
Redox-Mediated Rewiring of Signalling Pathways: The Role of a Cellular Clock in Brain Health and Disease

 , ,
, ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
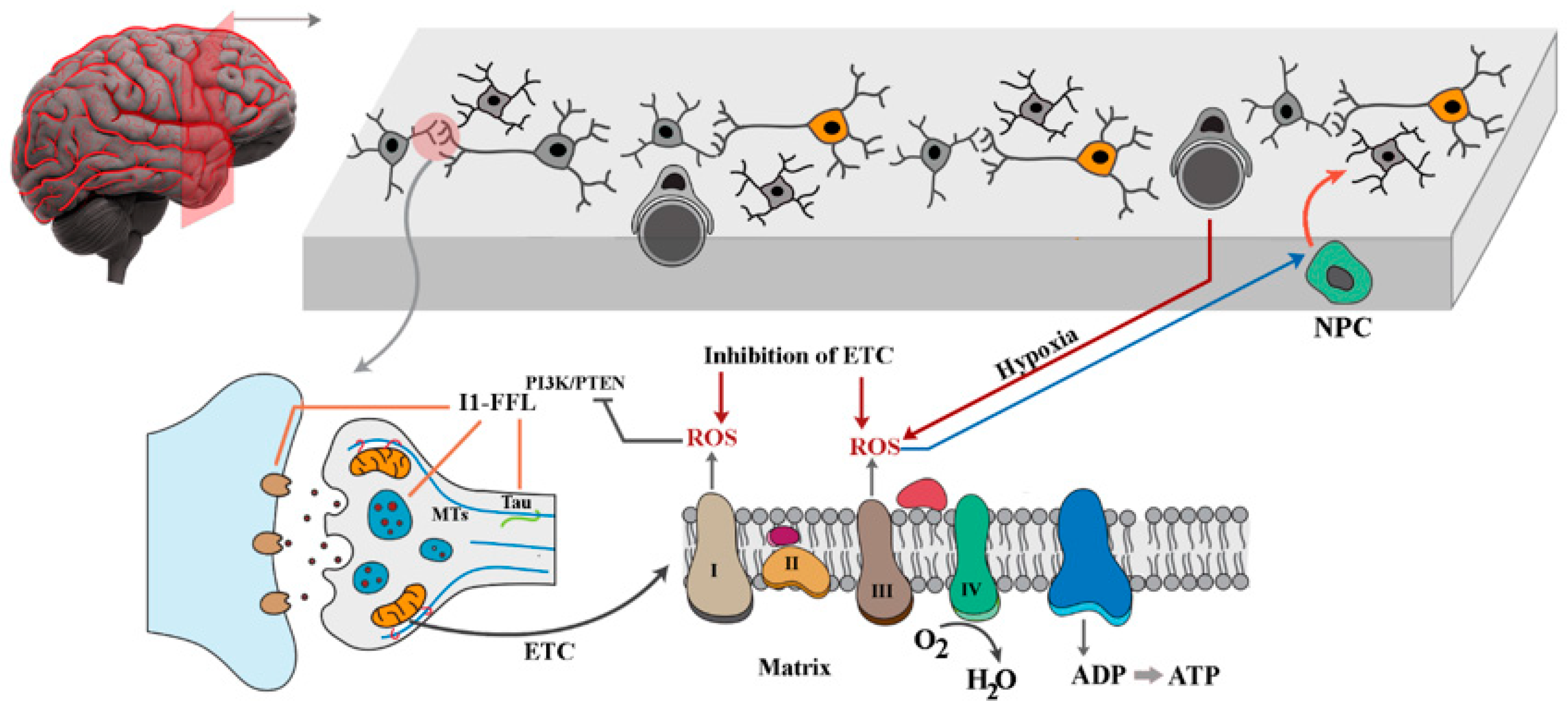{kind=link}
Abstract
:1. Introduction
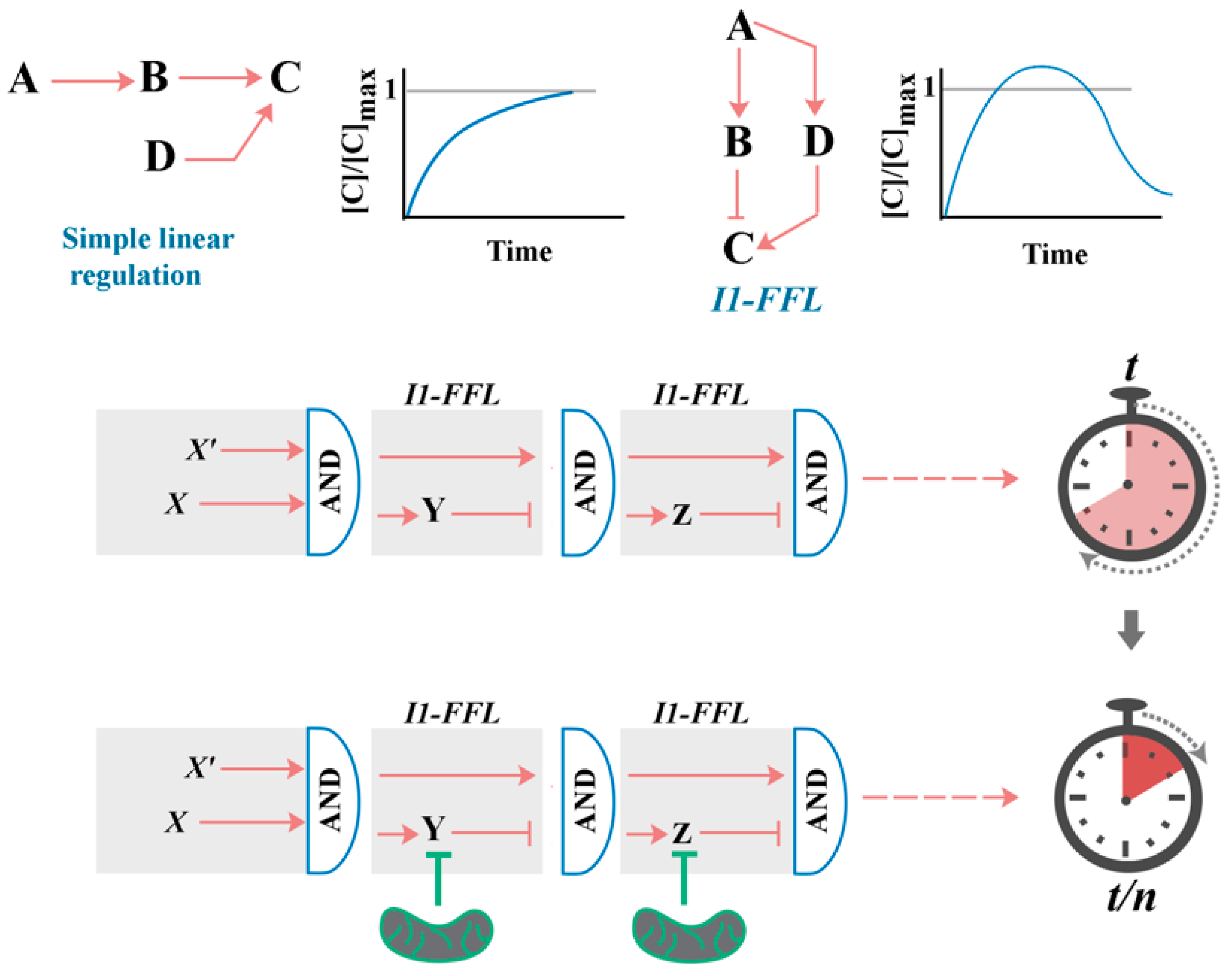
2. A Generic Blueprint for the “Cellular Clock”: The Role of Paradoxical Network Motifs in Programmability of Signalling Pathways
3. Metazoan Signal Transduction Pathways
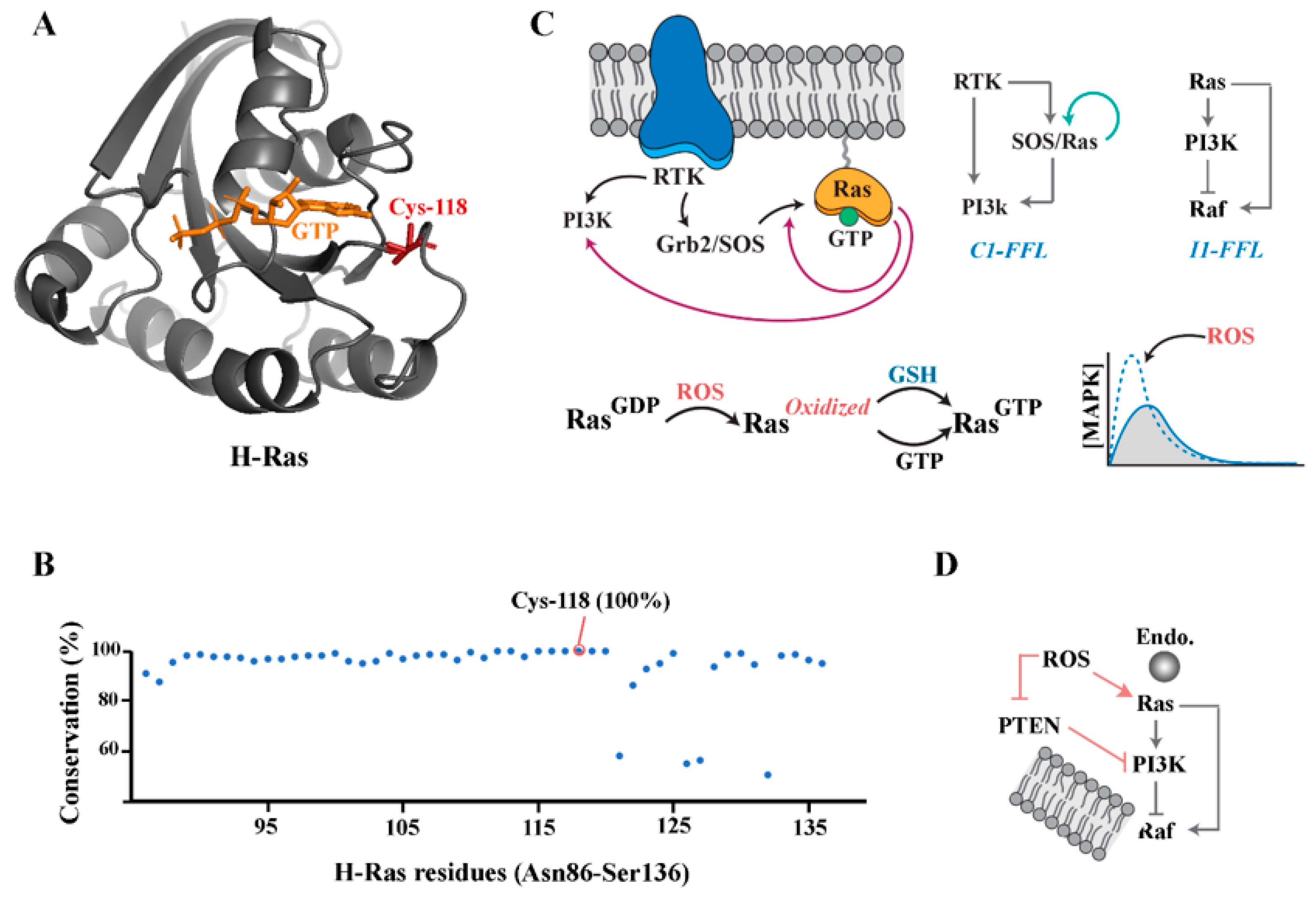
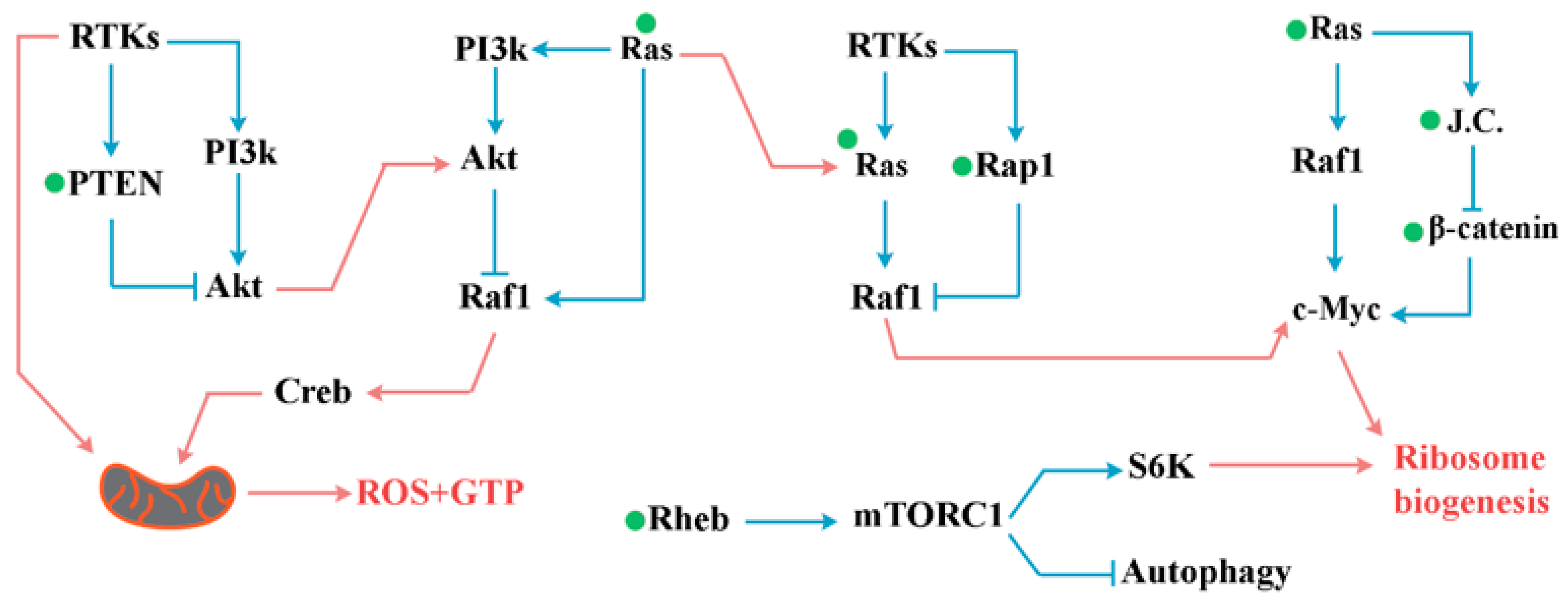
Small GTPases
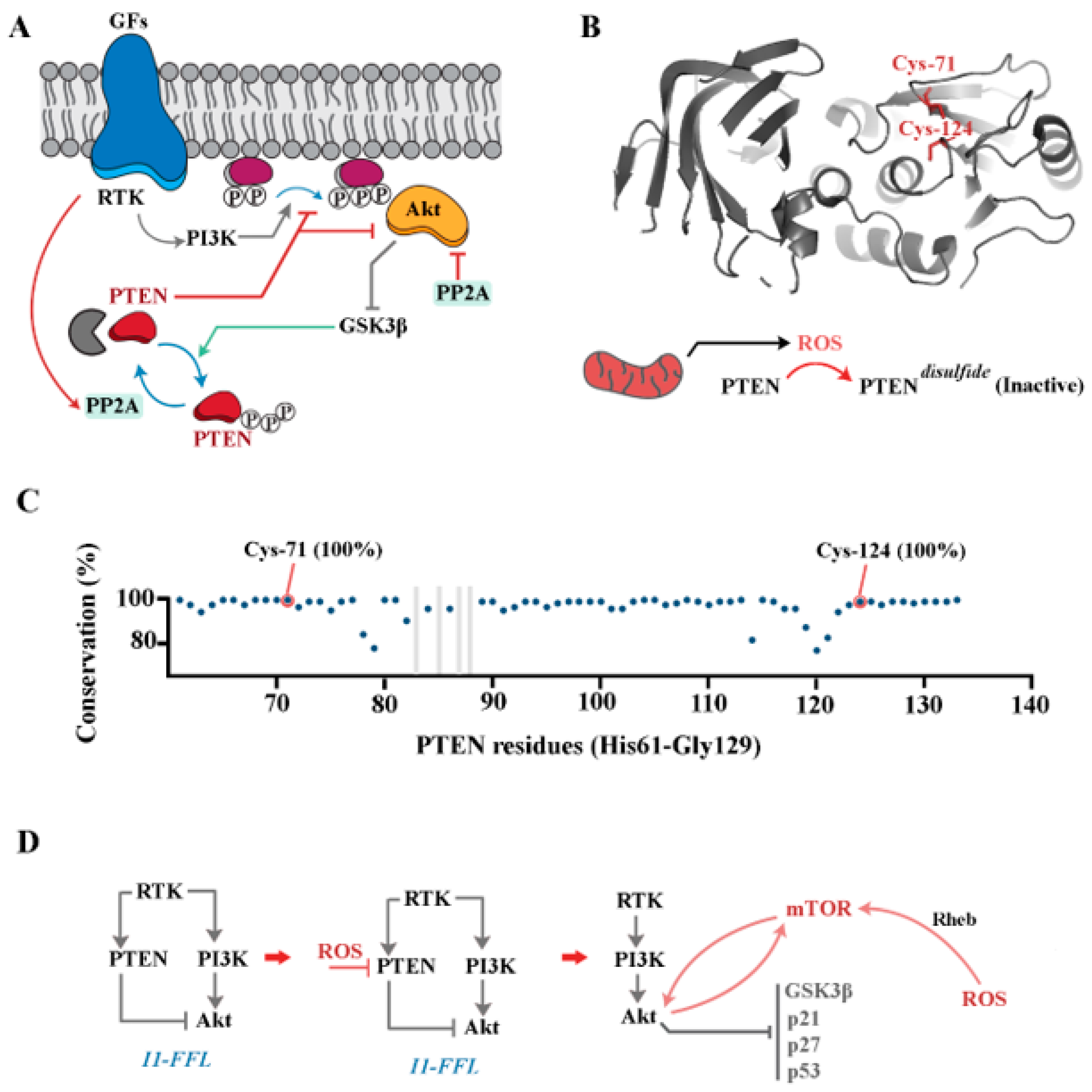
4. PI3K/Akt Pathway
5. Wnt/β-Catenin Pathway
6. Notch Signalling Pathway
7. Hypoxia-Inducible Factor (HIF)
8. JAK/STAT Pathway
9. NF-κB Signalling Pathway
10. The Ubiquitin System
11. A Blueprint for Redox-Mediated Resetting of the Cellular Clock
12. Mitochondria and the Cellular Clock
13. Reprogramming of the Cellular Clock: Application in Precision Medicine
14. Implication of Redox-Mediated Regulation of Cellular Clock in Brain Development and Disease
15. Conclusions
Funding
Conflicts of Interest
References
- Bufill, E.; Agustí, J.; Blesa, R. Human neoteny revisited: The case of synaptic plasticity. Am. J. Hum. Biol. 2011, 23, 729–739. [Google Scholar] [CrossRef] [PubMed]
- Denoël, M.; Joly, P. Neoteny and progenesis as two heterochronic processes involved in paedomorphosis in Triturus alpestris (Amphibia: Caudata). Proc. R. Soc. B Biol. Sci. 2000, 267, 1481–1485. [Google Scholar] [CrossRef] [PubMed]
- Rosenkilde, P.; Ussing, A.P. What mechanisms control neoteny and regulate induced metamorphosis in urodeles? Int. J. Dev. Biol. 1996, 40, 665–673. [Google Scholar]
- Safi, R.; Bertrand, S.; Marchand, O.; Duffraisse, M.; de Luze, A.; Vanacker, J.-M.; Maraninchi, M.; Margotat, A.; Demeneix, B.; Laudet, V. The Axolotl (Ambystoma mexicanum), a Neotenic Amphibian, Expresses Functional Thyroid Hormone Receptors. Endocrinology 2004, 145, 760–772. [Google Scholar] [CrossRef]
- Schwartz, J.H. Homeobox genes, fossils, and the origin of species. Anat. Rec. 1999, 257, 15–31. [Google Scholar] [CrossRef]
- Johnson, M.H. Functional brain development in humans. Nat. Rev. Neurosci. 2001, 2, 475–483. [Google Scholar] [CrossRef]
