
Common Reactivity and Properties of Heme Peroxidases: A DFT Study of Their Origin
 ,
,  , ,
, , 
Abstract
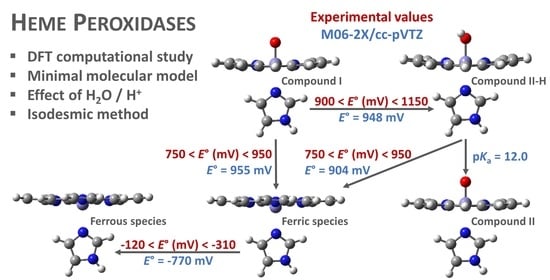
:
1. Introduction
2. Computational Details
2.1. Models
2.2. Methods
2.3. Calculation of Thermodynamic Parameters
3. Results and Discussion
3.1. Non-Protonated Species
3.1.1. Ferric and Ferrous Intermediates
3.1.2. Compound I and Compound II
3.2. Aquo Complexes
3.2.1. Ferric and Ferrous Intermediates
3.2.2. Compound I and Compound II
3.3. Protonated Species
3.3.1. Ferric and Ferrous Intermediates
3.3.2. Compound I and Compound II
3.4. Spectral Properties
3.5. Redox Properties
3.6. Adequacy of the Computational Molecular Model and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Ortiz de Montellano, P.R. Cytochrome P-450: Structure, Mechanism, and Biochemistry; Plenum Press: New York, NY, USA, 1986. [Google Scholar]
- Everse, J.; Everse, K.E.; Grisham, M.B. Peroxidases in Chemistry and Biology; CRC Press: Boca Raton, FL, USA, 1991. [Google Scholar]
- Zámocký, M.; Hofbauer, S.; Schaffner, I.; Gasselhuber, B.; Nicolussi, A.; Soudi, M.; Pirker, K.F.; Furtmüller, P.G.; Obinger, C. Independent evolution of four heme peroxidase superfamilies. Arch. Biochem. Biophys. 2015, 574, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Harris, D.L.; Loew, G.H. Identification of putative peroxide intermediates of peroxidases by electronic structure and spectra calculations. J. Am. Chem. Soc. 1996, 118, 10588–10594. [Google Scholar] [CrossRef]
- Loew, G.; Dupuis, M. Structure of a model transient peroxide intermediate of peroxidases by ab initio methods. J. Am. Chem. Soc. 1996, 118, 10584–10587. [Google Scholar] [CrossRef]
- Barea, G.; Maseras, F.; Lledós, A. Theoretical assessment on the viability of possible intermediates in the reaction mechanism of catalase and peroxidase models. J. Mol. Struct. Theochem 2003, 632, 323–333. [Google Scholar] [CrossRef]
- Poulos, T.L.; Kraut, J. The stereochemistry of peroxidase catalysis. J. Biol. Chem. 1980, 255, 8199–8205. [Google Scholar] [CrossRef] [PubMed]
- Ramos, D.R.; García, M.V.; Canle, L.M.; Santaballa, J.A.; Furtmüller, P.G.; Obinger, C. Myeloperoxidase-catalyzed taurine chlorination: Initial versus equilibrium rate. Arch. Biochem. Biophys. 2007, 466, 221–233. [Google Scholar] [CrossRef]
- Dunford, H.B. Heme Peroxidases; John Wiley & Sons, Inc.: New York, NY, USA, 1999. [Google Scholar]
- Spalteholtz, H.; Panasenko, O.M.; Arnhold, J. Formation of reactive halide species by myeloperoxidase and eosinophil peroxidase. Arch. Biochem. Biophys. 2006, 445, 225–234. [Google Scholar] [CrossRef]
- Vlasova, I.I.; Arnhold, J.; Osipov, A.N.; Panasenko, O.M. pH-dependent regulation of myeloperoxidase activity. Biochemistry 2006, 71, 667–677. [Google Scholar] [CrossRef]
- Ramos, D.R.; García, M.V.; Canle, L.M.; Santaballa, J.A.; Furtmüller, P.G.; Obinger, C. Myeloperoxidase-catalyzed chlorination: The quest for the active species. J. Inorg. Biochem. 2008, 102, 1300–1311. [Google Scholar] [CrossRef]
- Furtmüller, P.G.; Burner, U.; Obinger, C. Reaction of myeloperoxidase compound I with chloride, bromide, iodide, and thiocyanate. Biochemistry 1998, 37, 17923–17930. [Google Scholar] [CrossRef]
