
Pain in Hemophilia: Unexplored Role of Oxidative Stress


{kind=link}
Abstract
:1. Introduction
2. Pain in Hemophilia
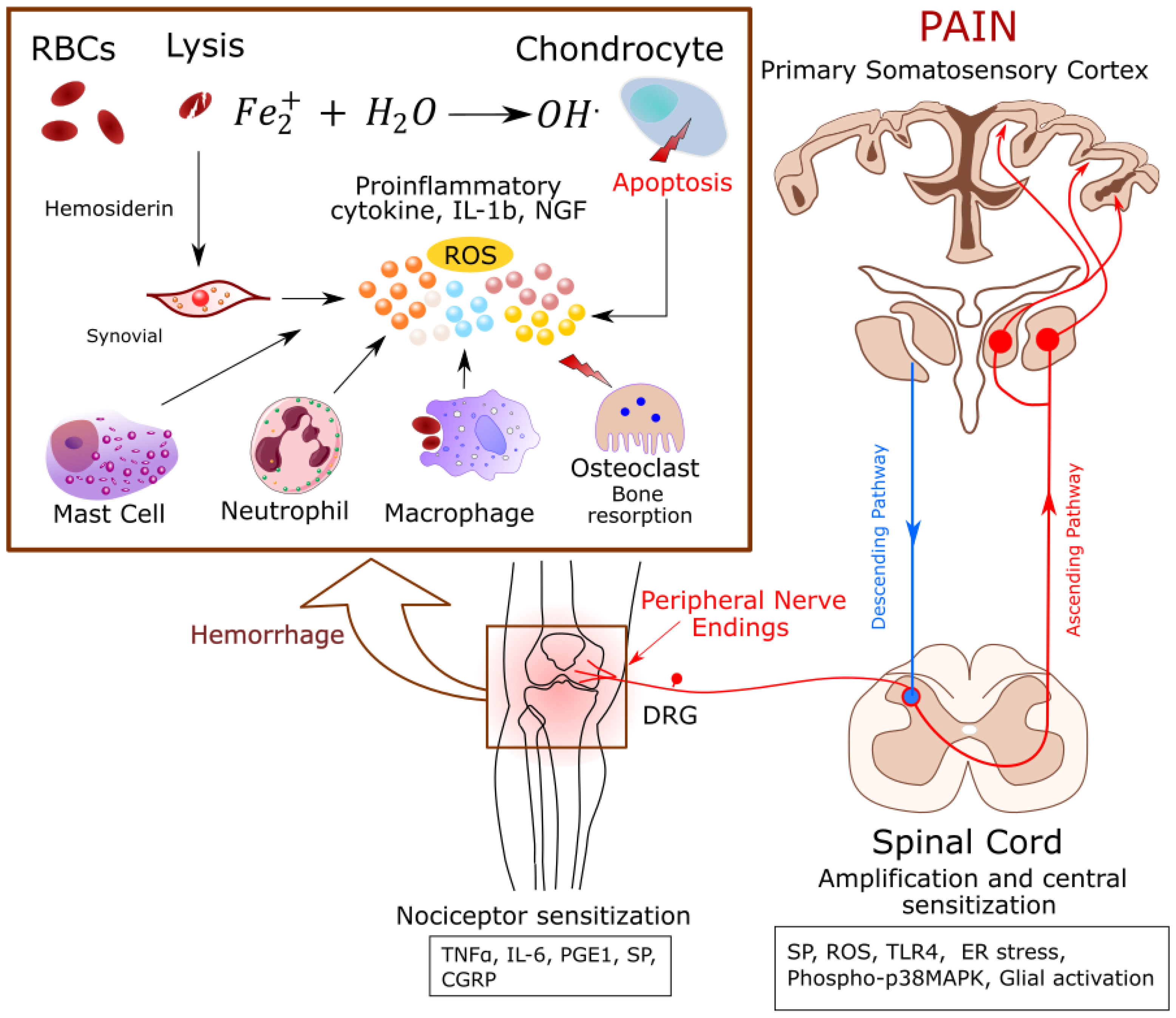
3. Hemarthropathy and Oxidative Stress, Two Faces of the Same Coin
3.1. Cell-Free Heme
3.2. Bone Destruction
3.3. Inflammation
3.4. Angiogensis
4. Oxidative Stress as a Contributor to Pain in Hemophilia
4.1. Nociceptive Pain
4.2. Neuropathic Pain
5. Novel Antioxidant Therapies for Chronic Pain in Hemophilia
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| PWH | Patients With Hemophilia |
| HRQoL | Health-Related Quality Of Life |
| pDq | Paindetect-Questionnaire |
| FLS | Fibroblast-Like Synoviocytes |
| IL | Interleukin |
| TNF-α | Tumor Necrosis Factor-Alpha |
| ROS | Reactive Oxygen Species |
| H2O2 | Hydrogen Peroxide |
| OA | Osteoarthritis |
| SCD | Sickle Cell Disease |
| MMPs | Matrix Metalloproteinases |
| NO | Nitric Oxide |
| NOX | Nadph Oxidase |
| O2− | Superoxide |
| SOD | Superoxide Dismutase |
| Cl− | Chloride |
| HOCl | Hypochlorous Acid |
| VEGF | Vascular Endothelial Growth Factor |
| RA | Rheumatoid Arthritis |
| CIA | Collagen-Induced Arthritis |
| PAR-1 | Protease-Activated Receptors |
| OPG | Osteoprotegerin |
| RANKL | RANK Ligand |
| NAC | N-Acetylcysteine |
| NSAIDs | Non-Steroidal Anti-Inflammatory Drugs |
| ONOOH | Peroxynitrite |
| LA | Linoleic Acid |
| AA | Arachidonic Acid |
| TRPV1 | Transient Receptor Potential Vanilloid 1 |
| OLAMs | Oxidized LA Metabolites |
| NGF | Nerve Growth Factor |
| COX2 | Cyclooxygenase-2 |
| PGE2 | Prostaglandin E2 |
| GPCR | G Protein-Coupled Receptors |
| EP1-4 | E Prostanoid Receptor Subtypes 1, 2, 3 And 4 |
| PGP 9.5 | Protein Gene Product 9.5 |
| CFA | Complete Freund’s Adjuvant |
| CGRP | Calcitonin Gene-Related Peptide |
| CCI | Chronic Constriction Injury |
| 4-HNE | 4-Hydroxynonenal |
| MDA | Malondialdehyde |
References
- Castaman, G.; Matino, D. Hemophilia A and B: Molecular and clinical similarities and differences. Haematologica 2019, 104, 1702–1709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulkarni, R.; Soucie, J.M.; Lusher, J.; Presley, R.; Shapiro, A.; Gill, J.; Manco-Johnson, M.; Koerper, M.; Mathew, P.; Abshire, T.; et al. Sites of initial bleeding episodes, mode of delivery and age of diagnosis in babies with haemophilia diagnosed before the age of 2 years: A report from The Centers for Disease Control and Prevention’s (CDC) Universal Data Collection (UDC) project. Haemophilia 2009, 15, 1281–1290. [Google Scholar] [CrossRef] [PubMed]
- Bolton-Maggs, P.H.; Pasi, K.J. Haemophilias A and B. Lancet 2003, 361, 1801–1809. [Google Scholar] [CrossRef]
- Pollmann, H.; Richter, H.; Ringkamp, H.; Jürgens, H. When are children diagnosed as having severe haemophilia and when do they start to bleed? A 10-year single-centre PUP study. Eur. J. Pediatrics 1999, 158, S166–S170. [Google Scholar] [CrossRef]
- Morfini, M.; Coppola, A.; Franchini, M.; Di Minno, G. Clinical use of factor VIII and factor IX concentrates. Blood Transfus. 2013, 11, s55–s63. [Google Scholar] [CrossRef]
- Rodriguez-Merchan, E.C. Musculoskeletal Complications of Hemophilia. HSS J.® 2010, 6, 37–42. [Google Scholar] [CrossRef]
- Franchini, M.; Mannucci, P.M. Co-morbidities and quality of life in elderly persons with haemophilia. Br. J. Haematol. 2010, 148, 522–533. [Google Scholar] [CrossRef]
- Philipp, C. The aging patient with hemophilia: Complications, comorbidities, and management issues. Hematol. Am. Soc. Hematol. Educ. Program 2010, 2010, 191–196. [Google Scholar] [CrossRef] [Green Version]
- Barr, R.D.; Saleh, M.; Furlong, W.; Horsman, J.; Sek, J.; Pai, M.; Walker, I. Health status and health-related quality of life associated with hemophilia. Am. J. Hematol. 2002, 71, 152–160. [Google Scholar] [CrossRef]
- van Genderen, F.R.; Westers, P.; Heijnen, L.; de Kleijn, P.; van den Berg, H.M.; Helders, P.J.; van Meeteren, N.L. Measuring patients’ perceptions on their functional abilities: Validation of the Haemophilia Activities List. Haemophilia 2006, 12, 36–46. [Google Scholar] [CrossRef]
- Auerswald, G.; Dolan, G.; Duffy, A.; Hermans, C.; Jiménez-Yuste, V.; Ljung, R.; Morfini, M.; Lambert, T.; Šalek, S.Z. Pain and pain management in haemophilia. Blood Coagul. Fibrinolysis 2016, 27, 845–854. [Google Scholar] [CrossRef] [Green Version]
- Witkop, M.; Santaella, M.; Nichols, C.D.; Lambing, A.Y.; Baumann, K.; Curtis, R.G.; Humphrey, C.; Humphries, T.J.; Newman, J.; Durben, N.; et al. Understanding the Pain Management Landscape Within the US Bleeding Disorder Community: A Multi-Center Survey. Pain Med. 2022, 23, 269–279. [Google Scholar] [CrossRef]
- Rambod, M.; Sharif, F.; Molazem, Z.; Khair, K.; von Mackensen, S. Health-Related Quality of Life and Psychological Aspects of Adults With Hemophilia in Iran. Clin. Appl. Thromb. Hemost. 2018, 24, 1073–1081. [Google Scholar] [CrossRef]
- Witkop, M.L.; Lambing, A.; Nichols, C.D.; Munn, J.E.; Anderson, T.L.; Tortella, B.J. Interrelationship between depression, anxiety, pain, and treatment adherence in hemophilia: Results from a US cross-sectional survey. Patient Prefer. Adherence 2019, 13, 1577–1587. [Google Scholar] [CrossRef] [Green Version]
- Witkop, M.; Wang, M.; Hernandez, G.; Recht, M.; Baumann, K.; Cooper, D.L. Impact of haemophilia on patients with mild-to-moderate disease: Results from the P-FiQ and B-HERO-S studies. Haemophilia 2021, 27, 8–16. [Google Scholar] [CrossRef]