- Miller, D.J.; Duka, T.; Stimpson, C.D.; Schapiro, S.J.; Baze, W.B.; McArthur, M.J.; Fobbs, A.J.; Sousa, A.M.M.; Šestan, N.; Wildman, D.E.; et al. Prolonged myelination in human neocortical evolution. Proc. Natl. Acad. Sci. USA 2012, 109, 16480–16485. [Google Scholar] [CrossRef] [PubMed]
- Somel, M.; Franz, H.; Yan, Z.; Lorenc, A.; Guo, S.; Giger, T.; Kelso, J.; Nickel, B.; Dannemann, M.; Bahn, S.; et al. Transcriptional neoteny in the human brain. Proc. Natl. Acad. Sci. USA 2009, 106, 5743–5748. [Google Scholar] [CrossRef]
- Rezaei-Lotfi, S.; Hunter, N.; Farahani, R.M. Coupled cycling programs multicellular self-organization of neural progenitors. Cell Cycle 2019, 18, 2040–2054. [Google Scholar] [CrossRef]
- Vujovic, F.; Hunter, N.; Farahani, R.M. Cellular self-organization: An overdrive in Cambrian diversity? BioEssays 2022, 44, e2200033. [Google Scholar] [CrossRef]
- Collinet, C.; Lecuit, T. Programmed and self-organized flow of information during morphogenesis. Nat. Rev. Mol. Cell Biol. 2021, 22, 245–265. [Google Scholar] [CrossRef] [PubMed]
- Eiraku, M.; Takata, N.; Ishibashi, H.; Kawada, M.; Sakakura, E.; Okuda, S.; Sekiguchi, K.; Adachi, T.; Sasai, Y. Self-organizing optic-cup morphogenesis in three-dimensional culture. Nature 2011, 472, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Toda, S.; Blauch, L.R.; Tang, S.K.Y.; Morsut, L.; Lim, W.A. Programming self-organizing multicellular structures with synthetic cell-cell signaling. Science 2018, 361, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Kashiwagi, A.; Hanada, H.; Yabuki, M.; Kanno, T.; Ishisaka, R.; Sasaki, J.; Inoue, M.; Utsumi, K. Thyroxine enhancement and the role of reactive oxygen species in tadpole tail apoptosis. Free. Radic. Biol. Med. 1999, 26, 1001–1009. [Google Scholar] [CrossRef] [PubMed]
- Dawson, A. Neoteny and the thyroid in ratites. Rev. Reprod. 1996, 1, 78–81. [Google Scholar] [CrossRef]
- Kezer, J. Thyroxin-Induced Metamorphosis of the Neotenic Salamanders Eurycea tynerensis and Eurycea neotenes. Copeia 1952, 4, 234–237. [Google Scholar] [CrossRef]
- Sinha, R.A.; You, S.-H.; Zhou, J.; Siddique, M.M.; Bay, B.-H.; Zhu, X.; Privalsky, M.L.; Cheng, S.-Y.; Stevens, R.D.; Summers, S.A.; et al. Thyroid hormone stimulates hepatic lipid catabolism via activation of autophagy. J. Clin. Investig. 2012, 122, 2428–2438. [Google Scholar] [CrossRef]
