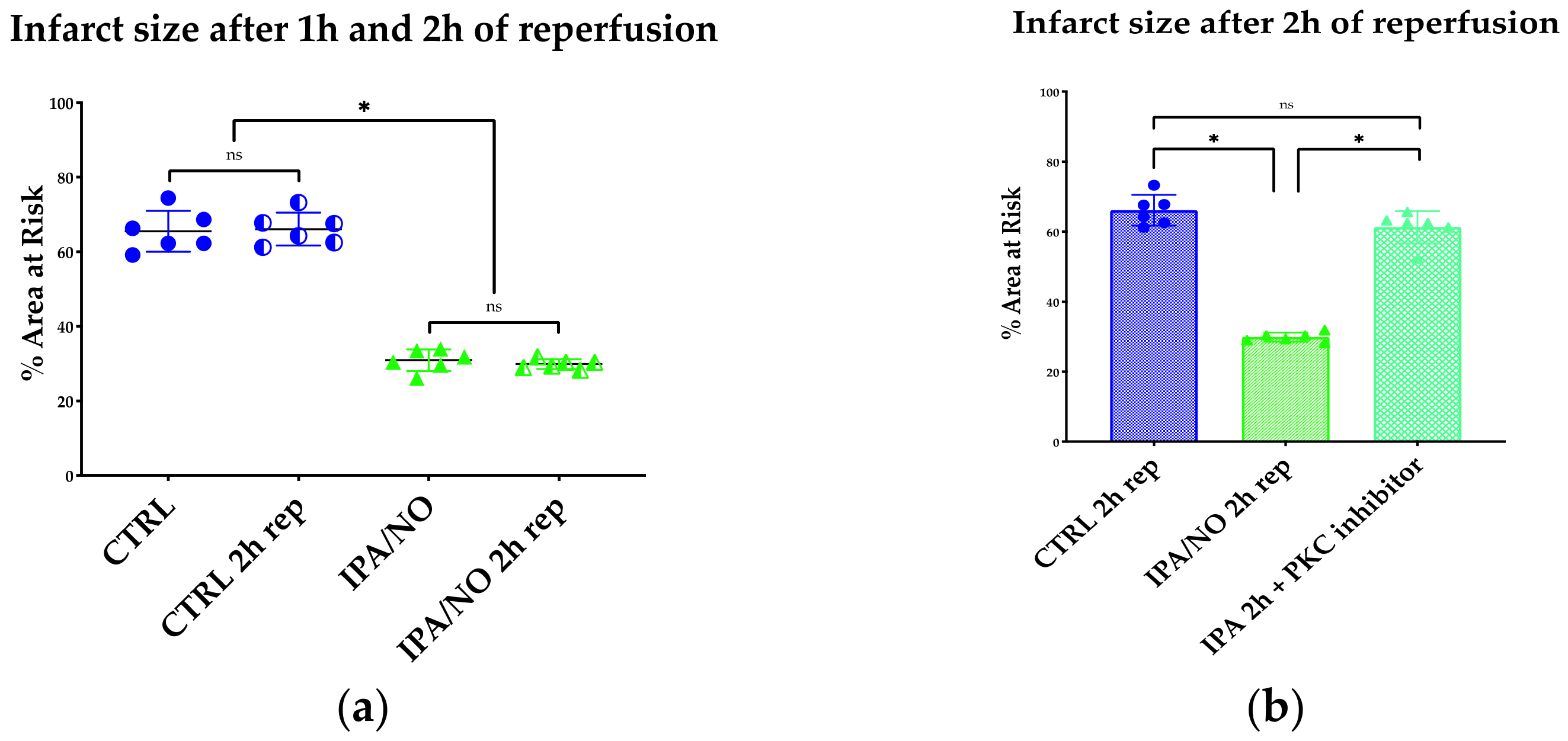
3.1. The HNO Donor, IPA/NO Limits Infarct Size and LV Dysfunction after I/R in Isolated Rat Hearts
3.2. IPA/NO Affords Long-Lasting Protection after I/R Injury
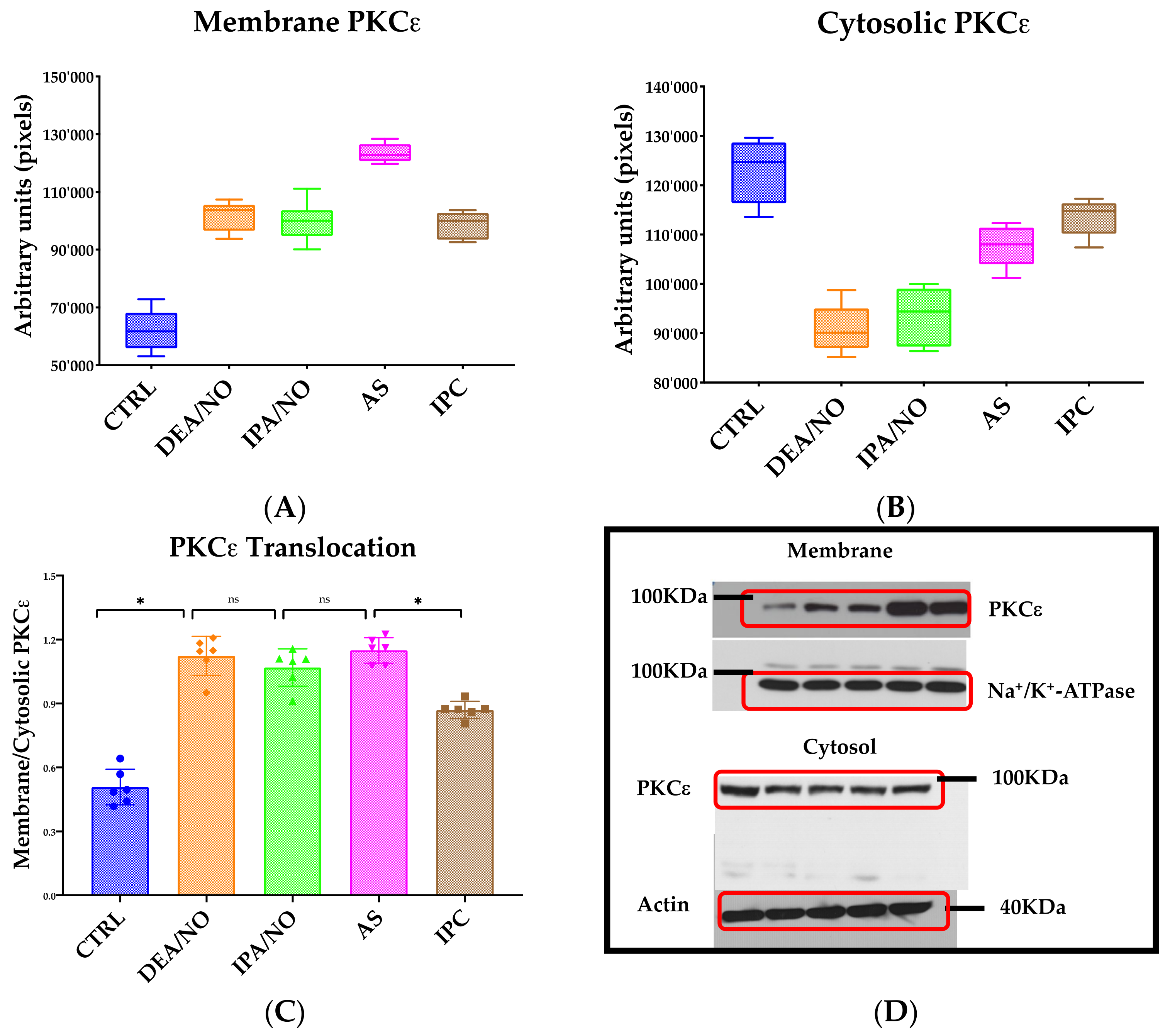
3.3. IPA/NO-Afforded Protection Involves PKCɛ Activation and Translocation
3.4. IPA/NO-Conferred Protection Is Not Mediated by Mito-KATP Channels
3.5. HNO Donors Enhance the mPTP ROS Threshold in Isolated Myocytes
4. Discussion
- -
- The pure HNO donor IPA/NO confers cardiac protection against I/R injury in a manner quantitatively similar to IPC, NO. donors, such as DEA/NO, and mixed HNO/nitrite releasers, such as AS;
- -
- HNO protection is mediated by PKCε-dependent signaling, but is independent from mito-KATP channel activation;
- -
- Mitochondrial protection triggered by HNO donors is the result of desensitizing the mPTP to ROS (enhancing its ROS threshold), which, in turn, delays opening of the pore, thus reducing cell damage as a result of ischemia-reperfusion injury.
