
Protective Effects of Therapeutic Neutrophil Depletion and Myeloperoxidase Inhibition on Left Ventricular Function and Remodeling in Myocardial Infarction
 , , , , , , and add
Show full author list
, , , , , , and add
Show full author list
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animal Studies and Ethics Statement
2.2. Left Anterior Descending Artery Ligation
- (1)
- Permanent left anterior descending artery (LAD) ligation (PI): After lateral thoracotomy of the fourth intercostal space, a suture (8/0 polypropylene suture, Polypro, CP Medical, Norcross, GA, USA) was placed around the LAD and the artery was ligated with a bow tie. Ischemia was visually confirmed by blanching of the left ventricular (LV) apex.
- (2)
- Ischemia and reperfusion (I/R): The LAD ligation was removed after 40 min to allow up to 21 days of reperfusion.
2.3. Ly6G Antibody Treatment
2.4. Flow Cytometry
2.5. MPO Plasma Level
2.6. Echocardiography
2.7. Pressure-Volume Loop Analyses (PV-Loop)
2.8. Assessment of Left Ventricular Fibrosis and Wall Thickness
2.9. Staining for Myocardial PMN Infiltration
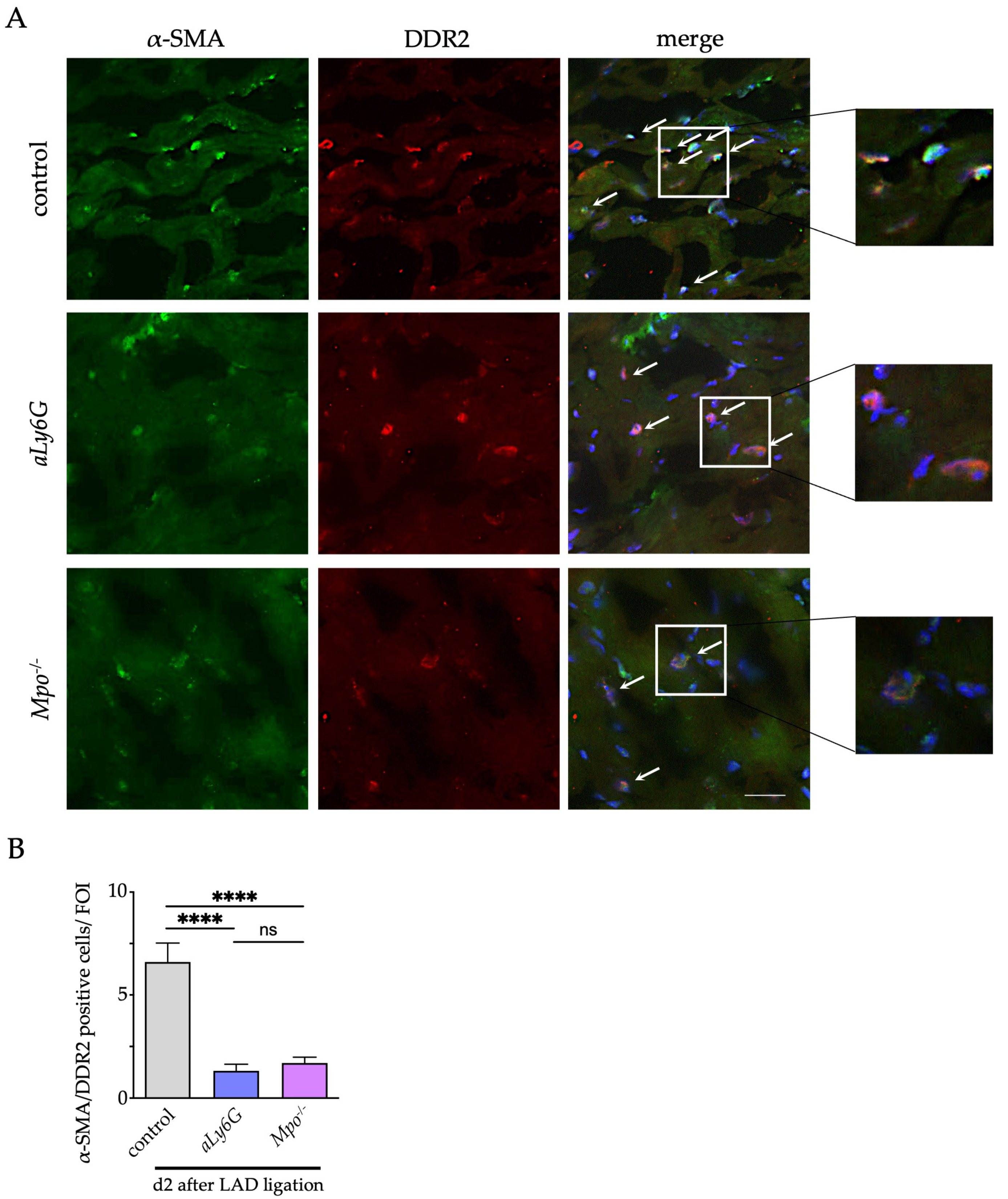
2.10. Immunofluorescence Staining for α-Smooth Muscle Actin, Discoidin Domain-Containing Receptor 2 and Connexin 43
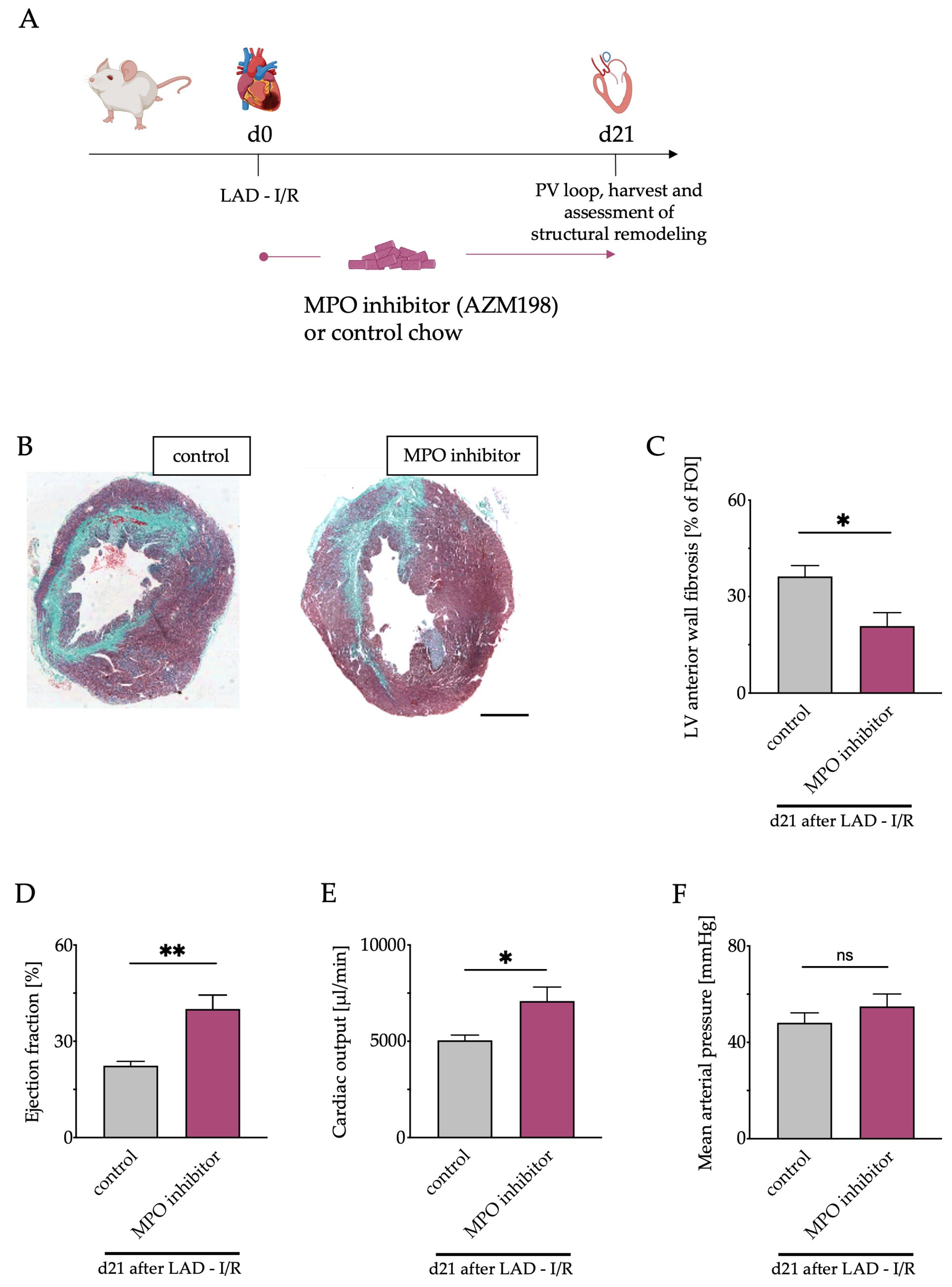
2.11. MPO Inhibitor Treatment
2.12. Statistics
3. Results
3.1. Inflammatory Response and Cardiac Neutrophil Infiltration
3.2. Influence of Neutrophils and MPO on Cardiac Function
3.3. Left Ventricular Fibrosis
3.4. Myofibroblast Accumulation
3.5. Connexin 43 in the Peri-Infarct Area
3.6. Pharmacological MPO Inhibition
3.7. Left Ventricular Fibrosis and Cardiac Function after Pharmacological MPO Inhibition
4. Discussion
4.1. Conclusions
4.2. Limitations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Heusch, G. Myocardial Ischaemia-Reperfusion Injury and Cardioprotection in Perspective. Nat. Rev. Cardiol. 2020, 17, 773–789. [Google Scholar] [CrossRef] [PubMed]
- Reed, G.W.; Rossi, J.E.; Cannon, C.P. Acute Myocardial Infarction. Lancet 2017, 389, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Savarese, G.; Becher, P.M.; Lund, L.H.; Seferovic, P.; Rosano, G.M.C.; Coats, A. Global Burden of Heart Failure: A Comprehensive and Updated Review of Epidemiology. Cardiovasc. Res. 2022, cvac013. [Google Scholar] [CrossRef] [PubMed]