- Gumiero, A.; Metcalfe, C.L.; Pearson, A.R.; Raven, E.L.; Moody, P.C.E. Nature of the ferryl heme in compounds I and II. J. Biol. Chem. 2011, 286, 1260–1268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sitter, A.J.; Reczek, C.M.; Terner, J. Heme-linked ionization of horseradish peroxidase compound II monitored by the resonance Raman Fe(IV)=O stretching vibration. J. Biol. Chem. 1985, 260, 7515–7522. [Google Scholar] [CrossRef] [PubMed]
- Kincaid, J.R.; Zheng, Y.; Al-Mustafa, J.; Czarnecki, K. Resonance Raman spectra of native and mesoheme-reconstituted horseradish peroxidase and their catalytic intermediates. J. Biol. Chem. 1996, 271, 28805–28811. [Google Scholar] [CrossRef] [Green Version]
- Green, M.T. Application of Badger’s rule to heme and non-heme iron-oxygen bonds: An examination of ferryl protonation states. J. Am. Chem. Soc. 2006, 128, 1902–1906. [Google Scholar] [CrossRef]
- Penner-Hahn, J.E.; Eble, K.S.; McMurry, T.J.; Renner, M.; Balch, A.L.; Groves, J.T.; Dawson, J.H.; Hodgson, K.O. Structural characterization of horseradish peroxidase using EXAFS spectroscopy. Evidence for Fe = O ligation in compounds I and II. J. Am. Chem. Soc. 1986, 108, 7819–7825. [Google Scholar] [CrossRef]
- Hough, M.A.; Antonyuk, S.V.; Strange, R.W.; Eady, R.R.; Hasnain, S.S. Crystallography with online optical and X-ray absorption spectroscopies demonstrates an ordered mechanism in copper nitrite reductase. J. Mol. Biol. 2008, 378, 353–361. [Google Scholar] [CrossRef]
- Hersleth, H.-P.; Andersson, K.K. How different oxidation states of crystalline myoglobin are influenced by X-rays. BBA-Proteins Proteom. 2011, 1814, 785–796. [Google Scholar] [CrossRef] [PubMed]
- Pfanzagl, V.; Beale, J.H.; Michlits, H.; Schmidt, D.; Gabler, T.; Obinger, C.; Djinović-Carugo, K.; Hofbauer, S. X-ray-induced photoreduction of heme metal centers rapidly induces active-site perturbations in a protein-independent manner. J. Biol. Chem. 2020, 295, 13488–13501. [Google Scholar] [CrossRef]
- Bonagura, C.A.; Bhaskar, B.; Shimizu, H.; Li, H.; Sundaramoorthy, M.; McRee, D.E.; Goodin, D.B.; Poulos, T.L. High-resolution crystal structures and spectroscopy of native and compound I cytochrome c peroxidase. Biochemistry 2003, 42, 5600–5608. [Google Scholar] [CrossRef]
- Chance, B.; Powers, L.; Ching, Y.; Poulos, T.; Schonbaum, G.R.; Yamazaki, I.; Paul, K.G. X-ray absorption studies of intermediates in peroxidase activity. Arch. Biochem. Biophys. 1984, 235, 596–611. [Google Scholar] [CrossRef]
- Stone, K.L.; Behan, R.K.; Green, M.T. Resonance Raman spectroscopy of chloroperoxidase compound II provides direct evidence for the existence of an iron(IV)–hydroxide. Proc. Natl. Acad. Sci. USA 2006, 103, 12307–12310. [Google Scholar] [CrossRef] [Green Version]
- Green, M.T.; Dawson, J.H.; Gray, H.B. Oxoiron(IV) in chloroperoxidase compound II is basic: Implications for P450 chemistry. Science 2004, 304, 1653–1656. [Google Scholar] [CrossRef]
