

YPL-001 Shows Various Beneficial Effects against Cigarette Smoke Extract-Induced Emphysema Formation: Anti-Inflammatory, Anti-Oxidative, and Anti-Apoptotic Effects


Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Reagents
2.2. CSE Preparation
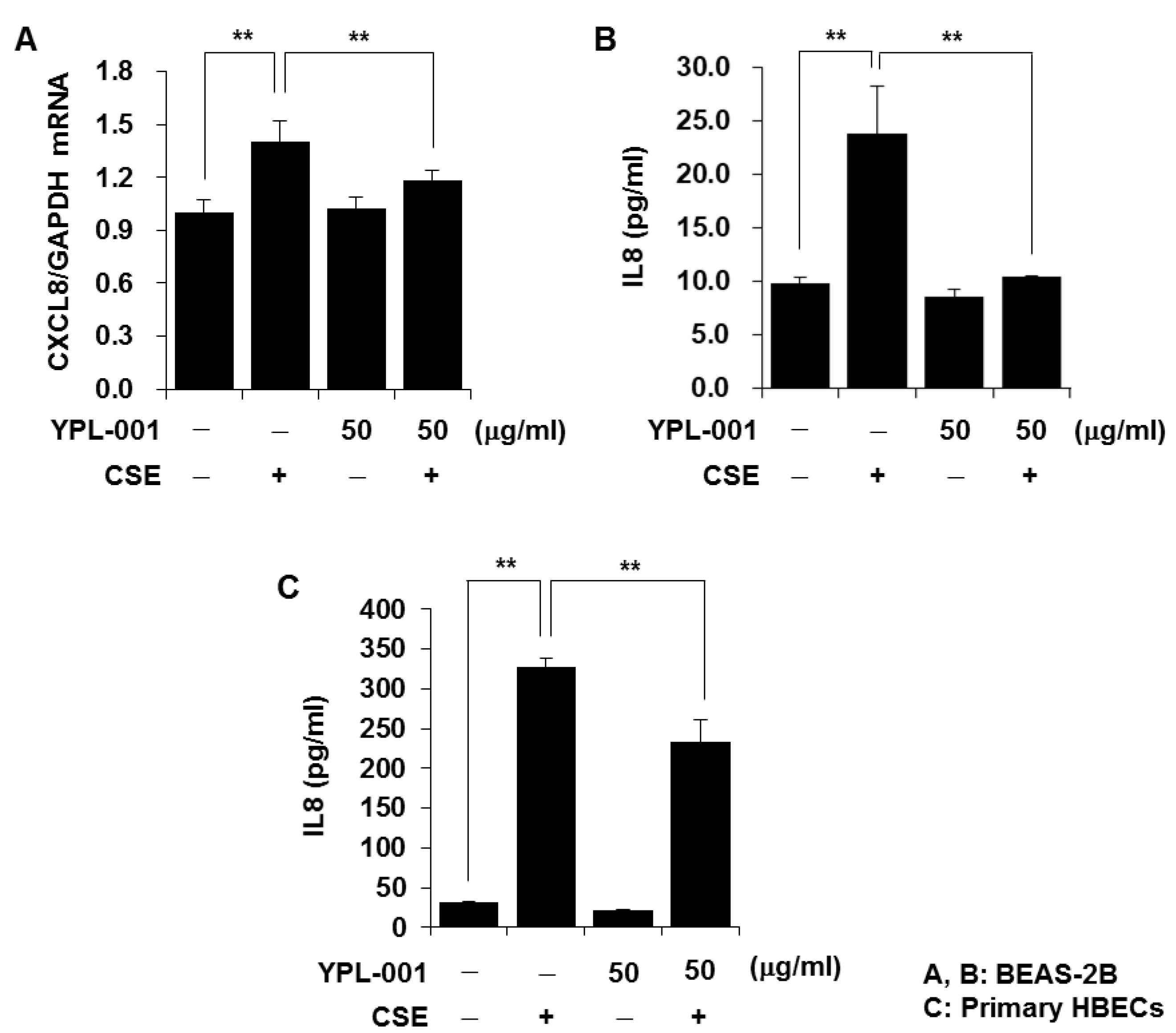
2.3. Determination of Cytokine Secretion
2.4. Quantitative Real-Time PCR
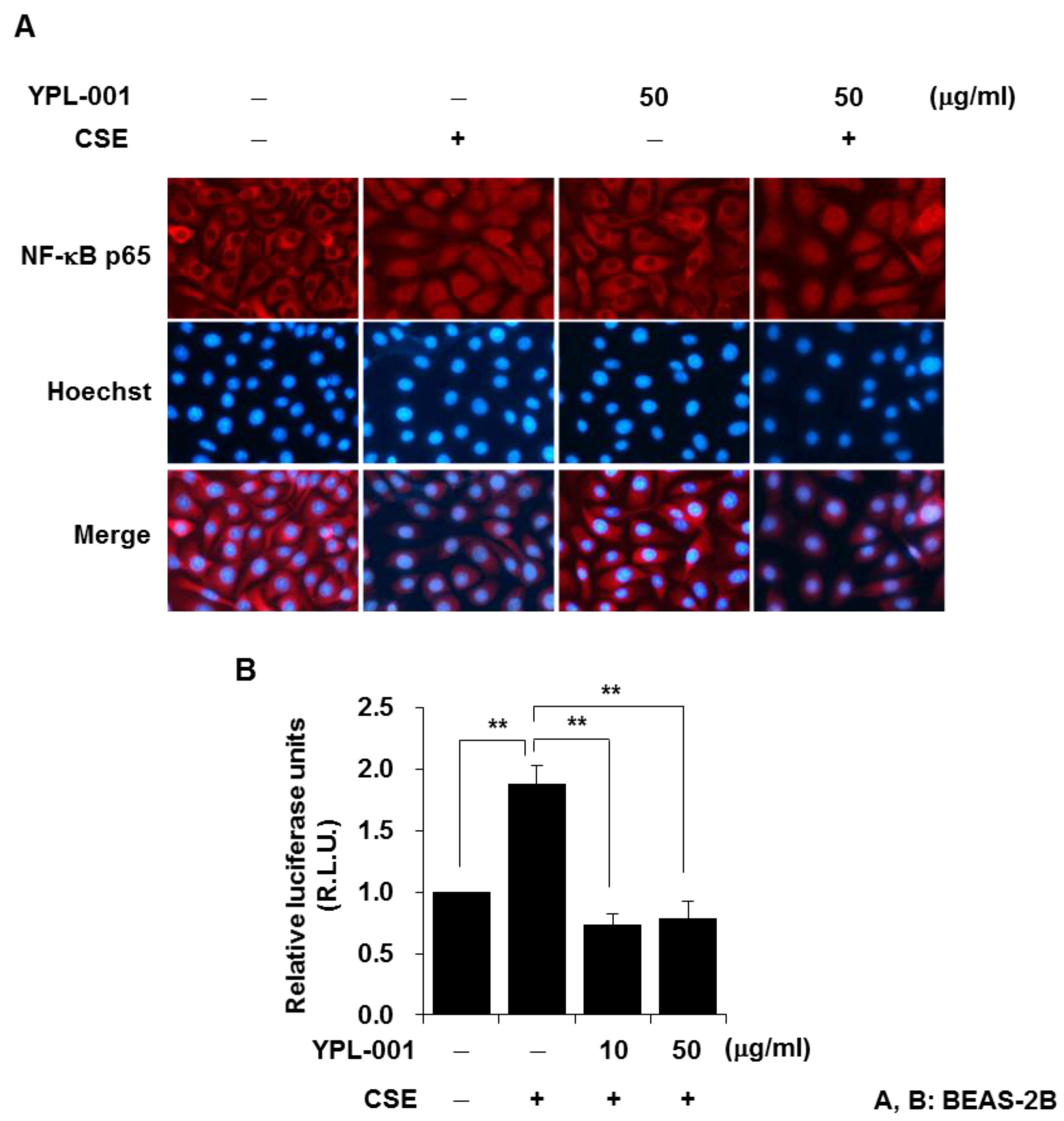
2.5. NF-κB p65 Immunofluorescent Staining
2.6. NF-κB Luciferase Activity Assay
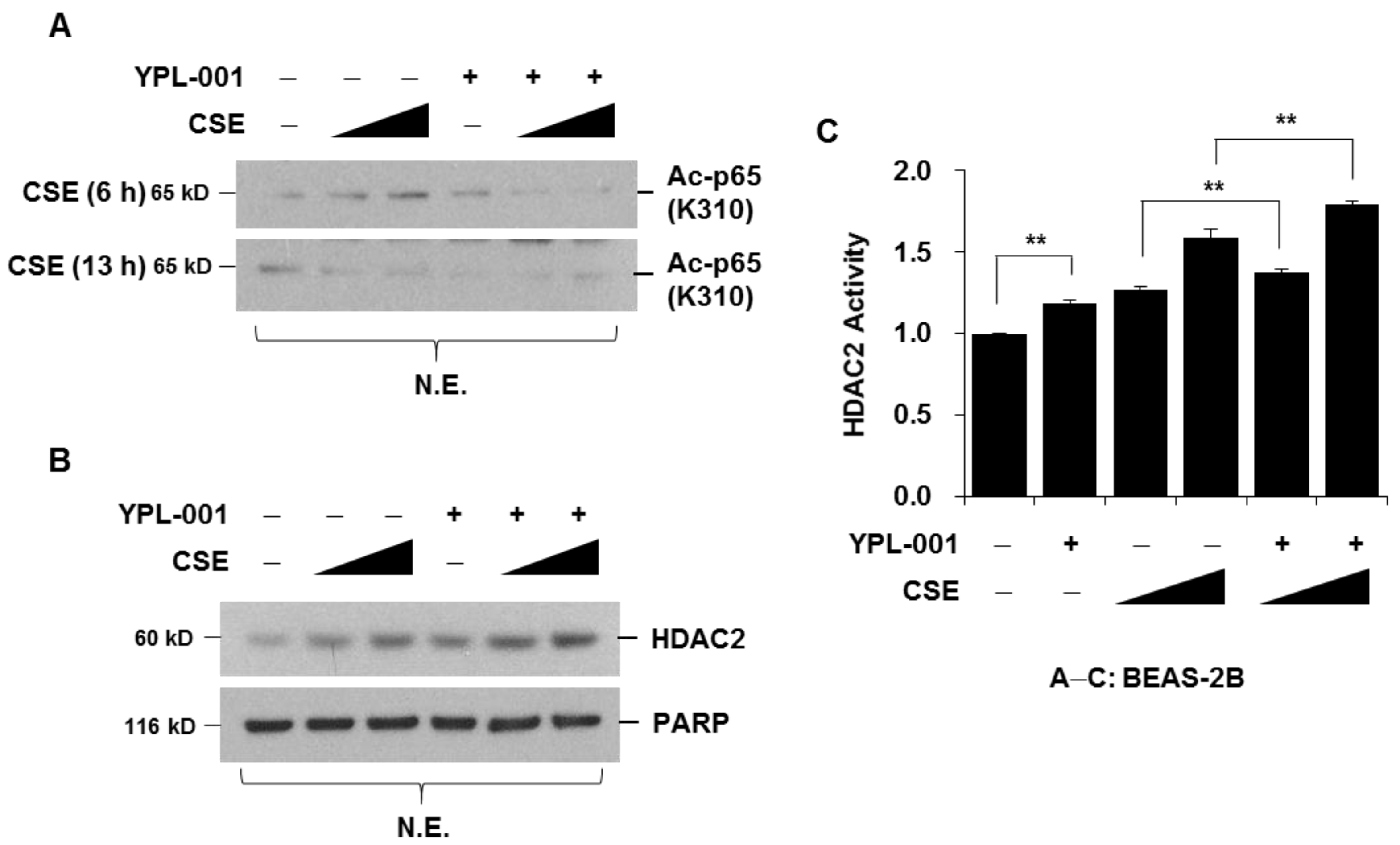
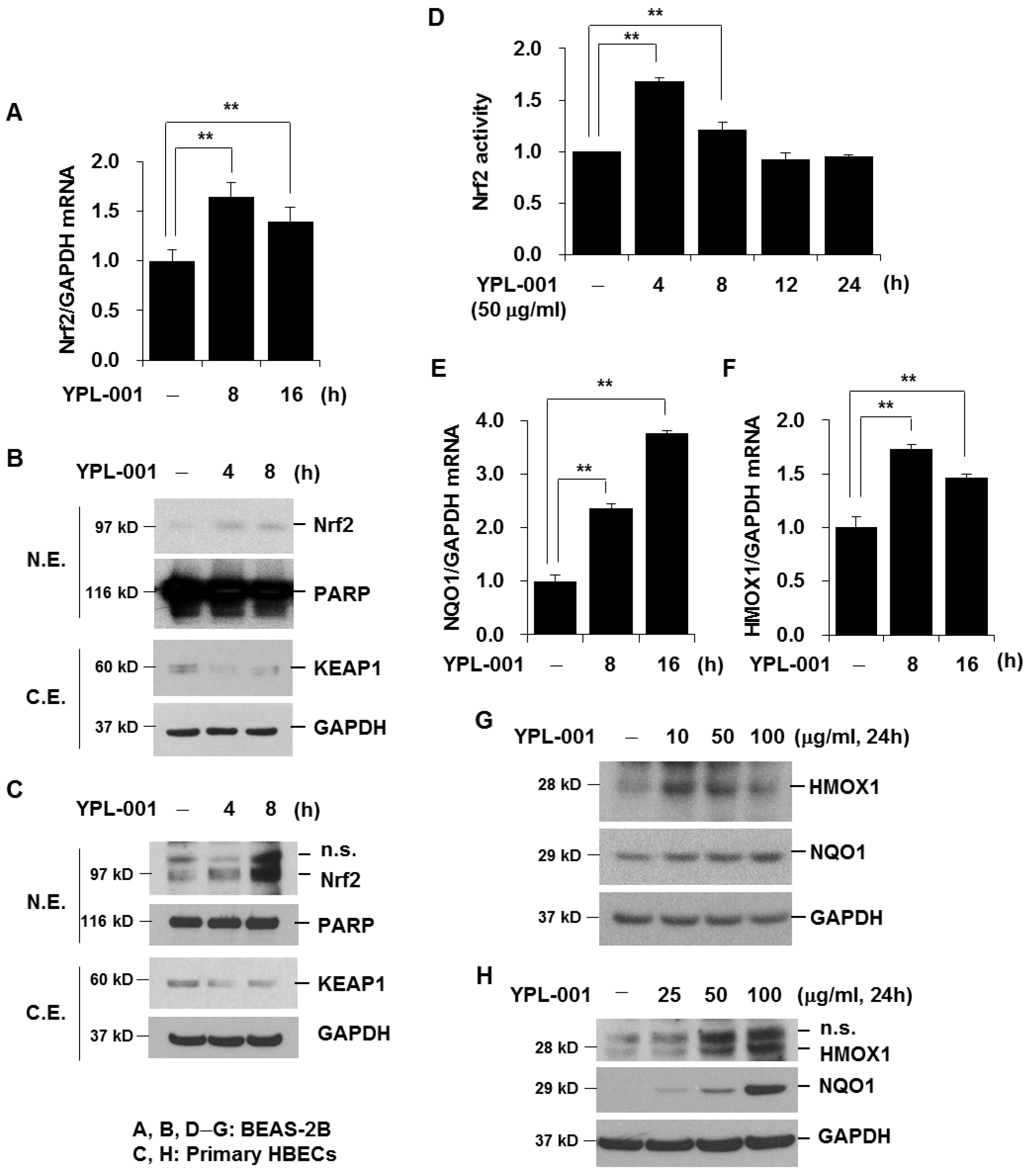
2.7. Protein Extraction and Western Blot Analysis
2.8. HDAC2 Activity Assay
2.9. Nrf2 Activity Assay
2.10. Lactate Dehydrogenase (LDH) Release Assay
2.11. Mouse Model
2.12. Measurement of Emphysema
2.13. Analysis of BALF
2.14. TUNEL Assay
2.15. Statistical Analysis
3. Results
3.1. YPL-001 Suppresses CSE-Induced IL8 Production
3.2. YPL-001 Suppresses Transcriptional Activity of NF-κB
3.3. YPL-001 Suppresses CSE-Induced Acetylation of NF-κB via Increase in HDAC2 Activity
3.4. YPL-001 Activates Nrf2
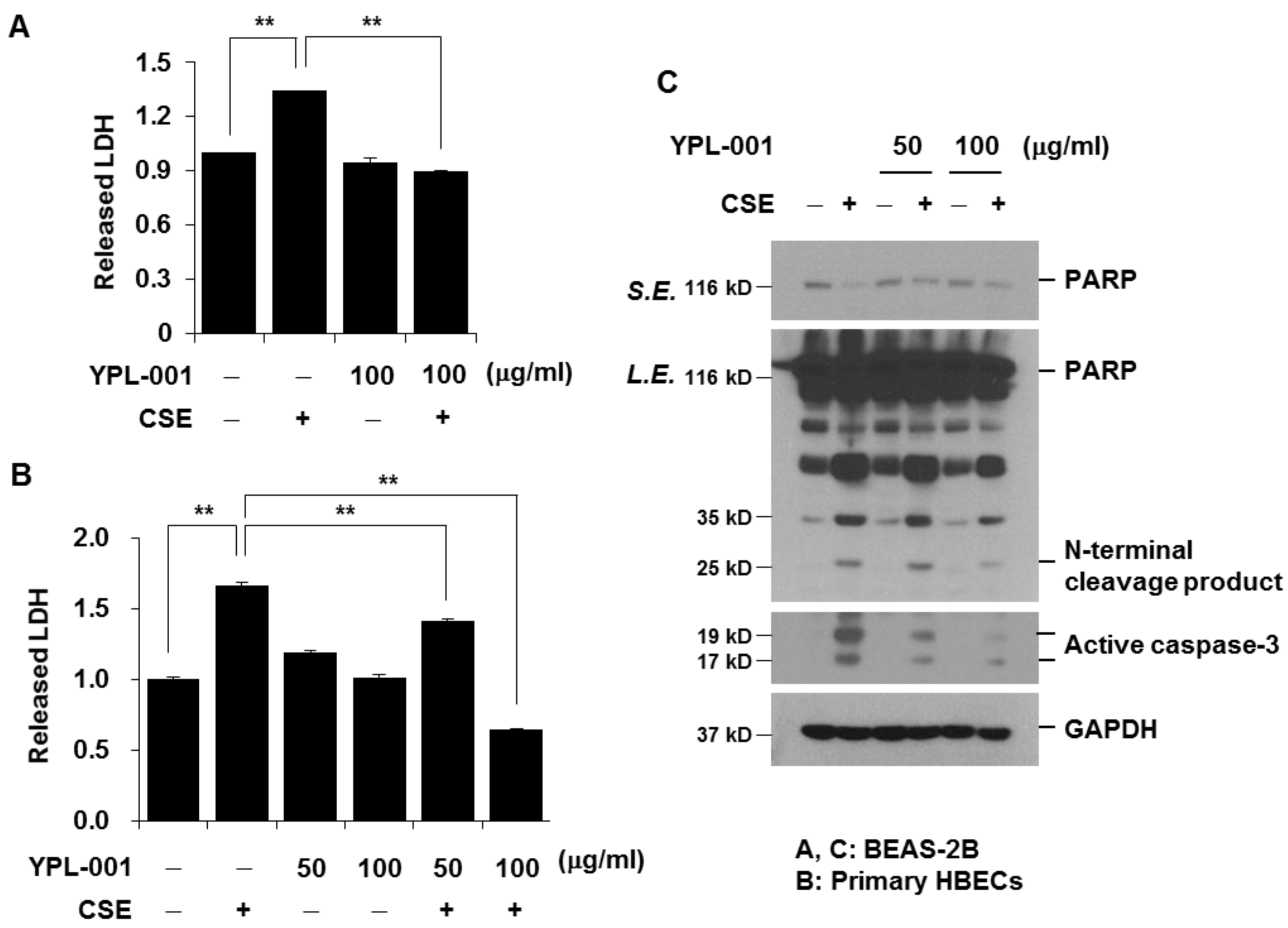
3.5. YPL-001 Suppresses CSE-Induced Apoptotic Cell Death
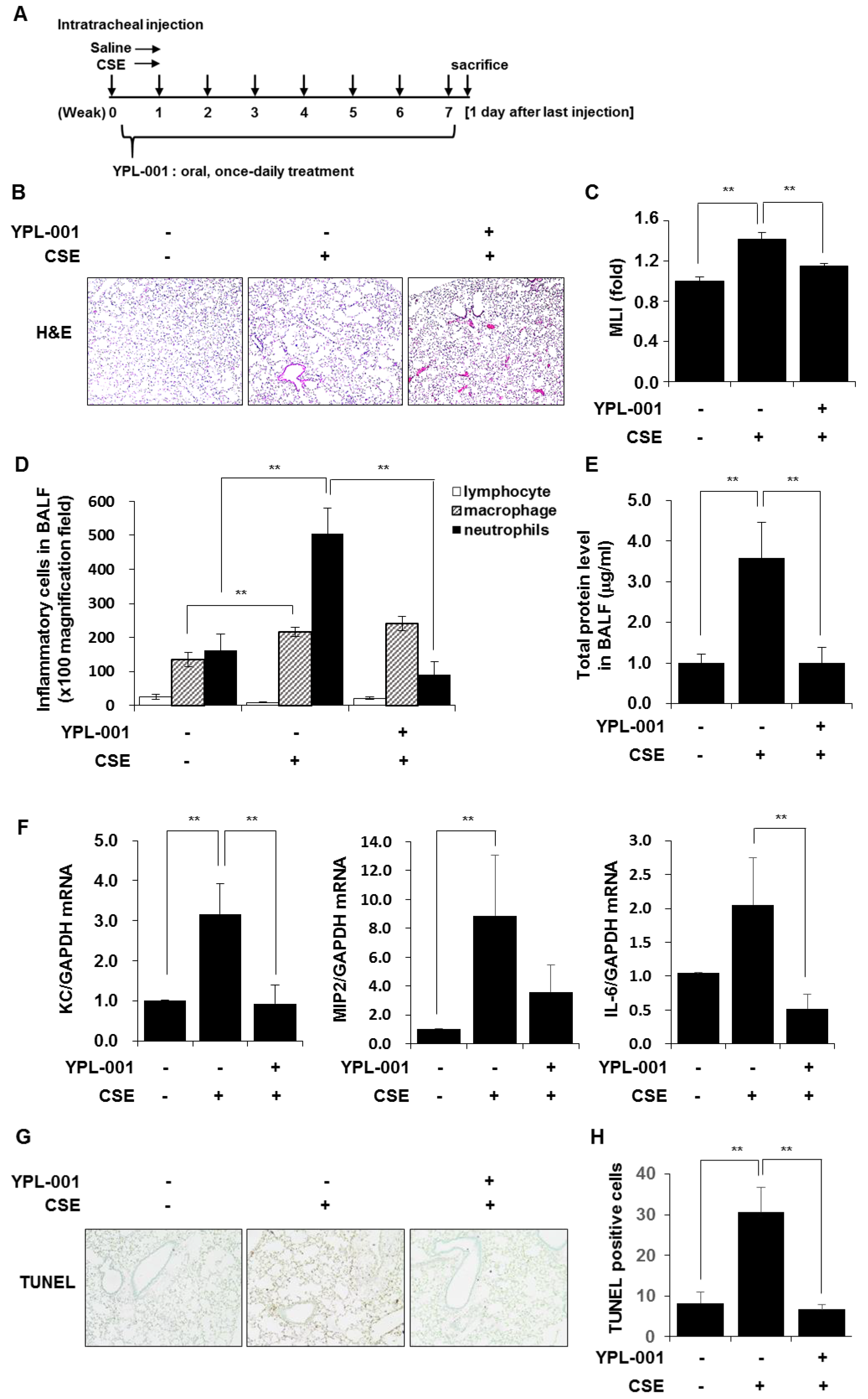