- Stromer, W.; Pabinger, I.; Ay, C.; Crevenna, R.; Donnerer, J.; Feistritzer, C.; Hemberger, S.; Likar, R.; Sevelda, F.; Thom, K.; et al. Pain management in hemophilia: Expert recommendations. Wien. Klin. Wochenschr. 2021, 133, 1042–1056. [Google Scholar] [CrossRef]
- Di Minno, M.N.; Santoro, C.; Corcione, A.; Di Minno, G.; Martinelli, M.; Mancuso, M.E.; Acone, B.; Molinari, A.C.; Passeri, E.V.; Rocino, A. Pain assessment and management in Italian Haemophilia Centres. Blood Transfus. 2021, 19, 335. [Google Scholar]
- Pinto, P.R.; Paredes, A.C.; Almeida, A. Pain prevalence, characteristics, and impact among people with hemophilia: Findings from the first Portuguese survey and implications for pain management. Pain Med. 2020, 21, 458–471. [Google Scholar] [CrossRef]
- Paredes, A.C.; Teixeira, P.; Almeida, A.; Pinto, P.R. Prevalence and Interference of Chronic Pain Among People With Hemophilia: A Systematic Review and Meta-Analysis. J. Pain 2021, 22, 1134–1145. [Google Scholar] [CrossRef]
- Stromer, W.; Messerer, B.; Crevenna, R.; Hemberger, S.H.; Jauk, B.; Schwarz, R.; Streif, W.; Thom, K.; Wagner, B.; Zwiauer, K.; et al. Pain therapy for children and adolescents with hemophilia: Recommendations by an expert panel. Schmerz 2018, 32, 404–418. [Google Scholar] [CrossRef]
- Witkop, M.; Neff, A.; Buckner, T.W.; Wang, M.; Batt, K.; Kessler, C.M.; Quon, D.; Boggio, L.; Recht, M.; Baumann, K.; et al. Self-reported prevalence, description and management of pain in adults with haemophilia: Methods, demographics and results from the Pain, Functional Impairment, and Quality of life (P-FiQ) study. Haemophilia 2017, 23, 556–565. [Google Scholar] [CrossRef]
- Srivastava, A.; Brewer, A.; Mauser-Bunschoten, E.; Key, N.; Kitchen, S.; Llinas, A.; Ludlam, C.; Mahlangu, J.; Mulder, K.; Poon, M. Guidelines for the management of hemophilia. Haemophilia 2013, 19, e1–e47. [Google Scholar] [CrossRef]
- van Genderen, F.R.; Fischer, K.; Heijnen, L.; de Kleijn, P.; van den Berg, H.M.; Helders, P.J.; van Meeteren, N.L. Pain and functional limitations in patients with severe haemophilia. Haemophilia 2006, 12, 147–153. [Google Scholar] [CrossRef]
- Witkop, M.; Lambing, A.; Kachalsky, E.; Divine, G.; Rushlow, D.; Dinnen, J. Assessment of acute and persistent pain management in patients with haemophilia. Haemophilia 2011, 17, 612–619. [Google Scholar] [CrossRef]
- Sidonio, R.F.; Mili, F.D.; Li, T.; Miller, C.H.; Hooper, W.C.; DeBaun, M.R.; Soucie, M. Females with FVIII and FIX deficiency have reduced joint range of motion. Am. J. Hematol. 2014, 89, 831–836. [Google Scholar] [CrossRef] [Green Version]
- Forneris, E.; Andreacchio, A.; Pollio, B.; Mannucci, C.; Franchini, M.; Mengoli, C.; Pagliarino, M.; Messina, M. Gait analysis in children with haemophilia: First Italian experience at the Turin Haemophilia Centre. Haemophilia 2016, 22, e184–e191. [Google Scholar] [CrossRef]
- Buranahirun, C.; Walsh, K.S.; Mrakotsky, C.; Croteau, S.E.; Rajpurkar, M.; Kearney, S.; Hannemann, C.; Wilkening, G.N.; Shapiro, K.A.; Cooper, D.L. Neuropsychological function in children with hemophilia: A review of the Hemophilia Growth and Development Study and introduction of the current eTHINK study. Pediatric Blood Cancer 2020, 67, e28004. [Google Scholar] [CrossRef]
- Schoenmakers, M.A.; Gulmans, V.A.; Helders, P.J.; van den Berg, H.M. Motor performance and disability in Dutch children with haemophilia: A comparison with their healthy peers. Haemophilia 2001, 7, 293–298. [Google Scholar] [CrossRef]
- Shapiro, A.D.; Donfield, S.M.; Lynn, H.S.; Cool, V.A.; Stehbens, J.A.; Hunsberger, S.L.; Tonetta, S.; Gomperts, E.D. Defining the impact of hemophilia: The Academic Achievement in Children with Hemophilia Study. Pediatrics 2001, 108, E105. [Google Scholar] [CrossRef] [Green Version]
- Krüger, S.; Hilberg, T. Neuropathic pain in patients with haemophilia, that is the question. Hamostaseologie 2015, 35, S5–S9. [Google Scholar] [CrossRef]
- Sophia Fox, A.J.; Bedi, A.; Rodeo, S.A. The basic science of articular cartilage: Structure, composition, and function. Sports Health 2009, 1, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Akkiraju, H.; Nohe, A. Role of Chondrocytes in Cartilage Formation, Progression of Osteoarthritis and Cartilage Regeneration. J. Dev. Biol. 2015, 3, 177–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, M.D. The normal synovium. Open Rheumatol. J. 2011, 5, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Di Nicola, V. Degenerative osteoarthritis a reversible chronic disease. Regen. Ther. 2020, 15, 149–160. [Google Scholar] [CrossRef]
- Melchiorre, D.; Manetti, M.; Matucci-Cerinic, M. Pathophysiology of Hemophilic Arthropathy. J. Clin. Med. 2017, 6, 63. [Google Scholar] [CrossRef]
- Knobe, K.; Berntorp, E. Haemophilia and joint disease: Pathophysiology, evaluation, and management. J. Comorb. 2011, 1, 51–59. [Google Scholar] [CrossRef] [Green Version]
- Valentino, L.A.; Hakobyan, N.; Enockson, C. Blood-induced joint disease: The confluence of dysregulated oncogenes, inflammatory signals, and angiogenic cues. In Seminars in Hematology; Elsevier: Amsterdam, The Netherlands, 2008; Volume 45, pp. S50–S57. [Google Scholar]
- Roosendaal, G.; Lafeber, F. Pathogenesis of haemophilic arthropathy. Haemophilia 2006, 12, 117–121. [Google Scholar] [CrossRef]
- Busso, N.; Morard, C.; Salvi, R.; Péclat, V.; So, A. Role of the tissue factor pathway in synovial inflammation. Arthritis Rheum. 2003, 48, 651–659. [Google Scholar] [CrossRef]