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Baines, C.P. How and when do myocytes die during ischemia and reperfusion: The late phase. J. Cardiovasc. Pharmacol. Ther. 2011, 106, 1190–1196. [Google Scholar] [CrossRef] [PubMed]
- Totzeck, M.; Hendgen-Cotta, U.B.; Rassaf, T. Nitrite-nitric oxide signaling and cardioprotection. In Mitochondrial Dynamics in Cardiovascular Medicine; Advances in Experimental Medicine and Biology; Springer: Berlin/Heidelberg, Germany, 2017; pp. 335–346. [Google Scholar] [CrossRef]
- Schulz, R.; Kelm, M.; Heusch, G. Nitric oxide in myocardial ischemia/reperfusion injury. Cardiovasc. Res. 2004, 61, 402–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massion, P.B.; Pelat, M.; Belge, C.; Balligand, J.L. Regulation of the mammalian heart function by nitric oxide. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 2005, 42, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Paolocci, N.; Biondi, R.; Bettini, M.; Lee, C.I.; Berlowitz, C.O.; Rossi, R.; Xia, Y.; Ambrosio, G.; L’Abbate, A.; Kass, D.A.; et al. Oxygen radical-mediated reduction in basal and agonist-evoked no release in isolated rat heart. J. Mol. Cell. Cardiol. 2001, 33, 671–679. [Google Scholar] [CrossRef] [PubMed]
- Miranda, K.M.; Paolocci, N.; Katori, T.; Thomas, D.D.; Ford, E.; Bartberger, M.D.; Espey, M.G.; Kass, D.A.; Feelisch, M.; Fukuto, J.M.; et al. A biochemical rationale for the discrete behavior of nitroxyl and nitric oxide in the cardiovascular system. Proc. Natl. Acad. Sci. USA 2003, 100, 9196–9201. [Google Scholar] [CrossRef] [Green Version]
- Wong, P.S.Y.; Hyun, J.; Fukuto, J.M.; Shirota, F.N.; DeMaster, E.G.; Shoeman, D.W.; Nagasawa, H.T. Reaction between S-Nitrosothiols and thiols: Generation of nitroxyl (HNO) and subsequent chemistry. Biochemistry 1998, 37, 5362–5371. [Google Scholar] [CrossRef]
- Hernandez-Resendiz, S.; Chinda, K.; Ong, S.-B.; Cabrera-Fuentes, H.; Zazueta, C.; Hausenloy, D.J. The Role of Redox Dysregulation in the Inflammatory Response to Acute Myocardial Ischaemia-reperfusion Injury—Adding Fuel to the Fire. Curr. Med. Chem. 2017, 25, 1275–1293. [Google Scholar] [CrossRef]
- Shiva, S.; Crawford, J.H.; Ramachandran, A.; Ceaser, E.K.; Hillson, T.; Brookes, P.S.; Patel, R.P.; Darley-Usmar, V.M. Mechanisms of the interaction of nitroxyl with mitochondria. Biochem. J. 2004, 379, 359–366. [Google Scholar] [CrossRef]
- Queliconi, B.B.; Wojtovich, A.P.; Nadtochiy, S.M.; Kowaltowski, A.J.; Brookes, P.S. Redox regulation of the mitochondrial KATP channel in cardioprotection. Biochim. Biophys. Acta Mol. Cell Res. 2011, 1813, 1309–1315. [Google Scholar] [CrossRef]
- Pagliaro, P.; Mancardi, D.; Rastaldo, R.; Penna, C.; Gattullo, D.; Miranda, K.M.; Feelisch, M.; Wink, D.A.; Kass, D.A.; Paolocci, N. Nitroxyl affords thiol-sensitive myocardial protective effects akin to early preconditioning. Free Radic. Biol. Med. 2003, 34, 33–43. [Google Scholar] [CrossRef]