3.6. YPL-001 Reduced CSE-Induced Emphysematous Formation via Multiple Mechanisms Such as Anti-Inflammatory and Anti-Apoptotic Effects
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Christenson, S.A.; Smith, B.M.; Bafadhel, M.; Putcha, N. Chronic obstructive pulmonary disease. Lancet 2022, 399, 2227–2242. [Google Scholar] [CrossRef] [PubMed]
- Agustí, A.; Hogg, J.C. Update on the Pathogenesis of Chronic Obstructive Pulmonary Disease. N. Engl. J. Med. 2019, 381, 1248–1256. [Google Scholar] [CrossRef] [PubMed]
- Hoenderdos, K.; Condliffe, A. The neutrophil in chronic obstructive pulmonary disease. Am. J. Respir. Cell. Mol. Biol. 2013, 48, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zheng, H.; Zhang, H.; Ma, W.; Wang, F.; Liu, C.; He, S. Increased interleukin (IL)-8 and decreased IL-17 production in chronic obstructive pulmonary disease (COPD) provoked by cigarette smoke. Cytokine 2011, 56, 717–725. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. Immunology of asthma and chronic obstructive pulmonary disease. Nat. Rev. Immunol. 2008, 8, 183–192. [Google Scholar] [CrossRef]
- Lee, K.H.; Lee, C.H.; Jeong, J.; Jang, A.H.; Yoo, C.G. Neutrophil Elastase Differentially Regulates Interleukin 8 (IL-8) and Vascular Endothelial Growth Factor (VEGF) Production by Cigarette Smoke Extract. J. Biol. Chem. 2015, 290, 28438–28445. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.R.; Chida, A.S.; Bauter, M.R.; Shafiq, N.; Seweryniak, K.; Maggirwar, S.B.; Kilty, I.; Rahman, I. Cigarette smoke induces proinflammatory cytokine release by activation of NF-kappaB and posttranslational modifications of histone deacetylase in macrophages. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 291, L46–L57. [Google Scholar] [CrossRef] [Green Version]
- Baldwin, A.S., Jr. The NF-kappa B and I kappa B proteins: New discoveries and insights. Annu. Rev. Immunol. 1996, 14, 649–683. [Google Scholar] [CrossRef] [Green Version]
- Barnes, P.J.; Karin, M. Nuclear factor-kappaB: A pivotal transcription factor in chronic inflammatory diseases. N. Engl. J. Med. 1997, 336, 1066–1071. [Google Scholar] [CrossRef]
- Ghosh, S.; May, M.J.; Kopp, E.B. NF-kappa B and Rel proteins: Evolutionarily conserved mediators of immune responses. Annu. Rev. Immunol. 1998, 16, 225–260. [Google Scholar] [CrossRef]