- Ziaeian, B.; Fonarow, G.C. Epidemiology and Aetiology of Heart Failure. Nat. Rev. Cardiol. 2016, 13, 368–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ong, S.-B.; Hernández-Reséndiz, S.; Crespo-Avilan, G.E.; Mukhametshina, R.T.; Kwek, X.-Y.; Cabrera-Fuentes, H.A.; Hausenloy, D.J. Inflammation Following Acute Myocardial Infarction: Multiple Players, Dynamic Roles, and Novel Therapeutic Opportunities. Pharmacol. Ther. 2018, 186, 73–87. [Google Scholar] [CrossRef] [PubMed]
- Rock, K.L.; Kono, H. The Inflammatory Response to Cell Death. Annu. Rev. Pathol. 2008, 3, 99–126. [Google Scholar] [CrossRef] [PubMed]
- Swirski, F.K.; Nahrendorf, M. Leukocyte Behavior in Atherosclerosis, Myocardial Infarction, and Heart Failure. Science 2013, 339, 161–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puhl, S.-L.; Steffens, S. Neutrophils in Post-Myocardial Infarction Inflammation: Damage vs. Resolution? Front. Cardiovasc. Med. 2019, 6, 25. [Google Scholar] [CrossRef] [Green Version]
- Vafadarnejad, E.; Rizzo, G.; Krampert, L.; Arampatzi, P.; Arias-Loza, A.-P.; Nazzal, Y.; Rizakou, A.; Knochenhauer, T.; Bandi, S.R.; Nugroho, V.A.; et al. Dynamics of Cardiac Neutrophil Diversity in Murine Myocardial Infarction. Circ. Res. 2020, 127, e232–e249. [Google Scholar] [CrossRef] [PubMed]
- Horckmans, M.; Ring, L.; Duchene, J.; Santovito, D.; Schloss, M.J.; Drechsler, M.; Weber, C.; Soehnlein, O.; Steffens, S. Neutrophils Orchestrate Post-Myocardial Infarction Healing by Polarizing Macrophages towards a Reparative Phenotype. Eur. Heart J. 2017, 38, 187–197. [Google Scholar] [CrossRef]
- Ali, M.; Pulli, B.; Courties, G.; Tricot, B.; Sebas, M.; Iwamoto, Y.; Hilgendorf, I.; Schob, S.; Dong, A.; Zheng, W.; et al. Myeloperoxidase Inhibition Improves Ventricular Function and Remodeling after Experimental Myocardial Infarction. JACC Basic Transl. Sci. 2016, 1, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Aratani, Y. Myeloperoxidase: Its Role for Host Defense, Inflammation, and Neutrophil Function. Arch. Biochem. Biophys. 2018, 640, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Eiserich, J.P.; Baldus, S.; Brennan, M.-L.; Ma, W.; Zhang, C.; Tousson, A.; Castro, L.; Lusis, A.J.; Nauseef, W.M.; White, C.R.; et al. Myeloperoxidase, a Leukocyte-Derived Vascular NO Oxidase. Science 2002, 296, 2391–2394. [Google Scholar] [CrossRef] [PubMed]