- Atashi, F.; Modarressi, A.; Pepper, M.S. The Role of Reactive Oxygen Species in Mesenchymal Stem Cell Adipogenic and Osteogenic Differentiation: A Review. Stem Cells Dev. 2015, 24, 1150–1163. [Google Scholar] [CrossRef]
- Özsoy, Ş.; Vujovic, F.; Simonian, M.; Valova, V.; Hunter, N.; Farahani, R.M. Cannibalized erythroblasts accelerate developmental neurogenesis by regulating mitochondrial dynamics. Cell Rep. 2021, 35, 108942. [Google Scholar] [CrossRef]
- Kirova, D.G.; Judasova, K.; Vorhauser, J.; Zerjatke, T.; Leung, J.K.; Glauche, I.; Mansfeld, J. A ROS-dependent mechanism promotes CDK2 phosphorylation to drive progression through S phase. Dev. Cell 2022, 57, 1712–1727.e9. [Google Scholar] [CrossRef]
- Farahani, R.; Rezaei-Lotfi, S.; Simonian, M.; Hunter, N. Bi-modal reprogramming of cell cycle by MiRNA-4673 amplifies human neurogenic capacity. Cell Cycle 2019, 18, 848–868. [Google Scholar] [CrossRef] [PubMed]
- Hart, Y.; Antebi, Y.E.; Mayo, A.E.; Friedman, N.; Alon, U. Design principles of cell circuits with paradoxical components. Proc. Natl. Acad. Sci. USA 2012, 109, 8346–8351. [Google Scholar] [CrossRef] [PubMed]
- Hart, Y.; Alon, U. The Utility of Paradoxical Components in Biological Circuits. Mol. Cell 2013, 49, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Mangan, S.; Alon, U. Structure and function of the feed-forward loop network motif. Proc. Natl. Acad. Sci. USA 2003, 100, 11980–11985. [Google Scholar] [CrossRef]
- Eichenberger, P.; Fujita, M.; Jensen, S.T.; Conlon, E.M.; Rudner, D.Z.; Wang, S.T.; Ferguson, C.; Haga, K.; Sato, T.; Liu, J.S.; et al. The Program of Gene Transcription for a Single Differentiating Cell Type during Sporulation in Bacillus subtilis. PLoS Biol. 2004, 2, e328. [Google Scholar] [CrossRef]
- Heo, J.; Campbell, S.L. Superoxide Anion Radical Modulates the Activity of Ras and Ras-related GTPases by a Radical-based Mechanism Similar to That of Nitric Oxide. J. Biol. Chem. 2005, 280, 12438–12445. [Google Scholar] [CrossRef]
- Heo, J.; Prutzman, K.C.; Mocanu, V.; Campbell, S.L. Mechanism of Free Radical Nitric Oxide-mediated Ras Guanine Nucleotide Dissociation. J. Mol. Biol. 2005, 346, 1423–1440. [Google Scholar] [CrossRef]