- Miranda, K.M.; Katori, T.; Torres De Holding, C.L.; Thomas, L.; Ridnour, L.A.; McLendon, W.J.; Cologna, S.M.; Dutton, A.S.; Champion, H.C.; Mancardi, D.; et al. Comparison of the NO and HNO donating properties of diazeniumdiolates: Primary amine adducts release HNO in vivo. J. Med. Chem. 2005, 48, 8220–8228. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, S.; Downey, J.M.; Ganote, C.E. Preconditioning of isolated rabbit cardiomyocytes: Induction by metabolic stress and blockade by the adenosine antagonist SPT and calphostin C, a protein kinase C inhibitor. Cardiovasc. Res. 1994, 28, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Dawn, B.; Bolli, R. Role of nitric oxide in myocardial preconditioning. Ann. N. Y. Acad. Sci. 2002, 962, 18–41. [Google Scholar] [CrossRef] [PubMed]
- Bouwman, R.A.; Musters, R.J.P.; Van Beek-Harmsen, B.J.; De Lange, J.J.; Boer, C. Reactive Oxygen Species Precede Protein Kinase C-δ Activation Independent of Adenosine Triphosphate-sensitive Mitochondrial Channel Opening in Sevoflurane-induced Cardioprotection. J. Am. Soc. Anesthesiol. 2004, 100, 506–514. [Google Scholar] [CrossRef]
- Miura, T.; Liu, Y.; Goto, M.; Tsuchida, A.; Mild, T.; Nakano, A.; Nishino, Y.; Ohnuma, Y.; Shimamoto, K. Mitochondrial ATP-sensitive K+ channels play a role in cardioprotection by Na+-H+ exchange inhibition against ischemia/reperfusion injury. J. Am. Coll. Cardiol. 2001, 37, 957–963. [Google Scholar] [CrossRef] [Green Version]
- Costa, A.D.T.; Garlid, K.D. Intramitochondrial signaling: Interactions among mitoKATP, PKCε, ROS, and MPT. Am. J. Physiol. Hearth Circ. Physiol. 2008, 295, H874–H882. [Google Scholar] [CrossRef] [Green Version]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [Green Version]
- Juhaszova, M.; Zorov, D.B.; Kim, S.H.; Pepe, S.; Fu, Q.; Fishbein, K.W.; Ziman, B.D.; Wang, S.; Ytrehus, K.; Antos, C.L.; et al. Glycogen synthase kinase-3β mediates convergence of protection signalling to inhibit the mitochondrial permeability transition pore. J. Clin. Investig. 2004, 113, 1535–1549. [Google Scholar] [CrossRef]
- Mancardi, D.; Tullio, F.; Crisafulli, A.; Rastaldo, R.; Folino, A.; Penna, C.; Pagliaro, P. Omega 3 has a beneficial effect on ischemia/reperfusion injury, but cannot reverse the effect of stressful forced exercise. Nutr. Metab. Cardiovasc. Dis. 2009, 19, 20–26. [Google Scholar] [CrossRef]
- Fukuto, J.M.; Jackson, M.I.; Kaludercic, N.; Paolocci, N. Examining Nitroxyl in Biological Systems. Methods Enzymol. 2008, 440, 411–431. [Google Scholar] [CrossRef]
- Ma, X.L.; Gao, F.; Liu, G.L.; Lopez, B.L.; Christopher, T.A.; Fukuto, J.M.; Wink, D.A.; Feelisch, M. Opposite effects of nitric oxide and nitroxyl on postischemic myocardial injury. Proc. Natl. Acad. Sci. USA 1999, 96, 14617–14622. [Google Scholar] [CrossRef] [Green Version]