- Yosca, T.H.; Behan, R.K.; Krest, C.M.; Onderko, E.L.; Langston, M.C.; Green, M.T. Setting an upper limit on the myoglobin iron(IV)hydroxide pKa: Insight into axial ligand tuning in heme protein catalysis. J. Am. Chem. Soc. 2014, 136, 9124–9131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casadei, C.M.; Gumiero, A.; Metcalfe, C.L.; Murphy, E.J.; Basran, J.; Concilio, M.G.; Teixeira, S.C.M.; Schrader, T.E.; Fielding, A.J.; Ostermann, A.; et al. Neutron cryo-crystallography captures the protonation state of ferryl heme in a peroxidase. Science 2014, 345, 193–197. [Google Scholar] [CrossRef]
- Kwon, H.; Basran, J.; Casadei, C.M.; Fielding, A.J.; Schrader, T.E.; Ostermann, A.; Devos, J.M.; Aller, P.; Blakeley, M.P.; Moody, P.C.E.; et al. Direct visualization of a Fe(IV)–OH intermediate in a heme enzyme. Nat. Commun. 2016, 7, 13445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Battistuzzi, G.; Stampler, J.; Bellei, M.; Vlasits, J.; Soudi, M.; Furtmüller, P.G.; Obinger, C. Influence of the covalent heme–protein bonds on the redox thermodynamics of human myeloperoxidase. Biochemistry 2011, 50, 7987–7994. [Google Scholar] [CrossRef] [PubMed]
- Maskill, H. The Investigation of Organic Reactions and Their Mechanisms; Blackwell Publishing: Oxford, UK, 2006. [Google Scholar] [CrossRef]
- Axe, F.U.; Waleh, A.; Chantranupong, L.; Loew, G.H. A comparative analysis of the active site properties of the resting states of cytochrome c peroxidase, metmyoglobin, and catalase. Int. J. Quantum Chem. 1989, 35, 181–191. [Google Scholar] [CrossRef]
- Filizola, M.; Loew, G.H. Role of protein environment in horseradish peroxidase compound I formation: Molecular dynamics simulations of horseradish peroxidase−HOOH complex. J. Am. Chem. Soc. 2000, 122, 18–25. [Google Scholar] [CrossRef]
- Devarajan, A.; Gaenko, A.V.; Ryde, U. Effect of covalent links on the structure, spectra, and redox properties of myeloperoxidase—A density functional study. J. Inorg. Biochem. 2008, 102, 1549–1557. [Google Scholar] [CrossRef]
- Lee, C.W.Z.; Mubarak, M.Q.E.; Green, A.P.; de Visser, S.P. How does replacement of the axial histidine ligand in cytochrome c peroxidase by Nδ-methyl histidine affect its properties and functions? A computational study. Int. J. Mol. Sci. 2020, 21, 7133. [Google Scholar] [CrossRef]
- Ramos, M.J.; Fernandes, P.A. Computational enzymatic catalysis. Acc. Chem. Res. 2008, 41, 689–698. [Google Scholar] [CrossRef]
- Zhang, Y.; Mao, J.; Godbout, N.; Oldfield, E. Mössbauer quadrupole splittings and electronic structure in heme proteins and model systems: A density functional theory investigation. J. Am. Chem. Soc. 2002, 124, 13921–13930. [Google Scholar] [CrossRef] [PubMed]
- Banci, L.; Bertini, I.; Gray, H.B.; Luchinat, C.; Reddig, T.; Rosato, A.; Turano, P. Solution structure of oxidized horse heart cytochrome c. Biochemistry 1997, 36, 9867–9877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rovira, C.; Kunc, K.; Hutter, J.; Ballone, P.; Parrinello, M. Equilibrium geometries and electronic structure of iron-porphyrin complexes: A density functional study. J. Phys. Chem. A 1997, 101, 8914–8925. [Google Scholar] [CrossRef]