- Iversen, L.; Tu, H.-L.; Lin, W.-C.; Christensen, S.M.; Abel, S.M.; Iwig, J.; Wu, H.-J.; Gureasko, J.; Rhodes, C.; Petit, R.S.; et al. Ras activation by SOS: Allosteric regulation by altered fluctuation dynamics. Science 2014, 345, 50–54. [Google Scholar] [CrossRef]
- Bar-Sagi, D. The Sos (Son of sevenless) protein. Trends Endocrinol. Metab. 1994, 5, 165–169. [Google Scholar] [CrossRef]
- Margarit, S.; Sondermann, H.; Hall, B.E.; Nagar, B.; Hoelz, A.; Pirruccello, M.; Bar-Sagi, D.; Kuriyan, J. Structural Evidence for Feedback Activation by Ras·GTP of the Ras-Specific Nucleotide Exchange Factor SOS. Cell 2003, 112, 685–695. [Google Scholar] [CrossRef]
- Das, J.; Ho, M.; Zikherman, J.; Govern, C.; Yang, M.; Weiss, A.; Chakraborty, A.K.; Roose, J.P. Digital Signaling and Hysteresis Characterize Ras Activation in Lymphoid Cells. Cell 2009, 136, 337–351. [Google Scholar] [CrossRef] [PubMed]
- Kolch, W.; Heidecker, G.; Lloyd, P.; Rapp, U.R. Raf-1 protein kinase is required for growth of induced NIH/3T3 cells. Nature 1991, 349, 426–428. [Google Scholar] [CrossRef]
- Sears, R.; Nuckolls, F.; Haura, E.; Taya, Y.; Tamai, K.; Nevins, J.R. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. J. Bone Jt. Surg. 2000, 14, 2501–2514. [Google Scholar] [CrossRef]
- Terrell, E.M.; Morrison, D.K. Ras-Mediated Activation of the Raf Family Kinases. Cold Spring Harb. Perspect. Med. 2018, 9, a033746. [Google Scholar] [CrossRef]
- Suire, S.; Hawkins, P.; Stephens, L. Activation of Phosphoinositide 3-Kinase γ by Ras. Curr. Biol. 2002, 12, 1068–1075. [Google Scholar] [CrossRef]
- Suire, S.; Condliffe, A.M.; Ferguson, G.J.; Ellson, C.D.; Guillou, H.; Davidson, K.; Welch, H.; Coadwell, J.; Turner, M.; Chilvers, E.R.; et al. Gβγs and the Ras binding domain of p110γ are both important regulators of PI3Kγ signalling in neutrophils. Nature 2006, 8, 1303–1309. [Google Scholar] [CrossRef]
- Inoki, K.; Li, Y.; Zhu, T.; Wu, J.; Guan, K.-L. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nature 2002, 4, 648–657. [Google Scholar] [CrossRef] [PubMed]
- Lu, S. Regulation of Hepatic Glutathione Synthesis. Semin. Liver Dis. 1998, 18, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.C. Regulation of glutathione synthesis. Mol. Asp. Med. 2001, 36, 95–116. [Google Scholar] [CrossRef]
- Lu, S.C. Regulation of glutathione synthesis. Mol. Asp. Med. 2009, 30, 42–59. [Google Scholar] [CrossRef]