- Pulles, A.E.; van Vulpen, L.F.D.; Coeleveld, K.; Mastbergen, S.C.; Schutgens, R.E.G.; Lafeber, F. On-demand treatment with the iron chelator deferasirox is ineffective in preventing blood-induced joint damage in haemophilic mice. Haemophilia 2021, 27, 648–656. [Google Scholar] [CrossRef]
- Aigner, T.; Soeder, S.; Haag, J. IL-1ß and BMPs-Interactive players of cartilage matrix degradation and regeneration. Eur. Cell Mater. 2006, 12, 49–56. [Google Scholar] [CrossRef]
- Mendonça, R.; Silveira, A.A.; Conran, N. Red cell DAMPs and inflammation. Inflamm. Res. 2016, 65, 665–678. [Google Scholar] [CrossRef]
- Hakobyan, N.; Kazarian, T.; Jabbar, A.A.; Jabbar, K.J.; Valentino, L.A. Pathobiology of hemophilic synovitis I: Overexpression of mdm2 oncogene. Blood 2004, 104, 2060–2064. [Google Scholar] [CrossRef] [Green Version]
- Wen, F.-Q.; Jabbar, A.A.; Chen, Y.-X.; Kazarian, T.; Patel, D.A.; Valentino, L.A. C-myc proto-oncogene expression in hemophilic synovitis: In vitro studies of the effects of iron and ceramide. Blood J. Am. Soc. Hematol. 2002, 100, 912–916. [Google Scholar] [CrossRef] [Green Version]
- Valentino, L. Blood-induced joint disease: The pathophysiology of hemophilic arthropathy. J. Thromb. Haemost. 2010, 8, 1895–1902. [Google Scholar] [CrossRef]
- Lafeber, F.; Miossec, P.; Valentino, L. Physiopathology of haemophilic arthropathy. Haemophilia 2008, 14, 3–9. [Google Scholar] [CrossRef]
- von Drygalski, A.; Barnes, R.F.; Jang, H.; Ma, Y.; Wong, J.H.; Berman, Z.; Du, J.; Chang, E.Y. Advanced magnetic resonance imaging of cartilage components in haemophilic joints reveals that cartilage hemosiderin correlates with joint deterioration. Haemophilia 2019, 25, 851–858. [Google Scholar] [CrossRef] [Green Version]
- Hooiveld, M.; Roosendaal, G.; Vianen, M.; van den Berg, M.; Bijlsma, J.; Lafeber, F. Blood-induced joint damage: Longterm effects in vitro and in vivo. J. Rheumatol. 2003, 30, 339–344. [Google Scholar]
- Hooiveld, M.J.; Roosendaal, G.; Van Den Berg, H.; Bijlsma, J.; Lafeber, F. Haemoglobin-derived iron-dependent hydroxyl radical formation in blood-induced joint damage: An in vitro study. Rheumatology 2003, 42, 784–790. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, J.; Sun, X.; Kang, R.; Tang, D. Ferroptosis: Process and function. Cell Death Differ. 2016, 23, 369–379. [Google Scholar] [CrossRef] [Green Version]
- Heli, H.; Mirtorabi, S.; Karimian, K. Advances in iron chelation: An update. Expert Opin. Ther. Pat. 2011, 21, 819–856. [Google Scholar] [CrossRef]
- Sousa, L.; Oliveira, M.M.; Pessôa, M.T.C.; Barbosa, L.A. Iron overload: Effects on cellular biochemistry. Clin. Chim. Acta 2020, 504, 180–189. [Google Scholar] [CrossRef]
- Jing, X.; Du, T.; Li, T.; Yang, X.; Wang, G.; Liu, X.; Jiang, Z.; Cui, X. The detrimental effect of iron on OA chondrocytes: Importance of pro-inflammatory cytokines induced iron influx and oxidative stress. J. Cell Mol. Med. 2021, 25, 5671–5680. [Google Scholar] [CrossRef]
- Wang, T.; He, C. Pro-inflammatory cytokines: The link between obesity and osteoarthritis. Cytokine Growth Factor Rev. 2018, 44, 38–50. [Google Scholar] [CrossRef]
- Cai, C.; Hu, W.; Chu, T. Interplay Between Iron Overload and Osteoarthritis: Clinical Significance and Cellular Mechanisms. Front. Cell Dev. Biol. 2022, 9, 817104. [Google Scholar] [CrossRef]
- Suantawee, T.; Tantavisut, S.; Adisakwattana, S.; Tanavalee, A.; Yuktanandana, P.; Anomasiri, W.; Deepaisarnsakul, B.; Honsawek, S. Oxidative stress, vitamin e, and antioxidant capacity in knee osteoarthritis. J. Clin. Diagn. Res. JCDR 2013, 7, 1855. [Google Scholar]
- Vanderhave, K.L.; Perkins, C.A.; Scannell, B.; Brighton, B.K. Orthopaedic manifestations of sickle cell disease. JAAOS J. Am. Acad. Orthop. Surg. 2018, 26, 94–101. [Google Scholar] [CrossRef]
- Sadat-Ali, M.; Sultan, O.; Al-Turki, H.; AlElq, A. Does high serum iron level induce low bone mass in sickle cell anemia? Biometals 2011, 24, 19–22. [Google Scholar] [CrossRef]
- Kiven, S.; Wang, Y.; Aich, A.; Argueta, D.A.; Lei, J.; Sagi, V.; Tennakoon, M.; Bedros, S.J.; Lambrecht, N.; Gupta, K. Spatiotemporal Alterations in Gait in Humanized Transgenic Sickle Mice. Front. Immunol. 2020, 11, 561947. [Google Scholar] [CrossRef]
- Katsarou, O.; Terpos, E.; Chatzismalis, P.; Provelengios, S.; Adraktas, T.; Hadjidakis, D.; Kouramba, A.; Karafoulidou, A. Increased bone resorption is implicated in the pathogenesis of bone loss in hemophiliacs: Correlations with hemophilic arthropathy and HIV infection. Ann. Hematol. 2010, 89, 67–74. [Google Scholar] [CrossRef] [Green Version]
- Christoforidis, A.; Economou, M.; Papadopoulou, E.; Kazantzidou, E.; Farmaki, E.; Tzimouli, V.; Tsatra, I.; Gompakis, N.; Athanassiou-Metaxa, M. Comparative study of dual energy X-ray absorptiometry and quantitative ultrasonography with the use of biochemical markers of bone turnover in boys with haemophilia. Haemophilia 2011, 17, e217–e222. [Google Scholar] [CrossRef]
- Pulles, A.E.; Mastbergen, S.C.; Schutgens, R.E.; Lafeber, F.P.; van Vulpen, L.F. Pathophysiology of hemophilic arthropathy and potential targets for therapy. Pharmacol. Res. 2017, 115, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Boyce, B.F.; Xing, L. Biology of RANK, RANKL, and osteoprotegerin. Arthritis Res. Ther. 2007, 9, S1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandooren, B.; Cantaert, T.; Noordenbos, T.; Tak, P.P.; Baeten, D. The abundant synovial expression of the RANK/RANKL/Osteoprotegerin system in peripheral spondylarthritis is partially disconnected from inflammation. Arthritis Rheum. 2008, 58, 718–729. [Google Scholar] [CrossRef] [PubMed]