- Boutten, A.; Goven, D.; Artaud-Macari, E.; Boczkowski, J.; Bonay, M. NRF2 targeting: A promising therapeutic strategy in chronic obstructive pulmonary disease. Trends Mol. Med. 2011, 17, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Rangasamy, T.; Cho, C.Y.; Thimmulappa, R.K.; Zhen, L.; Srisuma, S.S.; Kensler, T.W.; Yamamoto, M.; Petrache, I.; Tuder, R.M.; Biswal, S. Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke-induced emphysema in mice. J. Clin. Investig. 2004, 114, 1248–1259. [Google Scholar] [CrossRef] [PubMed]
- Raval, C.M.; Lee, P.J. Heme oxygenase-1 in lung disease. Curr. Drug Targets 2010, 11, 1532–1540. [Google Scholar] [CrossRef] [PubMed]
- Song, H.H.; Shin, I.S.; Woo, S.Y.; Lee, S.U.; Sung, M.H.; Ryu, H.W.; Kim, D.Y.; Ahn, K.S.; Lee, H.K.; Lee, D.; et al. Piscroside C, a novel iridoid glycoside isolated from Pseudolysimachion rotundum var. subinegrum suppresses airway inflammation induced by cigarette smooke. J. Ethnopharmacol. 2015, 170, 20–27. [Google Scholar] [CrossRef]
- Lee, K.H.; Jeong, J.; Koo, Y.J.; Jang, A.H.; Lee, C.H.; Yoo, C.G. Exogenous neutrophil elastase enters bronchial epithelial cells and suppresses cigarette smoke extract-induced heme oxygenase-1 by cleaving sirtuin 1. J. Biol. Chem. 2017, 292, 11970–11979. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.H.; Woo, J.; Kim, J.Y.; Lee, C.H.; Yoo, C.G. Cigarette smoke extract-induced downregulation of p300 is responsible for the impaired inflammatory cytokine response of macrophages. Cell. Signal. 2021, 85, 110050. [Google Scholar] [CrossRef]
- Drost, E.M.; Skwarski, K.M.; Sauleda, J.; Soler, N.; Roca, J.; Agusti, A.; MacNee, W. Oxidative stress and airway inflammation in severe exacerbations of COPD. Thorax 2005, 60, 293–300. [Google Scholar] [CrossRef] [Green Version]
- Roos, A.B.; Sandén, C.; Mori, M.; Bjermer, L.; Stampfli, M.R.; Erjefält, J.S. IL-17A Is Elevated in End-Stage Chronic Obstructive Pulmonary Disease and Contributes to Cigarette Smoke-induced Lymphoid Neogenesis. Am. J. Respir. Crit. Care Med. 2015, 191, 1232–1241. [Google Scholar] [CrossRef]
- Zhang, L.; Cheng, Z.; Liu, W.; Wu, K. Expression of interleukin (IL)-10, IL-17A and IL-22 in serum and sputum of stable chronic obstructive pulmonary disease patients. COPD 2013, 10, 459–465. [Google Scholar] [CrossRef]
- Hayden, M.S.; Ghosh, S. Shared principles in NF-kappaB signaling. Cell 2008, 132, 344–362. [Google Scholar] [CrossRef]
- Chen, L.F.; Mu, Y.; Greene, W.C. Acetylation of RelA at discrete sites regulates distinct nuclear functions of NF-kappaB. EMBO J. 2022, 21, 6539–6548. [Google Scholar] [CrossRef] [Green Version]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef]