- Lau, D.; Baldus, S. Myeloperoxidase and Its Contributory Role in Inflammatory Vascular Disease. Pharmacol. Ther. 2006, 111, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Askari, A.T.; Brennan, M.-L.; Zhou, X.; Drinko, J.; Morehead, A.; Thomas, J.D.; Topol, E.J.; Hazen, S.L.; Penn, M.S. Myeloperoxidase and Plasminogen Activator Inhibitor 1 Play a Central Role in Ventricular Remodeling after Myocardial Infarction. J. Exp. Med. 2003, 197, 615–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Rosen, H.; Madtes, D.K.; Shao, B.; Martin, T.R.; Heinecke, J.W.; Fu, X. Myeloperoxidase Inactivates TIMP-1 by Oxidizing Its N-Terminal Cysteine Residue: An oxidative mechanism for regulating proteolysis during inflammation. J. Biol. Chem. 2007, 282, 31826–31834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, M.J.; Hawkins, C.L. The Role of Myeloperoxidase in Biomolecule Modification, Chronic Inflammation, and Disease. Antioxid. Redox Signal. 2020, 32, 957–981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mollenhauer, M.; Friedrichs, K.; Lange, M.; Gesenberg, J.; Remane, L.; Kerkenpaß, C.; Krause, J.; Schneider, J.; Ravekes, T.; Maass, M.; et al. Myeloperoxidase Mediates Postischemic Arrhythmogenic Ventricular Remodeling. Circ. Res. 2017, 121, 56–70. [Google Scholar] [CrossRef]
- Lazarevic-Pasti, T.; Leskovac, A.; Vasic, V. Myeloperoxidase Inhibitors as Potential Drugs. Curr. Drug Metab. 2015, 16, 168–190. [Google Scholar] [CrossRef]
- Tidén, A.-K.; Sjögren, T.; Svensson, M.; Bernlind, A.; Senthilmohan, R.; Auchère, F.; Norman, H.; Markgren, P.-O.; Gustavsson, S.; Schmidt, S.; et al. 2-Thioxanthines Are Mechanism-Based Inactivators of Myeloperoxidase That Block Oxidative Stress during Inflammation. J. Biol. Chem. 2011, 286, 37578–37589. [Google Scholar] [CrossRef]
- Nelander, K.; Lagerstrom-Fermer, M.; Amilon, C.; Michaëlsson, E.; Heijer, M.; Kjaer, M.; Russell, M.; Han, D.; Lindstedt, E.-L.; Whatling, C.; et al. Early Clinical Experience with AZD4831, A Novel Myeloperoxidase Inhibitor, Developed for Patients with Heart Failure with Preserved Ejection Fraction. Clin. Transl. Sci. 2021, 14, 812–819. [Google Scholar] [CrossRef] [PubMed]