- Huang, C.; Chang, L.; Anderson, M.; Meister, A. Catalytic and regulatory properties of the heavy subunit of rat kidney gamma-glutamylcysteine synthetase. J. Biol. Chem. 1993, 268, 19675–19680. [Google Scholar] [CrossRef]
- Kim, S.K.; Woodcroft, K.J.; Khodadadeh, S.S.; Novak, R.F. Insulin Signaling Regulates γ-Glutamylcysteine Ligase Catalytic Subunit Expression in Primary Cultured Rat Hepatocytes. Experiment 2004, 311, 99–108. [Google Scholar] [CrossRef]
- Li, S. Regulation of glutathione in cardiac myocytes. J. Mol. Cell. Cardiol. 2003, 35, 1145–1152. [Google Scholar] [CrossRef]
- Stump, C.S.; Short, K.R.; Bigelow, M.L.; Schimke, J.M.; Nair, K.S. Effect of insulin on human skeletal muscle mitochondrial ATP production, protein synthesis, and mRNA transcripts. Proc. Natl. Acad. Sci. USA 2003, 100, 7996–8001. [Google Scholar] [CrossRef] [PubMed]
- Brand, M.D. The sites and topology of mitochondrial superoxide production. Exp. Gerontol. 2010, 45, 466–472. [Google Scholar] [CrossRef] [PubMed]
- St-Pierre, J.; Buckingham, J.A.; Roebuck, S.J.; Brand, M.D. Topology of Superoxide Production from Different Sites in the Mitochondrial Electron Transport Chain. J. Biol. Chem. 2002, 277, 44784–44790. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Magilnick, N.; Lee, C.; Kalmaz, D.; Ou, X.; Chan, J.Y.; Lu, S.C. Nrf1 and Nrf2 Regulate Rat Glutamate-Cysteine Ligase Catalytic Subunit Transcription Indirectly via NF-κB and AP-1. Mol. Cell. Biol. 2005, 25, 5933–5946. [Google Scholar] [CrossRef] [PubMed]
- Benassi, B.; Fanciulli, M.; Fiorentino, F.; Porrello, A.; Chiorino, G.; Loda, M.; Zupi, G.; Biroccio, A. c-Myc Phosphorylation Is Required for Cellular Response to Oxidative Stress. Mol. Cell 2006, 21, 509–519. [Google Scholar] [CrossRef]
- Kreuzer, J.; Viedt, C.; Brandes, R.P.; Seeger, F.; Rosenkranz, A.S.; Sauer, H.; Babich, A.; Nürnberg, B.; Kather, H.; Krieger-Brauer, H.I. Platelet-derived growth factor activates production of reactive oxygen species by NAD(P)H-oxidase in smooth muscle cells through Gi1,2. FASEB J. 2002, 17, 38–40. [Google Scholar] [CrossRef]
- Svegliati, S.; Cancello, R.; Sambo, P.; Luchetti, M.; Paroncini, P.; Orlandini, G.; Discepoli, G.; Paterno, R.; Santillo, M.; Cuozzo, C.; et al. Platelet-derived Growth Factor and Reactive Oxygen Species (ROS) Regulate Ras Protein Levels in Primary Human Fibroblasts via ERK1/2. J. Biol. Chem. 2005, 280, 36474–36482. [Google Scholar] [CrossRef]