- Ashburner, B.P.; Westerheide, S.D.; Baldwin, A.S., Jr. The p65 (RelA) subunit of NF-kappaB interacts with the histone deacetylase (HDAC) corepressors HDAC1 and HDAC2 to negatively regulate gene expression. Mol. Cell. Biol. 2001, 21, 7065–7077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.F.; Fischle, W.; Verdin, E.; Greene, W.C. Duration of nuclear NF-kappaB action regulated by reversible acetylation. Science 2001, 293, 1653–1657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, B.M.; Pavlisko, E.; Voynow, J.A. Pathogenic triad in COPD: Oxidative stress, protease-antiprotease imbalance, and inflammation. Int. J. Chron. Obstruct. Pulmon. Dis. 2011, 6, 413–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, L.; Wang, X.; Jin, M. Role of Airway Epithelial Cells in Development of Chronic Obstructive Pulmonary Disease. J. Epithel. Biol. Pharmacol. 2009, 2, 44–50. [Google Scholar] [CrossRef]
- Gründemann, C.; Garcia-Käufer, M.; Sauer, B.; Stangenberg, E.; Könczöl, M.; Merfort, I.; Zehl, M.; Huber, R. Traditionally used Veronica officinalis inhibits proinflammatory mediators via the NF-kappaB signalling pathway in a human lung cell line. J. Ethnopharmacol. 