- Rovira, C.; Parrinello, M. Factors influencing ligand binding properties of heme models: A first principles study of picket-fence and protoheme complexes. Chem. Eur. J. 1999, 5, 250–262. [Google Scholar] [CrossRef]
- Rovira, C.; Parrinello, M. Harmonic and anharmonic dynamics of Fe-CO and Fe-O2 in heme models. Biophys. J. 2000, 78, 93–100. [Google Scholar] [CrossRef] [Green Version]
- Strickland, N.; Harvey, J.N. Spin-forbidden ligand binding to the ferrous-heme group: Ab initio and DFT Studies. J. Phys. Chem. B 2007, 111, 841–852. [Google Scholar] [CrossRef]
- Zhang, K.; Chen, L.; Geng, D.; Lu, J.; Wu, J. Thermal stability mechanism via energy absorption by chemical bonds bending and stretching in free space and the interlayer reaction of layered molecular structure explosives. Phys. Chem. Chem. Phys. 2020, 22, 13248–13260. [Google Scholar] [CrossRef]
- Johansson, M.P.; Blomberg, M.R.A.; Sundholm, D.; Wikström, M. Change in electron and spin density upon electron tranfer to haem. Biochim. Biophys. Acta 2002, 1553, 183–187. [Google Scholar] [CrossRef] [Green Version]
- Johansson, M.P.; Sundholm, D.; Gerfen, G.; Wikström, M. The spin distribution in low-spin iron porphyrins. J. Am. Chem. Soc. 2002, 124, 11771–11780. [Google Scholar] [CrossRef]
- Johansson, M.P.; Sundholm, D. Spin and charge distribution in iron porphyrin models: A coupled cluster and density-functional study. J. Chem. Phys. 2004, 120, 3229–3236. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miehlich, B.; Savin, A.; Stoll, H.; Preuss, H. Results obtained with the correlation energy density functionals of Becke and Lee, Yang and Parr. Chem. Phys. Lett. 1989, 157, 200–206. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef] [Green Version]
- Ditchfield, R.; Hehre, W.J.; Pople, J.A. Self-consistent molecular-orbital methods. IX. An extended Gaussian-type basis for nolecular-orbital studies of organic molecules. J. Chem. Phys. 1971, 54, 724–728. [Google Scholar] [CrossRef]
- Dunning, T.H., Jr. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–283. [Google Scholar] [CrossRef]
- Tomasi, J.; Persico, M. Molecular interactions in solution: An overview of methods based on continuous distributions of the solvent. Chem. Rev. 1994, 94, 2027–2094. [Google Scholar] [CrossRef]
- Dennington, R.D.; Keith, T.A.; Millam, J.M. GaussView, Version 6.0.16; Semichem Inc.: Shawnee Mission, KS, USA, 2016. [Google Scholar]
- Glendening, E.D.; Reed, A.E.; Carpenter, J.E.; Weinhold, F. NBO Version 3.1; Gaussian Inc.: Pittsburgh, PA, USA, 2003. [Google Scholar]
- Carpenter, J.E.; Weinhold, F. Analysis of the geometry of the hydroxymethyl radical by the “different hybrids for different spins” natural bond orbital procedure. J. Mol. Struct. Theochem 1988, 169, 41–62. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. C.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Bartmess, J.E. Thermodynamics of the electron and the proton. J. Phys. Chem. 1994, 98, 6420–6424. [Google Scholar] [CrossRef]
- Tissandier, M.D.; Cowen, K.A.; Feng, W.Y.; Gundlach, E.; Cohen, M.H.; Earhart, A.D.; Coe, J.V.; Tuttle, T.R., Jr. The proton’s absolute aqueous enthalpy and Gibbs free energy of solvation from cluster-ion solvation data. J. Phys. Chem. A 1998, 102, 7787–7794. [Google Scholar] [CrossRef]