- Shi, Z.; Naowarojna, N.; Pan, Z.; Zou, Y. Multifaceted mechanisms mediating cystine starvation-induced ferroptosis. Nat. Commun. 2021, 12, 4792. [Google Scholar] [CrossRef] [PubMed]
- Mansfield, K.D.; Simon, M.C.; Keith, B.; Kan, W.-H.; Hsieh, C.-H.; Schwacha, M.G.; Choudhry, M.A.; Raju, R.; Bland, K.I.; Chaudry, I.H.; et al. Hypoxic reduction in cellular glutathione levels requires mitochondrial reactive oxygen species. J. Appl. Physiol. 2004, 97, 1358–1366. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, S.; Moelling, K. Phosphorylation and Regulation of Raf by Akt (Protein Kinase B). Science 1999, 286, 1741–1744. [Google Scholar] [CrossRef] [PubMed]
- Fritsch, R.; de Krijger, I.; Fritsch, K.; George, R.; Reason, B.; Kumar, M.S.; Diefenbacher, M.; Stamp, G.; Downward, J. RAS and RHO Families of GTPases Directly Regulate Distinct Phosphoinositide 3-Kinase Isoforms. Cell 2013, 153, 1050–1063. [Google Scholar] [CrossRef]
- Lee, T.; Yao, G.; Nevins, J.; You, L. Sensing and Integration of Erk and PI3K Signals by Myc. PLoS Comput. Biol. 2008, 4, e1000013. [Google Scholar] [CrossRef]
- Albeck, J.G.; Mills, G.B.; Brugge, J.S. Frequency-Modulated Pulses of ERK Activity Transmit Quantitative Proliferation Signals. Mol. Cell 2012, 49, 249–261. [Google Scholar] [CrossRef]
- Ender, P.; Gagliardi, P.A.; Dobrzyński, M.; Frismantiene, A.; Dessauges, C.; Höhener, T.; Jacques, M.-A.; Cohen, A.R.; Pertz, O. Spatiotemporal control of ERK pulse frequency coordinates fate decisions during mammary acinar morphogenesis. Dev. Cell 2022, 57, 2153–2167.e6. [Google Scholar] [CrossRef]
- Yeh, E.; Cunningham, M.; Arnold, H.; Chasse, D.; Monteith, T.; Ivaldi, G.; Hahn, W.C.; Stukenberg, P.T.; Shenolikar, S.; Uchida, T.; et al. A signalling pathway controlling c-Myc degradation that impacts oncogenic transformation of human cells. Nature 2004, 6, 308–318. [Google Scholar] [CrossRef]
- Bae, G.-U.; Seo, D.-W.; Kwon, H.-K.; Lee, H.Y.; Hong, S.; Lee, Z.-W.; Ha, K.-S.; Lee, H.-W.; Han, J.-W. Hydrogen Peroxide Activates p70S6k Signaling Pathway. J. Biol. Chem. 1999, 274, 32596–32602. [Google Scholar] [CrossRef]