- Saidenberg-Kermanac’h, N.; Cohen-Solal, M.; Bessis, N.; De Vernejoul, M.C.; Boissier, M.C. Role for osteoprotegerin in rheumatoid inflammation. Jt. Bone Spine 2004, 71, 9–13. [Google Scholar] [CrossRef]
- Kitaura, H.; Marahleh, A.; Ohori, F.; Noguchi, T.; Shen, W.R.; Qi, J.; Nara, Y.; Pramusita, A.; Kinjo, R.; Mizoguchi, I. Osteocyte-Related Cytokines Regulate Osteoclast Formation and Bone Resorption. Int. J. Mol. Sci. 2020, 21, 5169. [Google Scholar] [CrossRef] [PubMed]
- Ono, T.; Hayashi, M.; Sasaki, F.; Nakashima, T. RANKL biology: Bone metabolism, the immune system, and beyond. Inflamm. Regen. 2020, 40, 2. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.H.; Kong, Y.Y.; Penninger, J.M. Role of RANKL and RANK in bone loss and arthritis. Ann. Rheum. Dis. 2002, 61, ii32–ii39. [Google Scholar] [CrossRef] [Green Version]
- Boyce, B.F.; Xing, L. Functions of RANKL/RANK/OPG in bone modeling and remodeling. Arch. Biochem. Biophys. 2008, 473, 139–146. [Google Scholar] [CrossRef] [Green Version]
- Melchiorre, D.; Linari, S.; Manetti, M.; Romano, E.; Sofi, F.; Matucci-Cerinic, M.; Carulli, C.; Innocenti, M.; Ibba-Manneschi, L.; Castaman, G. Clinical, instrumental, serological and histological findings suggest that hemophilia B may be less severe than hemophilia A. Haematologica 2016, 101, 219–225. [Google Scholar] [CrossRef] [Green Version]
- Lean, J.M.; Jagger, C.J.; Kirstein, B.; Fuller, K.; Chambers, T.J. Hydrogen peroxide is essential for estrogen-deficiency bone loss and osteoclast formation. Endocrinology 2005, 146, 728–735. [Google Scholar] [CrossRef]
- Manolagas, S.C. From estrogen-centric to aging and oxidative stress: A revised perspective of the pathogenesis of osteoporosis. Endocr. Rev. 2010, 31, 266–300. [Google Scholar] [CrossRef] [Green Version]
- Baek, K.H.; Oh, K.W.; Lee, W.Y.; Lee, S.S.; Kim, M.K.; Kwon, H.S.; Rhee, E.J.; Han, J.H.; Song, K.H.; Cha, B.Y.; et al. Association of oxidative stress with postmenopausal osteoporosis and the effects of hydrogen peroxide on osteoclast formation in human bone marrow cell cultures. Calcif. Tissue Int. 2010, 87, 226–235. [Google Scholar] [CrossRef]
- Romagnoli, C.; Marcucci, G.; Favilli, F.; Zonefrati, R.; Mavilia, C.; Galli, G.; Tanini, A.; Iantomasi, T.; Brandi, M.L.; Vincenzini, M.T. Role of GSH/GSSG redox couple in osteogenic activity and osteoclastogenic markers of human osteoblast-like SaOS-2 cells. Febs. J. 2013, 280, 867–879. [Google Scholar] [CrossRef]
- Fontani, F.; Marcucci, G.; Iantomasi, T.; Brandi, M.L.; Vincenzini, M.T. Glutathione, N-acetylcysteine and lipoic acid down-regulate starvation-induced apoptosis, RANKL/OPG ratio and sclerostin in osteocytes: Involvement of JNK and ERK1/2 signalling. Calcif. Tissue Int. 2015, 96, 335–346. [Google Scholar] [CrossRef]
- Filaire, E.; Toumi, H. Reactive oxygen species and exercise on bone metabolism: Friend or enemy? Jt. Bone Spine 2012, 79, 341–346. [Google Scholar] [CrossRef]
- Domazetovic, V.; Marcucci, G.; Iantomasi, T.; Brandi, M.L.; Vincenzini, M.T. Oxidative stress in bone remodeling: Role of antioxidants. Clin. Cases Miner. Bone Metab. 2017, 14, 209–216. [Google Scholar] [CrossRef]
- Sendur, O.F.; Turan, Y.; Tastaban, E.; Serter, M. Antioxidant status in patients with osteoporosis: A controlled study. Jt. Bone Spine 2009, 76, 514–518. [Google Scholar] [CrossRef]
- Almeida, M.; Han, L.; Martin-Millan, M.; Plotkin, L.I.; Stewart, S.A.; Roberson, P.K.; Kousteni, S.; O’Brien, C.A.; Bellido, T.; Parfitt, A.M.; et al. Skeletal involution by age-associated oxidative stress and its acceleration by loss of sex steroids. J. Biol. Chem. 2007, 282, 27285–27297. [Google Scholar] [CrossRef] [Green Version]
- Maggio, D.; Barabani, M.; Pierandrei, M.; Polidori, M.C.; Catani, M.; Mecocci, P.; Senin, U.; Pacifici, R.; Cherubini, A. Marked decrease in plasma antioxidants in aged osteoporotic women: Results of a cross-sectional study. J. Clin. Endocrinol. Metab. 2003, 88, 1523–1527. [Google Scholar] [CrossRef] [Green Version]
- Hall, S.L.; Greendale, G.A. The relation of dietary vitamin C intake to bone mineral density: Results from the PEPI study. Calcif. Tissue Int. 1998, 63, 183–189. [Google Scholar] [CrossRef]
- Sanders, K.M.; Kotowicz, M.A.; Nicholson, G.C. Potential role of the antioxidant N-acetylcysteine in slowing bone resorption in early post-menopausal women: A pilot study. Transl. Res. 2007, 150, 215. [Google Scholar] [CrossRef]
- Morton, D.J.; Barrett-Connor, E.L.; Schneider, D.L. Vitamin C supplement use and bone mineral density in postmenopausal women. J. Bone Miner. Res. 2001, 16, 135–140. [Google Scholar] [CrossRef]
- Calcaterra, I.; Iannuzzo, G.; Dell’Aquila, F.; Di Minno, M.N.D. Pathophysiological Role of Synovitis in Hemophilic Arthropathy Development: A Two-Hit Hypothesis. Front. Physiol. 2020, 11, 541. [Google Scholar] [CrossRef]
- Øvlisen, K.; Kristensen, A.; Jensen, A.; Tranholm, M. IL-1β, IL-6, KC and MCP-1 are elevated in synovial fluid from haemophilic mice with experimentally induced haemarthrosis. Haemophilia 2009, 15, 802–810. [Google Scholar] [CrossRef]
- Rodriguez-Merchan, E. Haemophilic synovitis: Basic concepts. Haemophilia 2007, 13, 1–3. [Google Scholar] [CrossRef]