- Frezza, C.; Cipolat, S.; Scorrano, L. Organelle isolation: Functional mitochondria from mouse liver, muscle and cultured filroblasts. Nat. Protoc. 2007, 2, 287–295. [Google Scholar] [CrossRef] [Green Version]
- Capogrossi, M.C.; Kort, A.A.; Spurgeon, H.A.; Lakatta, E.G. Single adult rabbit and rat cardiac myocytes retain the Ca2+- and speciesdependent systolic and diastolic contractile properties of intact muscle. J. Gen. Physiol. 1986, 88, 589–613. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Filburn, C.R.; Klotz, L.O.; Zweier, J.L.; Sollott, S.J. Reactive oxygen species (ROS)-induced ROS release: A new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J. Exp. Med. 2000, 192, 1001–1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miranda, K.M.; Dutton, A.S.; Ridnour, L.A.; Foreman, C.A.; Ford, E.; Paolocci, N.; Katori, T.; Tocchetti, C.G.; Mancardi, D.; Thomas, D.D.; et al. Mechanism of aerobic decomposition of Angeli’s salt (Sodium Trioxodinitrate) at physiological pH. J. Am. Chem. Soc. 2005, 127, 722–731. [Google Scholar] [CrossRef] [PubMed]
- Salmon, D.J.; De Holding, C.L.T.; Thomas, L.; Peterson, K.V.; Goodman, G.P.; Saavedra, J.E.; Srinivasan, A.; Davies, K.M.; Keefer, L.K.; Miranda, K.M. HNO and NO release from a primary amine-based diazeniumdiolate as a function of pH. Inorg. Chem. 2011, 50, 3262–3270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hausenloy, D.J.; Barrabes, J.A.; Bøtker, H.E.; Davidson, S.M.; Di Lisa, F.; Downey, J.; Engstrom, T.; Ferdinandy, P.; Carbrera-Fuentes, H.A.; Heusch, G.; et al. Ischaemic conditioning and targeting reperfusion injury: A 30 year voyage of discovery. Basic Res. Cardiol. 2016, 111, 70. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Li, Q.; Xuan, Y.-T.; Wu, W.-J.; Tan, W.; Slezak, J.; Zhu, X.; Tomlin, A.; Bolli, R. Exercise-induced late preconditioning in mice is triggered by eNOS-dependent generation of nitric oxide and activation of PKCε and is mediated by increased iNOS activity. Int. J. Cardiol. 2021, 340, 68–78. [Google Scholar] [CrossRef]
- Uecker, M.; Da Silva, R.; Grampp, T.; Pasch, T.; Schaub, M.C.; Zaugg, M. Translocation of protein kinase C isoforms to subcellular targets in ischemic and anesthetic preconditioning. Anesthesiology 2003, 99, 138–147. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Kudo, M.; Xu, M.; Ayub, A.; Ashraf, M. Mitochondrial KATP channel as an end effector of cardioprotection during late preconditioning: Triggering role of nitric oxide. J. Mol. Cell. Cardiol. 2001, 33, 2037–2046. [Google Scholar] [CrossRef]
- Brown, D.A.; Chicco, A.J.; Jew, K.N.; Johnson, M.S.; Lynch, J.M.; Watson, P.A.; Moore, R.L. Cardioprotection afforded by chronic exercise is mediated by the sarcolemmal, and not the mitochondrial, isoform of the KATP channel in the rat. J. Physiol. 2005, 569, 913–924. [Google Scholar] [CrossRef] [PubMed]