2013, 145, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.U.; Sung, M.H.; Ryu, H.W.; Lee, J.; Kim, H.S.; In, H.J.; Ahn, K.S.; Lee, H.J.; Lee, H.K.; Shin, D.H.; et al. Verproside inhibits TNF-α-induced MUC5AC expression through suppression of the TNF-α/NF-κB pathway in human airway epithelial cells. Cytokine 2016, 77, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Chavey, C.; Mühlbauer, M.; Bossard, C.; Freund, A.; Durand, S.; Jorgensen, C.; Jobin, C.; Lazennec, G. Interleukin-8 expression is regulated by histone deacetylases through the nuclear factor-kappaB pathway in breast cancer. Mol. Pharmacol. 2008, 74, 1359–1366. [Google Scholar] [CrossRef]
- Gao, W.; Li, L.; Wang, Y.; Zhang, S.; Adcock, I.M.; Barnes, P.J.; Huang, M.; Yao, X. Bronchial epithelial cells: The key effector cells in the pathogenesis of chronic obstructive pulmonary disease? Respirology 2015, 20, 722–729. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Active Compounds | |
|---|---|
| 1 | Verproside |
| 2 | Picroside II |
| 3 | 6-O-veratroylcatalpol |
| 4 | Catalposide |
| 5 | Minecoside |
| 6 | Verminoside |
| 7 | Isovanillylcatalpol |