- Hitchon, C.A.; El-Gabalawy, H.S. Oxidation in rheumatoid arthritis. Arthritis Res. Ther. 2004, 6, 265–278. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, G.T.; Green, E.R.; Mecsas, J. Neutrophils to the ROScue: Mechanisms of NADPH Oxidase Activation and Bacterial Resistance. Front. Cell. Infect. Microbiol. 2017, 7, 373. [Google Scholar] [CrossRef]
- Miao, L.; St Clair, D.K. Regulation of superoxide dismutase genes: Implications in disease. Free Radic. Biol. Med. 2009, 47, 344–356. [Google Scholar] [CrossRef] [Green Version]
- Morgan, M.J.; Liu, Z.G. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [Green Version]
- Davies, M.J.; Hawkins, C.L. The Role of Myeloperoxidase in Biomolecule Modification, Chronic Inflammation, and Disease. Antioxid. Redox Signal. 2020, 32, 957–981. [Google Scholar] [CrossRef] [Green Version]
- Acharya, S.S.; Kaplan, R.N.; Macdonald, D.; Fabiyi, O.T.; DiMichele, D.; Lyden, D. Neoangiogenesis contributes to the development of hemophilic synovitis. Blood J. Am. Soc. Hematol. 2011, 117, 2484–2493. [Google Scholar]
- Acharya, S.S. Exploration of the pathogenesis of haemophilic joint arthropathy: Understanding implications for optimal clinical management. Br. J. Haematol. 2012, 156, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Zetterberg, E.; Palmblad, J.; Wallensten, R.; Morfini, M.; Melchiorre, D.; Holmström, M. Angiogenesis is increased in advanced haemophilic joint disease and characterised by normal pericyte coverage. Eur. J. Haematol. 2014, 92, 256–262. [Google Scholar] [PubMed]
- Etherington, P.J.; Winlove, P.; Taylor, P.; Paleolog, E.; Miotla, J.M. VEGF release is associated with reduced oxygen tensions in experimental inflammatory arthritis. Clin. Exp. Rheumatol. 2002, 20, 799–805. [Google Scholar]
- Biniecka, M.; Connolly, M.; Gao, W.; Ng, C.T.; Balogh, E.; Gogarty, M.; Santos, L.; Murphy, E.; Brayden, D.; Veale, D.J.; et al. Redox-Mediated Angiogenesis in the Hypoxic Joint of Inflammatory Arthritis. Arthritis Rheumatol. 2014, 66, 3300–3310. [Google Scholar] [CrossRef] [Green Version]
- Chenevier-Gobeaux, C.; Simonneau, C.; Lemarechal, H.; Bonnefont-Rousselot, D.; Poiraudeau, S.; Rannou, F.; Anract, P.; Borderie, D. Hypoxia induces nitric oxide synthase in rheumatoid synoviocytes: Consequences on NADPH oxidase regulation. Free. Radic. Res. 2012, 46, 628–636. [Google Scholar] [CrossRef]
- Bayraktutan, U.; Blayney, L.; Shah, A.M. Molecular characterization and localization of the NAD (P) H oxidase components gp91-phox and p22-phox in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1903–1911. [Google Scholar] [CrossRef] [Green Version]
- Haruna, Y.; Morita, Y.; Komai, N.; Yada, T.; Sakuta, T.; Tomita, N.; Fox, D.A.; Kashihara, N. Endothelial dysfunction in rat adjuvant-induced arthritis: Vascular superoxide production by NAD(P)H oxidase and uncoupled endothelial nitric oxide synthase. Arthritis Rheum 2006, 54, 1847–1855. [Google Scholar] [CrossRef] [Green Version]
- Ray, R.; Shah, A.M. NADPH oxidase and endothelial cell function. Clin. Sci. 2005, 109, 217–226. [Google Scholar] [CrossRef] [Green Version]
- Frey, R.S.; Ushio-Fukai, M.; Malik, A.B. NADPH oxidase-dependent signaling in endothelial cells: Role in physiology and pathophysiology. Antioxid. Redox Signal. 2009, 11, 791–810. [Google Scholar] [CrossRef]
- Manea, A. NADPH oxidase-derived reactive oxygen species: Involvement in vascular physiology and pathology. Cell Tissue Res. 2010, 342, 325–339. [Google Scholar] [CrossRef]
- Simonini, G.; Matucci Cerinic, M.; Cimaz, R.; Anichini, M.; Cesaretti, S.; Zoppi, M.; Generini, S.; Falcini, F. Evidence for immune activation against oxidized lipoproteins in inactive phases of juvenile chronic arthritis. J. Rheumatol. 2001, 28, 198–203. [Google Scholar]
- Taylor, P.C.; Sivakumar, B. Hypoxia and angiogenesis in rheumatoid arthritis. Curr. Opin. Rheumatol. 2005, 17, 293–298. [Google Scholar] [CrossRef]
- Garrido-Urbani, S.; Jemelin, S.; Deffert, C.; Carnesecchi, S.; Basset, O.; Szyndralewiez, C.; Heitz, F.; Page, P.; Montet, X.; Michalik, L.; et al. Targeting vascular NADPH oxidase 1 blocks tumor angiogenesis through a PPARα mediated mechanism. PLoS ONE 2011, 6, e14665. [Google Scholar] [CrossRef]
- Colavitti, R.; Pani, G.; Bedogni, B.; Anzevino, R.; Borrello, S.; Waltenberger, J.; Galeotti, T. Reactive oxygen species as downstream mediators of angiogenic signaling by vascular endothelial growth factor receptor-2/KDR. J. Biol. Chem. 2002, 277, 3101–3108. [Google Scholar] [CrossRef] [Green Version]
- Roussel, N.A.; Chantrain, V.A.; Foubert, A.; Lambert, C.; Hermans, C.; Meeus, M.; Guillaume, S.; Lecouvet, F.; Krüger, S.; Hilberg, T.; et al. Gaining more insight into ankle pain in haemophilia: A study exploring pain, structural and functional evaluation of the ankle joint. Haemophilia 2022, 28, 480–490. [Google Scholar] [CrossRef]
- Humphries, T.J.; Kessler, C.M. Managing chronic pain in adults with haemophilia: Current status and call to action. Haemophilia 2015, 21, 41–51. [Google Scholar] [CrossRef]
- Holstein, K.; Klamroth, R.; Richards, M.; Carvalho, M.; Pérez-Garrido, R.; Gringeri, A. Pain management in patients with haemophilia: A European survey. Haemophilia 2012, 18, 743–752. [Google Scholar] [CrossRef] [Green Version]
- Humphries, T.J.; Kessler, C.M. The challenge of pain evaluation in haemophilia: Can pain evaluation and quantification be improved by using pain instruments from other clinical situations? Haemophilia 2013, 19, 181–187. [Google Scholar] [CrossRef]