- Ohtani, H.; Katoh, H.; Tanaka, T.; Saotome, M.; Urushida, T.; Satoh, H.; Hayashi, H. Effects of nitric oxide on mitochondrial permeability transition pore and thiol-mediated responses in cardiac myocytes. Nitric Oxide Biol. Chem. 2012, 26, 95–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paolocci, N.; Saavedra, W.F.; Miranda, K.M.; Martignani, C.; Isoda, T.; Hare, J.M.; Espey, M.G.; Fukuto, J.M.; Feelisch, M.; Winkt, D.A.; et al. Nitroxyl anion exerts redox-sensitive positive cardiac inotropy in vivo by calcitonin gene-related peptide signaling. Proc. Natl. Acad. Sci. USA 2001, 98, 10463–10468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paolocci, N.; Katori, T.; Champion, H.C.; John, M.E.S.; Miranda, K.M.; Fukuto, J.M.; Wink, D.A.; Kass, D.A. Positive inotropic and lusitropic effects of HNO/NO- in failing hearts: Independence from β-adrenergic signaling. Proc. Natl. Acad. Sci. USA 2003, 100, 5537–5542. [Google Scholar] [CrossRef] [Green Version]
- Cowart, D.; Venuti, R.P.; Lynch, K.; Guptill, J.T.; Noveck, R.J.; Foo, S.Y. A Phase 1 Randomized Study of Single Intravenous Infusions of the Novel Nitroxyl Donor BMS-986231 in Healthy Volunteers. J. Clin. Pharmacol. 2019, 59, 717–730. [Google Scholar] [CrossRef]
- Tocchetti, C.G.; Mercurio, V.; Maack, C. The multifaceted mechanisms of nitroxyl in heart failure: Inodilator or ‘only’ vasodilator? Eur. J. Heart Fail. 2021, 23, 1156–1159. [Google Scholar] [CrossRef]
- Wojcik, B.; Knapp, M.; Gorski, J. Non-ischemic heart preconditioning. J. Physiol. Pharmacol. 2018, 69, 173–184. [Google Scholar] [CrossRef]
- Javador, S.A.; Clarke, S.; Das, M.; Griffiths, E.J.; Lim, K.H.H.; Halestrap, A.P. Ischaemic preconditioning inhibits opening of mitochondrial permeability transition pores in the reperfused rat heart. J. Physiol. 2003, 549, 513–524. [Google Scholar] [CrossRef]
- Miranda, K.M.; Espey, M.G.; Yamada, K.; Krishna, M.; Ludwick, N.; Kim, S.; Jourd’heuil, D.; Grisham, M.B.; Feelisch, M.; Fukuto, J.M.; et al. Unique oxidative mechanisms for the reactive nitrogen oxide species, nitroxyl anion. J. Biol. Chem. 2001, 276, 1720–1727. [Google Scholar] [CrossRef] [Green Version]
- Smulik, R.; Debski, D.; Zielonka, J.; Michalowski, B.; Adamus, J.; Marcinek, A.; Kalyanaraman, B.; Sikora, A. Nitroxyl (HNO) reacts with molecular oxygen and forms peroxynitrite at physiological ph: Biological implications. J. Biol. Chem. 2014, 289, 35570–35581. [Google Scholar] [CrossRef] [Green Version]
- Fukuto, J.M. A recent history of nitroxyl chemistry, pharmacology and therapeutic potential. Br. J. Pharmacol. 2019, 176, 135–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohnuma, Y.; Miura, T.; Miki, T.; Tanno, M.; Kuno, A.; Tsuchida, A.; Shimamoto, K. Opening of mitochondrial KATP channel occurs downstream of PKC-ε activation in the mechanism of preconditioning. Am. J. Physiol. Hear. Circ. Physiol. 2002, 283, H440–H447. [Google Scholar] [CrossRef] [PubMed]