- Inghardt, T.; Antonsson, T.; Ericsson, C.; Hovdal, D.; Johannesson, P.; Johansson, C.; Jurva, U.; Kajanus, J.; Kull, B.; Michaëlsson, E.; et al. Discovery of AZD4831, a Mechanism-Based Irreversible Inhibitor of Myeloperoxidase, As a Potential Treatment for Heart Failure with Preserved Ejection Fraction. J. Med. Chem. 2022, 65, 11485–11496. [Google Scholar] [CrossRef] [PubMed]
- Rashid, I.; Maghzal, G.J.; Chen, Y.-C.; Cheng, D.; Talib, J.; Newington, D.; Ren, M.; Vajandar, S.K.; Searle, A.; Maluenda, A.; et al. Myeloperoxidase Is a Potential Molecular Imaging and Therapeutic Target for the Identification and Stabilization of High-Risk Atherosclerotic Plaque. Eur. Heart J. 2018, 39, 3301–3310. [Google Scholar] [CrossRef] [Green Version]
- Klinke, A.; Berghausen, E.; Friedrichs, K.; Molz, S.; Lau, D.; Remane, L.; Berlin, M.; Kaltwasser, C.; Adam, M.; Mehrkens, D.; et al. Myeloperoxidase Aggravates Pulmonary Arterial Hypertension by Activation of Vascular Rho-Kinase. JCI Insight 2018, 3, 97530. [Google Scholar] [CrossRef] [PubMed]
- Tiyerili, V.; Camara, B.; Becher, M.U.; Schrickel, J.W.; Lütjohann, D.; Mollenhauer, M.; Baldus, S.; Nickenig, G.; Andrié, R.P. Neutrophil-Derived Myeloperoxidase Promotes Atherogenesis and Neointima Formation in Mice. Int. J. Cardiol. 2016, 204, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Brennan, M.L.; Anderson, M.M.; Shih, D.M.; Qu, X.D.; Wang, X.; Mehta, A.C.; Lim, L.L.; Shi, W.; Hazen, S.L.; Jacob, J.S.; et al. Increased Atherosclerosis in Myeloperoxidase-Deficient Mice. J. Clin. Investig. 2001, 107, 419–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanno, S.; Lerner, D.L.; Schuessler, R.B.; Betsuyaku, T.; Yamada, K.A.; Saffitz, J.E.; Kovacs, A. Echocardiographic Evaluation of Ventricular Remodeling in a Mouse Model of Myocardial Infarction. J. Am. Soc. Echocardiogr. 2002, 15, 601–609. [Google Scholar] [CrossRef]
- Ghanem, A.; Troatz, C.; Elhafi, N.; Dewald, O.; Heeschen, C.; Nickenig, G.; Stypmann, J.; Tiemann, K. Quantitation of Myocardial Borderzone Using Reconstructive 3-D Echocardiography after Chronic Infarction in Rats—Incremental Value of Low-Dose Dobutamine. Ultrasound Med. Biol. 2008, 34, 559–566. [Google Scholar] [CrossRef]