- Topol, I.A.; Tawa, G.J.; Burt, S.K.; Rashin, A.A. On the structure and thermodynamics of solvated monoatomic ions using a hybrid solvation model. J. Chem. Phys. 1999, 111, 10998–11014. [Google Scholar] [CrossRef]
- Ho, J.; Coote, M.L.; Cramer, C.J.; Truhlar, D.G. Organic electrochemistry. In Theoretical Calculation of Reduction Potentials, 5th ed.; Revised and Expanded Edition; Hammerich, O., Speiser, B., Eds.; CRC Press: Boca Raton, FL, USA, 2015. [Google Scholar] [CrossRef]
- IUPAC. Compendium of Chemical Terminology (the "Gold Book"), 2nd ed.; Blackwell Scientific Publications: Oxford, UK, 1997. [Google Scholar]
- Isse, A.A.; Gennaro, A. Absolute potential of the standard hydrogen electrode and the problem of interconversion of potentials in different solvents. J. Phys. Chem. B 2010, 114, 7894–7899. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, K.; Becker, U. Computational redox potential predictions: Applications to inorganic and organic aqueous complexes, and complexes adsorbed to mineral surfaces. Minerals 2014, 4, 345–387. [Google Scholar] [CrossRef]
- CRC Handbook of Chemistry and Physics, 95th ed.; Haynes, W.M. (Ed.) CRC Press: Boca Raton, FL, USA, 2014. [Google Scholar]
- Sastre, S.; Casasnovas, R.; Muñoz, F.; Frau, J. Isodesmic reaction for accurate theoretical pKa calculations of amino acids and peptides. Phys. Chem. Chem. Phys. 2016, 18, 11202–11212. [Google Scholar] [CrossRef]
- Dutra, F.R.; Silva, C.D.S.; Custodio, R. On the accuracy of the direct method to calculate pKa from electronic structure calculations. J. Phys. Chem. A 2021, 125, 65–73. [Google Scholar] [CrossRef]
- Battistuzzi, G.; Bellei, M.; Bortolotti, C.A.; Sola, M. Redox properties of heme peroxidases. Arch. Biochem. Biophys. 2010, 500, 21–36. [Google Scholar] [CrossRef]
- Namazian, M.; Norouzi, P.; Ranjbar, R. Prediction of electrode potentials of some quinone derivatives in acetonitrile. J. Mol. Struct. Theochem 2003, 625, 235–241. [Google Scholar] [CrossRef]
- Wiberg, K.B. Application of the pople-santry-segal CNDO method to the cyclopropylcarbinyl and cyclobutyl cation and to bicyclobutane. Tetrahedron 1968, 24, 1083–1096. [Google Scholar] [CrossRef]
- Berglund, G.I.; Carlsson, G.H.; Smith, A.T.; Szöke, H.; Henriksen, A.; Hajdu, J. The catalytic pathway of horseradish peroxidase at high resolution. Nature 2002, 417, 463–468. [Google Scholar] [CrossRef]
- Carlsson, G.H.; Nicholls, P.; Svistunenko, D.; Berglund, G.I.; Hajdu, J. Complexes of horseradish peroxidase with formate, acetate, and carbon monoxide. Biochemistry 2005, 44, 635–642. [Google Scholar] [CrossRef] [PubMed]
- Sharp, K.H.; Mewies, M.; Moody, P.C.E.; Raven, E.L. Crystal structure of the ascorbate peroxidase-ascorbate complex. Nat. Struct. Mol. Biol. 2003, 10, 303–307. [Google Scholar] [CrossRef]
- Singh, A.K.; Singh, N.; Sharma, S.; Singh, S.B.; Kaur, P.; Bhushan, A.; Srinivasan, A.; Singh, T.P. Crystal structure of lactoperoxidase at 2.4 Å resolution. J. Mol. Biol. 2008, 376, 1060–1075. [Google Scholar] [CrossRef] [PubMed]