- Koop, S.M.; ten Klooster, P.M.; Vonkeman, H.E.; Steunebrink, L.M.; van de Laar, M.A. Neuropathic-like pain features and cross-sectional associations in rheumatoid arthritis. Arthritis Res. Ther. 2015, 17, 237. [Google Scholar] [CrossRef] [Green Version]
- Eitner, A.; Hofmann, G.O.; Schaible, H.-G. Mechanisms of Osteoarthritic Pain. Studies in Humans and Experimental Models. Front. Mol. Neurosci. 2017, 10, 349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colloca, L.; Ludman, T.; Bouhassira, D.; Baron, R.; Dickenson, A.H.; Yarnitsky, D.; Freeman, R.; Truini, A.; Attal, N.; Finnerup, N.B.; et al. Neuropathic pain. Nat. Rev. Dis. Primers 2017, 3, 17002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicholson, B. Differential diagnosis: Nociceptive and neuropathic pain. Am. J. Manag. Care 2006, 12, S256–S262. [Google Scholar] [PubMed]
- St John Smith, E. Advances in understanding nociception and neuropathic pain. J. Neurol. 2018, 265, 231–238. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.H.; Seo, H.J.; Abdi, S.; Huh, B. All about pain pharmacology: What pain physicians should know. Korean J. Pain 2020, 33, 108–120. [Google Scholar] [CrossRef] [Green Version]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxid. Med. Cell Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef]
- Hendrix, J.; Nijs, J.; Ickmans, K.; Godderis, L.; Ghosh, M.; Polli, A. The Interplay between Oxidative Stress, Exercise, and Pain in Health and Disease: Potential Role of Autonomic Regulation and Epigenetic Mechanisms. Antioxidants 2020, 9, 1166. [Google Scholar] [CrossRef]
- Quiñonez-Flores, C.M.; González-Chávez, S.A.; Del Río Nájera, D.; Pacheco-Tena, C. Oxidative Stress Relevance in the Pathogenesis of the Rheumatoid Arthritis: A Systematic Review. Biomed. Res. Int. 2016, 2016, 6097417. [Google Scholar] [CrossRef] [Green Version]
- Telen, M.J.; Malik, P.; Vercellotti, G.M. Therapeutic strategies for sickle cell disease: Towards a multi-agent approach. Nat. Rev. Drug. Discov. 2019, 18, 139–158. [Google Scholar] [CrossRef]
- Wauquier, F.; Leotoing, L.; Coxam, V.; Guicheux, J.; Wittrant, Y. Oxidative stress in bone remodelling and disease. Trends Mol. Med. 2009, 15, 468–477. [Google Scholar] [CrossRef]
- La Hausse De Lalouviere, L.; Morice, O.; Fitzgerald, M. Altered sensory innervation and pain hypersensitivity in a model of young painful arthritic joints: Short- and long-term effects. Inflamm. Res. 2021, 70, 483–493. [Google Scholar] [CrossRef]
- Syx, D.; Tran, P.B.; Miller, R.E.; Malfait, A.M. Peripheral Mechanisms Contributing to Osteoarthritis Pain. Curr. Rheumatol. Rep. 2018, 20, 9. [Google Scholar] [CrossRef]
- Klafke, J.Z.; da Silva, M.A.; Rossato, M.F.; de Prá, S.D.; Rigo, F.K.; Walker, C.I.; Bochi, G.V.; Moresco, R.N.; Ferreira, J.; Trevisan, G. Acute and chronic nociceptive phases observed in a rat hind paw ischemia/reperfusion model depend on different mechanisms. Pflugers Arch. 2016, 468, 229–241. [Google Scholar] [CrossRef]
- Pan, L.; Yu, L.; Wang, L.; He, J.; Sun, J.; Wang, X.; Wang, H.; Bai, Z.; Feng, H.; Pei, H. Inflammatory stimuli promote oxidative stress in pancreatic acinar cells via Toll-like receptor 4/nuclear factor-κB pathway. Int. J. Mol. Med. 2018, 42, 3582–3590. [Google Scholar] [CrossRef] [Green Version]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive oxygen species in inflammation and tissue injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef] [Green Version]
- Zahan, O.M.; Serban, O.; Gherman, C.; Fodor, D. The evaluation of oxidative stress in osteoarthritis. Med. Pharm. Rep. 2020, 93, 12–22. [Google Scholar] [CrossRef]
- Bevan, S.; Quallo, T.; Andersson, D.A. TRPV1. In Mammalian Transient Receptor Potential (TRP) Cation Channels: Volume I.; Nilius, B., Flockerzi, V., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 207–245. [Google Scholar]
- Patwardhan, A.M.; Akopian, A.N.; Ruparel, N.B.; Diogenes, A.; Weintraub, S.T.; Uhlson, C.; Murphy, R.C.; Hargreaves, K.M. Heat generates oxidized linoleic acid metabolites that activate TRPV1 and produce pain in rodents. J. Clin. Investig. 2010, 120, 1617–1626. [Google Scholar] [CrossRef]
- Mills, C.D.; Nguyen, T.; Tanga, F.Y.; Zhong, C.; Gauvin, D.M.; Mikusa, J.; Gomez, E.J.; Salyers, A.K.; Bannon, A.W. Characterization of nerve growth factor-induced mechanical and thermal hypersensitivity in rats. Eur. J. Pain 2013, 17, 469–479. [Google Scholar] [CrossRef]
- Eskander, M.A.; Ruparel, S.; Green, D.P.; Chen, P.B.; Por, E.D.; Jeske, N.A.; Gao, X.; Flores, E.R.; Hargreaves, K.M. Persistent Nociception Triggered by Nerve Growth Factor (NGF) Is Mediated by TRPV1 and Oxidative Mechanisms. J. Neurosci. 2015, 35, 8593–8603. [Google Scholar] [CrossRef] [Green Version]
- St-Jacques, B.; Ma, W. Peripheral prostaglandin E2 prolongs the sensitization of nociceptive dorsal root ganglion neurons possibly by facilitating the synthesis and anterograde axonal trafficking of EP4 receptors. Exp. Neurol. 2014, 261, 354–366. [Google Scholar] [CrossRef]