- Guyton, K.Z.; Liu, Y.; Gorospe, M.; Xu, Q.; Holbrook, N.J. Activation of Mitogen-activated Protein Kinase by H2O2. J. Biol. Chem. 1996, 271, 4138–4142. [Google Scholar] [CrossRef]
- Muller, J.M.; Cahill, M.A.; Rupec, R.A.; Baeuerle, P.A.; Nordheim, A. Antioxidants as Well as Oxidants Activate C-fos Via Ras-Dependent Activation of Extracellular-Signal-Regulated Kinase 2 and Elk-1. JBIC J. Biol. Inorg. Chem. 1997, 244, 45–52. [Google Scholar] [CrossRef]
- Wilmer, W.A.; Tan, L.C.; Dickerson, J.A.; Danne, M.; Rovin, B.H. Interleukin-1β Induction of Mitogen-activated Protein Kinases in Human Mesangial Cells. J. Biol. Chem. 1997, 272, 10877–10881. [Google Scholar] [CrossRef]
- Roy, S.; Wyse, B.; Hancock, J.F. H-Ras Signaling and K-Ras Signaling Are Differentially Dependent on Endocytosis. Mol. Cell. Biol. 2002, 22, 5128–5140. [Google Scholar] [CrossRef] [PubMed]
- Oakley, F.D.; Abbott, D.; Li, Q.; Engelhardt, J.F.; Devarie-Baez, N.O.; Lopez, E.I.S.; Furdui, C.M.; Bilan, D.S.; Belousov, V.V.; Bendjersi, F.Z.; et al. Signaling Components of Redox Active Endosomes: The Redoxosomes. Antioxid. Redox Signal. 2009, 11, 1313–1333. [Google Scholar] [CrossRef]
- Yoshida, S.; Hong, S.; Suzuki, T.; Nada, S.; Mannan, A.M.; Wang, J.; Okada, M.; Guan, K.-L.; Inoki, K. Redox Regulates Mammalian Target of Rapamycin Complex 1 (mTORC1) Activity by Modulating the TSC1/TSC2-Rheb GTPase Pathway. J. Biol. Chem. 2011, 286, 32651–32660. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Jiang, X.; Li, B.; Yang, H.J.; Miller, M.; Yang, A.; Dhar, A.; Pavletich, N.P. Mechanisms of mTORC1 activation by RHEB and inhibition by PRAS40. Nature 2017, 552, 368–373. [Google Scholar] [CrossRef]
- Abraham, R.T. PI 3-kinase related kinases: ‘big’ players in stress-induced signaling pathways. DNA Repair 2004, 3, 883–887. [Google Scholar] [CrossRef]
- Hay, N.; Sonenberg, N. Upstream and Downstream of mTOR. Genes Dev. 2004, 18, 1926–1945. [Google Scholar] [CrossRef]
- Wullschleger, S.; Loewith, R.; Hall, M.N. TOR Signaling in Growth and Metabolism. Cell 2006, 124, 471–484. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and Regulation of Akt/PKB by the Rictor-mTOR Complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [PubMed]