| 8 | Catalpol |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, K.-H.; Woo, J.; Kim, J.; Lee, C.-H.; Yoo, C.-G. YPL-001 Shows Various Beneficial Effects against Cigarette Smoke Extract-Induced Emphysema Formation: Anti-Inflammatory, Anti-Oxidative, and Anti-Apoptotic Effects. Antioxidants 2023, 12, 15. https://doi.org/10.3390/antiox12010015
Lee K-H, Woo J, Kim J, Lee C-H, Yoo C-G. YPL-001 Shows Various Beneficial Effects against Cigarette Smoke Extract-Induced Emphysema Formation: Anti-Inflammatory, Anti-Oxidative, and Anti-Apoptotic Effects. Antioxidants. 2023; 12(1):15. https://doi.org/10.3390/antiox12010015
Chicago/Turabian StyleLee, Kyoung-Hee, Jisu Woo, Jiyeon Kim, Chang-Hoon Lee, and Chul-Gyu Yoo. 2023. "YPL-001 Shows Various Beneficial Effects against Cigarette Smoke Extract-Induced Emphysema Formation: Anti-Inflammatory, Anti-Oxidative, and Anti-Apoptotic Effects" Antioxidants 12, no. 1: 15. https://doi.org/10.3390/antiox12010015
APA StyleLee, K. -H., Woo, J., Kim, J., Lee, C. -H., & Yoo, C. -G. (2023). YPL-001 Shows Various Beneficial Effects against Cigarette Smoke Extract-Induced Emphysema Formation: Anti-Inflammatory, Anti-Oxidative, and Anti-Apoptotic Effects. Antioxidants, 12(1), 15. https://doi.org/10.3390/antiox12010015