- Frangogiannis, N.G.; Smith, C.W.; Entman, M.L. The Inflammatory Response in Myocardial Infarction. Cardiovasc. Res. 2002, 53, 31–47. [Google Scholar] [CrossRef]
- Konstam, M.A.; Kramer, D.G.; Patel, A.R.; Maron, M.S.; Udelson, J.E. Left Ventricular Remodeling in Heart Failure. JACC Cardiovasc. Imaging 2011, 4, 98–108. [Google Scholar] [CrossRef]
- van den Borne, S.W.M.; Diez, J.; Blankesteijn, W.M.; Verjans, J.; Hofstra, L.; Narula, J. Myocardial Remodeling after Infarction: The Role of Myofibroblasts. Nat. Rev. Cardiol. 2010, 7, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Roell, W.; Klein, A.M.; Breitbach, M.; Becker, T.S.; Parikh, A.; Lee, J.; Zimmermann, K.; Reining, S.; Gabris, B.; Ottersbach, A.; et al. Overexpression of Cx43 in Cells of the Myocardial Scar: Correction of Post-Infarct Arrhythmias through Heterotypic Cell-Cell Coupling. Sci. Rep. 2018, 8, 7145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Wang, H.; Li, J. Inflammation and Inflammatory Cells in Myocardial Infarction and Reperfusion Injury: A Double-Edged Sword. Clin. Med. Insights Cardiol. 2016, 10, 79–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Midwinter, R.G.; Vissers, M.C.; Winterbourn, C.C. Hypochlorous Acid Stimulation of the Mitogen-Activated Protein Kinase Pathway Enhances Cell Survival. Arch. Biochem. Biophys. 2001, 394, 13–20. [Google Scholar] [CrossRef]
- Zaman, S.; Kovoor, P. Sudden Cardiac Death Early after Myocardial Infarction. Circulation 2014, 129, 2426–2435. [Google Scholar] [CrossRef]
- Eloff, B.C.; Lerner, D.L.; Yamada, K.A.; Schuessler, R.B.; Saffitz, J.E.; Rosenbaum, D.S. High Resolution Optical Mapping Reveals Conduction Slowing in Connexin43 Deficient Mice. Cardiovasc. Res. 2001, 51, 681–690. [Google Scholar] [CrossRef] [Green Version]
- Poelzing, S.; Rosenbaum, D.S. Altered Connexin43 Expression Produces Arrhythmia Substrate in Heart Failure. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H1762–H1770. [Google Scholar] [CrossRef]
- Fu, X.; Kassim, S.Y.; Parks, W.C.; Heinecke, J.W. Hypochlorous Acid Oxygenates the Cysteine Switch Domain of Pro-Matrilysin (MMP-7). A Mechanism for Matrix Metalloproteinase Activation and Atherosclerotic Plaque Rupture by Myeloperoxidase. J. Biol. Chem. 2001, 276, 41279–41287. [Google Scholar] [CrossRef] [Green Version]