- Fiedler, T.J.; Davey, C.A.; Fenna, R.E. X-ray crystal structure and characterization of halide-binding sites of human myeloperoxidase at 1.8 Å resolution. J. Biol. Chem. 2000, 275, 11964–11971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galstyan, A.S.; Zarić, S.D.; Knapp, E.W. Computational studies on imidazole heme conformations. J. Biol. Inorg. Chem. 2005, 10, 343–354. [Google Scholar] [CrossRef]
- Mulliken, R.S. Electronic population analysis on LCAO–MO molecular wave functions. I. J. Chem. Phys. 1955, 23, 1833–1840. [Google Scholar] [CrossRef] [Green Version]
- Murray-Rust, P.; Glusker, J.P. Directional hydrogen bonding to sp2- and sp3-hybridized oxygen atoms and its relevance to ligand-macromolecule interactions. J. Am. Chem. Soc. 1984, 106, 1018–1025. [Google Scholar] [CrossRef]
- Chiavarino, B.; Crestoni, M.E.; Fornarini, S.; Rovira, C. Protonated heme. Chem. Eur. J. 2007, 13, 776–785. [Google Scholar] [CrossRef]
- Hashimoto, S.; Nakajima, R.; Yamazaki, I.; Tatsuno, Y.; Kitagawa, T. Oxygen exchange between the Fe(IV)=O heme and bulk water for the A2 isozyme of horseradish peroxidase. FEBS Lett. 1986, 208, 305–307. [Google Scholar] [CrossRef] [Green Version]
- Oertling, W.A.; Hoogland, H.; Babcock, G.T.; Wever, R. Identification and properties of an oxoferryl structure in myeloperoxidase compound II. Biochemistry 1988, 27, 5395–5400. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Dunford, H.B. Hammett ρσ correlation for reactions of lactoperoxidase compound II with phenols. Can. J. Chem. 1993, 71, 1990–1994. [Google Scholar] [CrossRef]
- Boaz, N.C.; Bell, S.R.; Groves, J.T. Ferryl protonation in oxoiron(IV) porphyrins and its role in oxygen transfer. J. Am. Chem. Soc. 2015, 137, 2875–2885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sivaraja, M.; Goodin, D.B.; Smith, M.; Hoffman, B.M. Identification by ENDOR of Trp191 as the free-radical site in cytochrome c peroxidase compound ES. Science 1989, 245, 738–740. [Google Scholar] [CrossRef] [PubMed]
- Fenna, R.E. Myeloperoxidase. In Handbook of Metalloproteins; Messerschmidt, A., Huber, R., Poulos, T., Wieghardt, K., Eds.; John Wiley & Sons: Chichester, UK, 2001; Volume 1, pp. 211–221. [Google Scholar]
- Parac, M.; Grimme, S. A TDDFT study of the lowest excitation energies of polycyclic aromatic hydrocarbons. Chem. Phys. 2003, 292, 11–21. [Google Scholar] [CrossRef]
- Jacquemin, D.; Perpète, E.A.; Scuseria, G.E.; Ciofini, I.; Adamo, C. TD-DFT performance for the visible absorption spectra of organic dyes: Conventional versus long-range hybrids. J. Chem. Theory Comput. 2008, 4, 123–135. [Google Scholar] [CrossRef]
- Battistuzzi, G.; Bellei, M.; Zederbauer, M.; Furtmüller, P.G.; Sola, M.; Obinger, C. Redox thermodynamics of the Fe(III)/Fe(II) couple of human myeloperoxidase in its high-spin and low-spin forms. Biochemistry 2006, 45, 12750–12755. [Google Scholar] [CrossRef]
- Furtmüller, P.G.; Arnhold, J.; Jantschko, W.; Pichler, H.; Obinger, C. Redox properties of the couples compound I/compound II and compound II/native enzyme of human myeloperoxidase. Biochem. Biophys. Res. Commun. 2003, 301, 551–557. [Google Scholar] [CrossRef]