- La Hausse de Lalouvière, L.; Ioannou, Y.; Fitzgerald, M. Neural mechanisms underlying the pain of juvenile idiopathic arthritis. Nat. Rev. Rheumatol. 2014, 10, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Ji, R.R.; Chamessian, A.; Zhang, Y.Q. Pain regulation by non-neuronal cells and inflammation. Science 2016, 354, 572–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwok, C.H.T.; Learoyd, A.E.; Canet-Pons, J.; Trang, T.; Fitzgerald, M. Spinal interleukin-6 contributes to central sensitisation and persistent pain hypersensitivity in a model of juvenile idiopathic arthritis. Brain Behav. Immun. 2020, 90, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Gopal, S.; Barnes, R.F.W.; Cooke, E.J.; Zhou, J.Y.; Levin, I.; Emery, P.; Hughes, T.H.; Karsdal, M.A.; Manon-Jensen, T.; von Drygalski, A. Systemic vascular basement membrane markers linked to synovial vascular remodeling are biomarkers of hemarthrosis in patients with hemophilia. J. Thromb. Haemost. 2021, 19, 1200–1211. [Google Scholar] [CrossRef]
- Krüger, S.; Hilberg, T. Understanding the pain profile in patients with haemophilia: Impaired descending pain inhibition as measured by conditioned pain modulation. Haemophilia 2020, 26, 236–242. [Google Scholar] [CrossRef] [Green Version]
- Ghilardi, J.R.; Freeman, K.T.; Jimenez-Andrade, J.M.; Coughlin, K.A.; Kaczmarska, M.J.; Castaneda-Corral, G.; Bloom, A.P.; Kuskowski, M.A.; Mantyh, P.W. Neuroplasticity of sensory and sympathetic nerve fibers in a mouse model of a painful arthritic joint. Arthritis Rheum. 2012, 64, 2223–2232. [Google Scholar] [CrossRef]
- Longo, G.; Osikowicz, M.; Ribeiro-da-Silva, A. Sympathetic fiber sprouting in inflamed joints and adjacent skin contributes to pain-related behavior in arthritis. J. Neurosci. 2013, 33, 10066–10074. [Google Scholar] [CrossRef]
- Ashraf, S.; Wibberley, H.; Mapp, P.I.; Hill, R.; Wilson, D.; Walsh, D.A. Increased vascular penetration and nerve growth in the meniscus: A potential source of pain in osteoarthritis. Ann. Rheum. Dis. 2011, 70, 523–529. [Google Scholar] [CrossRef]
- Carrasco, C.; Naziroǧlu, M.; Rodríguez, A.B.; Pariente, J.A. Neuropathic Pain: Delving into the Oxidative Origin and the Possible Implication of Transient Receptor Potential Channels. Front. Physiol. 2018, 9, 95. [Google Scholar] [CrossRef]
- Bennett, G.J.; Xie, Y.K. A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain 1988, 33, 87–107. [Google Scholar] [CrossRef]
- Naik, A.K.; Tandan, S.K.; Dudhgaonkar, S.P.; Jadhav, S.H.; Kataria, M.; Prakash, V.R.; Kumar, D. Role of oxidative stress in pathophysiology of peripheral neuropathy and modulation by N-acetyl-L-cysteine in rats. Eur. J. Pain 2006, 10, 573–579. [Google Scholar] [CrossRef]
- Yowtak, J.; Lee, K.Y.; Kim, H.Y.; Wang, J.; Kim, H.K.; Chung, K.; Chung, J.M. Reactive oxygen species contribute to neuropathic pain by reducing spinal GABA release. Pain 2011, 152, 844–852. [Google Scholar] [CrossRef] [Green Version]
- De Logu, F.; Nassini, R.; Materazzi, S.; Carvalho Gonçalves, M.; Nosi, D.; Rossi Degl’Innocenti, D.; Marone, I.M.; Ferreira, J.; Li Puma, S.; Benemei, S. Schwann cell TRPA1 mediates neuroinflammation that sustains macrophage-dependent neuropathic pain in mice. Nat. Commun. 2017, 8, 1–16. [Google Scholar] [CrossRef]
- Grace, P.M.; Gaudet, A.D.; Staikopoulos, V.; Maier, S.F.; Hutchinson, M.R.; Salvemini, D.; Watkins, L.R. Nitroxidative Signaling Mechanisms in Pathological Pain. Trends Neurosci. 2016, 39, 862–879. [Google Scholar] [CrossRef] [Green Version]
- Salvemini, D.; Little, J.W.; Doyle, T.; Neumann, W.L. Roles of reactive oxygen and nitrogen species in pain. Free. Radic. Biol. Med. 2011, 51, 951–966. [Google Scholar] [CrossRef] [Green Version]
- Areti, A.; Yerra, V.G.; Naidu, V.; Kumar, A. Oxidative stress and nerve damage: Role in chemotherapy induced peripheral neuropathy. Redox Biol. 2014, 2, 289–295. [Google Scholar] [CrossRef] [Green Version]
- Hongpaisan, J.; Winters, C.A.; Andrews, S.B. Strong calcium entry activates mitochondrial superoxide generation, upregulating kinase signaling in hippocampal neurons. J. Neurosci. 2004, 24, 10878–10887. [Google Scholar] [CrossRef] [Green Version]
- Lai, B.; Zhang, L.; Dong, L.-Y.; Zhu, Y.-H.; Sun, F.-Y.; Zheng, P. Impact of inhibition of Qo site of mitochondrial complex III with myxothiazol on persistent sodium currents via superoxide and protein kinase C in rat hippocampal CA1 cells. Neurobiol. Dis. 2006, 21, 206–216. [Google Scholar] [CrossRef]
- Fang, L.; Wu, J.; Lin, Q.; Willis, W.D. Protein kinases regulate the phosphorylation of the GluR1 subunit of AMPA receptors of spinal cord in rats following noxious stimulation. Mol. Brain Res. 2003, 118, 160–165. [Google Scholar] [CrossRef]
- Fischer, M.J.; Reeh, P.W. Sensitization to heat through G-protein-coupled receptor pathways in the isolated sciatic mouse nerve. Eur. J. Neurosci. 2007, 25, 3570–3575. [Google Scholar] [CrossRef]
- Sculptoreanu, A.; Aura Kullmann, F.; De Groat, W.C. Neurokinin 2 receptor-mediated activation of protein kinase C modulates capsaicin responses in DRG neurons from adult rats. Eur. J. Neurosci. 2008, 27, 3171–3181. [Google Scholar] [CrossRef] [Green Version]
- Kohno, T.; Wang, H.; Amaya, F.; Brenner, G.J.; Cheng, J.-K.; Ji, R.-R.; Woolf, C.J. Bradykinin enhances AMPA and NMDA receptor activity in spinal cord dorsal horn neurons by activating multiple kinases to produce pain hypersensitivity. J. Neurosci. 2008, 28, 4533–4540. [Google Scholar] [CrossRef]
- Li, K.-C.; Zheng, J.-H.; Chen, J. Involvement of spinal protein kinase C in induction and maintenance of both persistent spontaneous flinching reflex and contralateral heat hyperalgesia induced by subcutaneous bee venom in the conscious rat. Neurosci. Lett. 2000, 285, 103–106. [Google Scholar] [CrossRef]