- Baldus, S.; Heeschen, C.; Meinertz, T.; Zeiher, A.M.; Eiserich, J.P.; Münzel, T.; Simoons, M.L.; Hamm, C.W.; CAPTURE Investigators. Myeloperoxidase Serum Levels Predict Risk in Patients with Acute Coronary Syndromes. Circulation 2003, 108, 1440–1445. [Google Scholar] [CrossRef] [Green Version]
- Rudolph, V.; Andrié, R.P.; Rudolph, T.K.; Friedrichs, K.; Klinke, A.; Hirsch-Hoffmann, B.; Schwoerer, A.P.; Lau, D.; Fu, X.; Klingel, K.; et al. Myeloperoxidase Acts as a Profibrotic Mediator of Atrial Fibrillation. Nat. Med. 2010, 16, 470–474. [Google Scholar] [CrossRef]
- Sultan, A.; Wörmann, J.; Lüker, J.; Bruck, J.-H.V.D.; Plenge, T.; Rudolph, V.; Klinke, A.; Heijman, J.; Mollenhauer, M.; Ravekes, T.; et al. Significance of Myeloperoxidase Plasma Levels as a Predictor for Cardiac Resynchronization Therapy Response. Clin. Res. Cardiol. 2021, 110, 1173–1180. [Google Scholar] [CrossRef] [PubMed]
- Janus, S.E.; Hajjari, J.; Chami, T.; Karnib, M.; Al-Kindi, S.G.; Rashid, I. Myeloperoxidase Is Independently Associated with Incident Heart Failure in Patients with Coronary Artery Disease and Kidney Disease. Curr. Probl. Cardiol. 2021, 47, 101080. [Google Scholar] [CrossRef] [PubMed]
- Gan, L.-M.; Lagerström-Fermér, M.; Ericsson, H.; Nelander, K.; Lindstedt, E.-L.; Michaëlsson, E.; Kjaer, M.; Heijer, M.; Whatling, C.; Fuhr, R. Safety, Tolerability, Pharmacokinetics and Effect on Serum Uric Acid of the Myeloperoxidase Inhibitor AZD4831 in a Randomized, Placebo-Controlled, Phase I Study in Healthy Volunteers. Br. J. Clin. Pharmacol. 2019, 85, 762–770. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, T.K.; Wipper, S.; Reiter, B.; Rudolph, V.; Coym, A.; Detter, C.; Lau, D.; Klinke, A.; Friedrichs, K.; Rau, T.; et al. Myeloperoxidase Deficiency Preserves Vasomotor Function in Humans. Eur. Heart J. 2012, 33, 1625–1634. [Google Scholar] [CrossRef] [PubMed]
- Moncada, S.; Radomski, M.W.; Palmer, R.M.J. Endothelium-Derived Relaxing Factor: Identification as Nitric Oxide and Role in the Control of Vascular Tone and Platelet Function. Biochem. Pharmacol. 1988, 37, 2495–2501. [Google Scholar] [CrossRef]
- Varin, R.; Mulder, P.; Tamion, F.; Richard, V.; Henry, J.P.; Lallemand, F.; Lerebours, G.; Thuillez, C. Improvement of Endothelial Function by Chronic Angiotensin-Converting Enzyme Inhibition in Heart Failure: Role of Nitric Oxide, Prostanoids, Oxidant Stress, and Bradykinin. Circulation 2000, 102, 351–356. [Google Scholar] [CrossRef] [Green Version]