- Furtmüller, P.G.; Arnhold, J.; Jantschko, W.; Zederbauer, M.; Jakopitsch, C.; Obinger, C. Standard reduction potentials of all couples of the peroxidase cycle of lactoperoxidase. J. Inorg. Biochem. 2005, 99, 1220–1229. [Google Scholar] [CrossRef]
- Zederbauer, M.; Furtmüller, P.G.; Brogioni, S.; Jakopitsch, C.; Smulevich, G.; Obinger, C. Heme to protein linkages in mammalian peroxidases: Impact on spectroscopic, redox and catalytic properties. Nat. Prod. Rep. 2007, 24, 571–584. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C.; Vissers, M.C.; Kettle, A.J. Myeloperoxidase. Curr. Opin. Hematol. 2000, 7, 53–58. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Spin | ΔG°/kJ mol−1 |
|---|---|---|
| Fe(III)-PO | 1/2 | 49.73 |
| 5/2 | 0.00 | |
| Fe(III)-PO-H2O | 1/2 | −12.44 |
| 5/2 | 0.86 | |
| Fe(III)-PO-H | 1/2 | 269.21 |
| 5/2 | 307.07 | |
| Fe(II)-PO | 0 | 46.38 |
| 1 | 59.96 | |
| 2 | 0.00 | |
| Fe(II)-PO-H2O | 0 | 25.66 |
| 1 | 63.38 | |
| 2 | 5.13 | |
| Fe(II)-PO-H | 0 | 231.01 |
| 1 | 179.50 | |
| 2 | 221.61 | |
| PO-I | 1/2 | 0.00 |
| 5/2 | 49.61 | |
| PO-I-H2O | 1/2 | 9.95 |
| 5/2 | 59.44 | |
| PO-I-H | 1/2 | 24.06 |
| 5/2 | 86.53 | |
| PO-II | 1 | 0.00 |
| 2 | 56.93 | |
| PO-II-H2O | 1 | 4.53 |
| 2 | 62.60 | |
| PO-II-H | 1 | −48.66 |
| 2 | −9.19 |
| Species | Spin | ΔG°A-H2O(aq) | Spin | pKa1 | pKa2 |
|---|---|---|---|---|---|
| Fe(III)-PO | 5/2–1/2 | 12.44 | 5/2–1/2 | −47.2 | −60.7 |
| Fe(II)-PO | 2–2 | −5.12 | 2–1 | −31.4 | −45.0 |
| PO-I | 1/2–1/2 | −9.95 | 1/2–1/2 | −4.2 | −17.7 |
| PO-II | 1–1 | −4.53 | 1–1 | 8.5 | −5.0 |
| Reduction Half-Reaction | ||||||
|---|---|---|---|---|---|---|
| Fe(III)-PO/Fe(II)-PO (5/2–2) | PO-I/Fe(III)-PO (1/2–5/2) | PO-I/PO-II (1/2–1) | PO-I/PO-II-H (1/2–1) | PO-II/Fe(III)-PO (1–5/2) | PO-II-H/Fe(III)-PO (1–5/2) | |
| Direct method | ||||||
| B3LYP/cc-pVDZ | 377 | 1066 | 741 | 1245 | 1391 | 886 |
| M06-2X/cc-pVTZ | 662 | 2358 | 1120 | 2380 | 3596 | 2336 |
| Isodesmic method | ||||||
| B3LYP/cc-pVDZ | 237 | 798 | 601 | 1105 | 1251 | 747 |
| M06-2X/cc-pVTZ | −770 | 955 | −312 | 948 | 2164 | 904 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramos, D.R.; Furtmüller, P.G.; Obinger, C.; Peña-Gallego, Á.; Pérez-Juste, I.; Santaballa, J.A. Common Reactivity and Properties of Heme Peroxidases: A DFT Study of Their Origin. Antioxidants 2023, 12, 303. https://doi.org/10.3390/antiox12020303
Ramos DR, Furtmüller PG, Obinger C, Peña-Gallego Á, Pérez-Juste I, Santaballa JA. Common Reactivity and Properties of Heme Peroxidases: A DFT Study of Their Origin. Antioxidants. 2023; 12(2):303. https://doi.org/10.3390/antiox12020303
Chicago/Turabian StyleRamos, Daniel R., Paul G. Furtmüller, Christian Obinger, Ángeles Peña-Gallego, Ignacio Pérez-Juste, and J. Arturo Santaballa. 2023. "Common Reactivity and Properties of Heme Peroxidases: A DFT Study of Their Origin" Antioxidants 12, no. 2: 303. https://doi.org/10.3390/antiox12020303
APA StyleRamos, D. R., Furtmüller, P. G., Obinger, C., Peña-Gallego, Á., Pérez-Juste, I., & Santaballa, J. A. (2023). Common Reactivity and Properties of Heme Peroxidases: A DFT Study of Their Origin. Antioxidants, 12(2), 303. https://doi.org/10.3390/antiox12020303