- Grabowska, W.; Sikora, E.; Bielak-Zmijewska, A. Sirtuins, a promising target in slowing down the ageing process. Biogerontology 2017, 18, 447–476. [Google Scholar] [CrossRef] [Green Version]
- Ilari, S.; Giancotti, L.A.; Lauro, F.; Gliozzi, M.; Malafoglia, V.; Palma, E.; Tafani, M.; Russo, M.A.; Tomino, C.; Fini, M.; et al. Natural Antioxidant Control of Neuropathic Pain-Exploring the Role of Mitochondrial SIRT3 Pathway. Antioxidants 2020, 9, 1103. [Google Scholar] [CrossRef]
- Song, F.H.; Liu, D.Q.; Zhou, Y.Q.; Mei, W. SIRT1: A promising therapeutic target for chronic pain. CNS Neurosci. Ther. 2022, 28, 818–828. [Google Scholar] [CrossRef]
- Cheng, A.; Yang, Y.; Zhou, Y.; Maharana, C.; Lu, D.; Peng, W.; Liu, Y.; Wan, R.; Marosi, K.; Misiak, M. Mitochondrial SIRT3 mediates adaptive responses of neurons to exercise and metabolic and excitatory challenges. Cell Metab. 2016, 23, 128–142. [Google Scholar] [CrossRef] [Green Version]
- Ilari, S.; Giancotti, L.A.; Lauro, F.; Dagostino, C.; Gliozzi, M.; Malafoglia, V.; Sansone, L.; Palma, E.; Tafani, M.; Russo, M.A. Antioxidant modulation of sirtuin 3 during acute inflammatory pain: The ROS control. Pharmacol. Res. 2020, 157, 104851. [Google Scholar] [CrossRef]
- Gao, J.; Zheng, Z.; Gu, Q.; Chen, X.; Liu, X.; Xu, X. Deacetylation of MnSOD by PARP-regulated SIRT3 protects retinal capillary endothelial cells from hyperglycemia-induced damage. Biochem. Biophys. Res. Commun. 2016, 472, 425–431. [Google Scholar] [CrossRef]
- Muscoli, C.; Lauro, F.; Dagostino, C.; Ilari, S.; Giancotti, L.; Gliozzi, M.; Costa, N.; Carresi, C.; Musolino, V.; Casale, F. Olea Europea-derived phenolic products attenuate antinociceptive morphine tolerance: An innovative strategic approach to treat cancer pain. J. Biol. Regul. Homeost. Agents 2014, 28, 105–116. [Google Scholar]
- Lauro, F.; Giancotti, L.A.; Ilari, S.; Dagostino, C.; Gliozzi, M.; Morabito, C.; Malafoglia, V.; Raffaeli, W.; Muraca, M.; Goffredo, B.M. Inhibition of spinal oxidative stress by bergamot polyphenolic fraction attenuates the development of morphine induced tolerance and hyperalgesia in mice. PLoS ONE 2016, 11, e0156039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, I.; Chung, S. Dietary polyphenols, deacetylases and chromatin remodeling in inflammation. J. Nutrigenet. Nutr. 2010, 3, 220–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, Q.; Dong, W.; Zhang, L.; Yang, X. Activating Sirt1 by resveratrol suppresses Nav1.7 expression in DRG through miR-182 and alleviates neuropathic pain in rats. Channels 2020, 14, 69–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benameur, T.; Soleti, R.; Panaro, M.A.; La Torre, M.E.; Monda, V.; Messina, G.; Porro, C. Curcumin as Prospective Anti-Aging Natural Compound: Focus on Brain. Molecules 2021, 26, 4794. [Google Scholar] [CrossRef] [PubMed]
- Basu, P.; Basu, A. In Vitro and In Vivo Effects of Flavonoids on Peripheral Neuropathic Pain. Molecules 2020, 25, 1171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akbar, S.; Subhan, F.; Karim, N.; Shahid, M.; Ahmad, N.; Ali, G.; Mahmood, W.; Fawad, K. 6-Methoxyflavanone attenuates mechanical allodynia and vulvodynia in the streptozotocin-induced diabetic neuropathic pain. Biomed. Pharmacother. 2016, 84, 962–971. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.S.; Mamun, A.A.; Rahman, M.A.; Kabir, M.T.; Alkahtani, S.; Alanazi, I.S.; Perveen, A.; Ashraf, G.M.; Bin-Jumah, M.N.; Abdel-Daim, M.M. Exploring the Promise of Flavonoids to Combat Neuropathic Pain: From Molecular Mechanisms to Therapeutic Implications. Front. Neurosci. 2020, 14, 478. [Google Scholar] [CrossRef]
- Yamashita, S.; Matsuzawa, Y. Where are we with probucol: A new life for an old drug? Atherosclerosis 2009, 207, 16–23. [Google Scholar] [CrossRef]
- Derangula, K.; Javalgekar, M.; Kumar Arruri, V.; Gundu, C.; Kumar Kalvala, A.; Kumar, A. Probucol attenuates NF-κB/NLRP3 signalling and augments Nrf-2 mediated antioxidant defence in nerve injury induced neuropathic pain. Int. Immunopharmacol. 2022, 102, 108397. [Google Scholar] [CrossRef]
- Basu, P.; Averitt, D.L.; Maier, C.; Basu, A. The Effects of Nuclear Factor Erythroid 2 (NFE2)-Related Factor 2 (Nrf2) Activation in Preclinical Models of Peripheral Neuropathic Pain. Antioxidants 2022, 11, 430. [Google Scholar] [CrossRef]
- Santos, M.C.Q.; Silva, T.; Silva, F.; Siebert, C.; Kroth, A.; Silveira, E.M.S.; Wyse, A.T.S.; Partata, W.A. Effects of vitamin D administration on nociception and spinal cord pro-oxidant and antioxidant markers in a rat model of neuropathic pain. Braz. J. Med. Biol. Res. 2021, 54, e11207. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fouda, R.; Argueta, D.A.; Gupta, K. Pain in Hemophilia: Unexplored Role of Oxidative Stress. Antioxidants 2022, 11, 1113. https://doi.org/10.3390/antiox11061113
Fouda R, Argueta DA, Gupta K. Pain in Hemophilia: Unexplored Role of Oxidative Stress. Antioxidants. 2022; 11(6):1113. https://doi.org/10.3390/antiox11061113
Chicago/Turabian StyleFouda, Raghda, Donovan A. Argueta, and Kalpna Gupta. 2022. "Pain in Hemophilia: Unexplored Role of Oxidative Stress" Antioxidants 11, no. 6: 1113. https://doi.org/10.3390/antiox11061113
APA StyleFouda, R., Argueta, D. A., & Gupta, K. (2022). Pain in Hemophilia: Unexplored Role of Oxidative Stress. Antioxidants, 11(6), 1113. https://doi.org/10.3390/antiox11061113