- Hartupee, J.; Mann, D.L. Neurohormonal Activation in Heart Failure with Reduced Ejection Fraction. Nat. Rev. Cardiol. 2017, 14, 30–38. [Google Scholar] [CrossRef] [Green Version]
- Pinho-Gomes, A.C.; Rahimi, K. Management of Blood Pressure in Heart Failure. Heart 2019, 105, 589–595. [Google Scholar] [CrossRef]
- Schultz, J.; Kaminker, K. Myeloperoxidase of the Leucocyte of Normal Human Blood. I. Content and Localization. Arch. Biochem. Biophys. 1962, 96, 465–467. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guthoff, H.; Hof, A.; Klinke, A.; Maaß, M.; Konradi, J.; Mehrkens, D.; Geißen, S.; Nettersheim, F.S.; Braumann, S.; Michaelsson, E.; et al. Protective Effects of Therapeutic Neutrophil Depletion and Myeloperoxidase Inhibition on Left Ventricular Function and Remodeling in Myocardial Infarction. Antioxidants 2023, 12, 33. https://doi.org/10.3390/antiox12010033
Guthoff H, Hof A, Klinke A, Maaß M, Konradi J, Mehrkens D, Geißen S, Nettersheim FS, Braumann S, Michaelsson E, et al. Protective Effects of Therapeutic Neutrophil Depletion and Myeloperoxidase Inhibition on Left Ventricular Function and Remodeling in Myocardial Infarction. Antioxidants. 2023; 12(1):33. https://doi.org/10.3390/antiox12010033
Chicago/Turabian StyleGuthoff, Henning, Alexander Hof, Anna Klinke, Martina Maaß, Jürgen Konradi, Dennis Mehrkens, Simon Geißen, Felix S. Nettersheim, Simon Braumann, Erik Michaelsson, and et al. 2023. "Protective Effects of Therapeutic Neutrophil Depletion and Myeloperoxidase Inhibition on Left Ventricular Function and Remodeling in Myocardial Infarction" Antioxidants 12, no. 1: 33. https://doi.org/10.3390/antiox12010033
APA StyleGuthoff, H., Hof, A., Klinke, A., Maaß, M., Konradi, J., Mehrkens, D., Geißen, S., Nettersheim, F. S., Braumann, S., Michaelsson, E., Nies, R. J., Lee, S., Redzinski, M. -C., Peters, V. B. M., Nemade, H. N., von Stein, P., Winkels, H., Rudolph, V., Baldus, S., ... Mollenhauer, M. (2023). Protective Effects of Therapeutic Neutrophil Depletion and Myeloperoxidase Inhibition on Left Ventricular Function and Remodeling in Myocardial Infarction. Antioxidants, 12(1), 33. https://doi.org/10.3390/antiox12010033