- Obal, D.; Dettwiler, S.; Favoccia, C.; Scharbatke, H.; Preckel, B.; Schlack, W. The influence of mitochondrial KATP-channels in the cardioprotection of preconditioning and postconditioning by sevoflurane in the rat in vivo. Anesth. Analg. 2005, 101, 1252–1260. [Google Scholar] [CrossRef] [PubMed]
- Wojtovich, A.P.; Williams, D.M.; Karcz, M.K.; Lopes, C.M.B.; Gray, D.A.; Nehrke, K.W.; Brookes, P.S. A novel mitochondrial KATP channel assay. Circ. Res. 2010, 106, 1190–1196. [Google Scholar] [CrossRef] [Green Version]
- Halestrap, A.P.; Clarke, S.J.; Javadov, S.A. Mitochondrial permeability transition pore opening during myocardial reperfusion—A target for cardioprotection. Cardiovasc. Res. 2004, 61, 372–385. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CPP (mmHg) | SIST (mmHg) | EDP (mmHg) | dP/dt Max (mmHg/s) | Heart Weight (g) | |
|---|---|---|---|---|---|
| Control | 87 ± 7 | 99 ± 6 | 8 ± 2 | 1.783 ± 289 | 1.77 ± 0.14 |
| DEA/NO | 83 ± 6 | 97 ± 8 | 7 ± 2 | 2.020 ± 351 | 1.70 ± 0.15 |
| DEA/NO + 5HD | 86 ± 9 | 95 ± 4 | 9 ± 2 | 1.596 ± 246 | 1.70 ± 0.22 |
| IPA/NO | 82 ± 6 | 100 ± 5 | 9 ± 2 | 1.545 ± 238 | 1.67 ± 0.08 |
| IPA/NO + 5HD | 88 ± 6 | 100 ± 3 | 8 ± 3 | 1.856 ± 374 | 1.63 ± 0.23 |
| AS | 85 ± 9 | 101 ± 4 | 10 ± 3 | 1.921 ± 208 | 1.70 ± 0.10 |
| AS + 5HD | 82 ± 8 | 98 ± 10 | 10 ± 4 | 1.595 ± 109 | 1.74 ± 0.15 |
| IPC | 83 ± 6 | 99 ± 5 | 9 ± 6 | 1.852 ± 298 | 1.74 ± 0.10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mancardi, D.; Pagliaro, P.; Ridnour, L.A.; Tocchetti, C.G.; Miranda, K.; Juhaszova, M.; Sollott, S.J.; Wink, D.A.; Paolocci, N. HNO Protects the Myocardium against Reperfusion Injury, Inhibiting the mPTP Opening via PKCε Activation. Antioxidants 2022, 11, 382. https://doi.org/10.3390/antiox11020382
Mancardi D, Pagliaro P, Ridnour LA, Tocchetti CG, Miranda K, Juhaszova M, Sollott SJ, Wink DA, Paolocci N. HNO Protects the Myocardium against Reperfusion Injury, Inhibiting the mPTP Opening via PKCε Activation. Antioxidants. 2022; 11(2):382. https://doi.org/10.3390/antiox11020382
Chicago/Turabian StyleMancardi, Daniele, Pasquale Pagliaro, Lisa A. Ridnour, Carlo G. Tocchetti, Katrina Miranda, Magdalena Juhaszova, Steven J. Sollott, David A. Wink, and Nazareno Paolocci. 2022. "HNO Protects the Myocardium against Reperfusion Injury, Inhibiting the mPTP Opening via PKCε Activation" Antioxidants 11, no. 2: 382. https://doi.org/10.3390/antiox11020382
APA StyleMancardi, D., Pagliaro, P., Ridnour, L. A., Tocchetti, C. G., Miranda, K., Juhaszova, M., Sollott, S. J., Wink, D. A., & Paolocci, N. (2022). HNO Protects the Myocardium against Reperfusion Injury, Inhibiting the mPTP Opening via PKCε Activation. Antioxidants, 11(2), 382. https://doi.org/10.3390/antiox11020382