- Garami, A.; Zwartkruis, F.J.; Nobukuni, T.; Joaquin, M.; Roccio, M.; Stocker, H.; Kozma, S.C.; Hafen, E.; Bos, J.L.; Thomas, G. Insulin Activation of Rheb, a Mediator of mTOR/S6K/4E-BP Signaling, Is Inhibited by TSC1 and 2. Mol. Cell 2003, 11, 1457–1466. [Google Scholar] [CrossRef]
- Inoki, K.; Li, Y.; Xu, T.; Guan, K.-L. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. J. Bone Jt. Surg. 2003, 17, 1829–1834. [Google Scholar] [CrossRef] [PubMed]
- Tee, A.R.; Fingar, D.C.; Manning, B.D.; Kwiatkowski, D.J.; Cantley, L.C.; Blenis, J. Tuberous sclerosis complex-1 and -2 gene products function together to inhibit mammalian target of rapamycin (mTOR)-mediated downstream signaling. Proc. Natl. Acad. Sci. USA 2002, 99, 13571–13576. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Ouyang, H.; Zhu, T.; Lindvall, C.; Wang, Y.; Zhang, X.; Yang, Q.; Bennett, C.; Harada, Y.; Stankunas, K.; et al. TSC2 Integrates Wnt and Energy Signals via a Coordinated Phosphorylation by AMPK and GSK3 to Regulate Cell Growth. Cell 2006, 126, 955–968. [Google Scholar] [CrossRef] [PubMed]
- Leslie, N.R.; Bennett, D.; Lindsay, Y.E.; Stewart, H.; Gray, A.; Downes, C. Redox regulation of PI 3-kinase signalling via inactivation of PTEN. EMBO J. 2003, 22, 5501–5510. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, T.; Nishiyama, M.; Moroishi, T.; Kawamura, A.; Nakayama, K.I. FBXL5 Inactivation in Mouse Brain Induces Aberrant Proliferation of Neural Stem Progenitor Cells. Mol. Cell. Biol. 2017, 37, e00470-16. [Google Scholar] [CrossRef]
- Yang, M.; Yang, S.-L.; Herrlinger, S.; Liang, C.; Dzieciatkowska, M.; Hansen, K.C.; Desai, R.; Nagy, A.; Niswander, L.; Moss, E.G.; et al. Lin28 promotes the proliferative capacity of neural progenitor cells in brain development. Development 2015, 142, 1616–1627. [Google Scholar] [CrossRef]
- Stringari, C.; Nourse, J.L.; Flanagan, L.A.; Gratton, E. Phasor Fluorescence Lifetime Microscopy of Free and Protein-Bound NADH Reveals Neural Stem Cell Differentiation Potential. PLoS ONE 2012, 7, e48014. [Google Scholar] [CrossRef]
- Ito, K.; Suda, T. Metabolic requirements for the maintenance of self-renewing stem cells. Nat. Rev. Mol. Cell Biol. 2014, 15, 243–256. [Google Scholar] [CrossRef]
- Xiao, W.; Loscalzo, J. Metabolic Responses to Reductive Stress. Antioxid. Redox Signal. 2020, 32, 1330–1347. [Google Scholar] [CrossRef]
- Nanba, D.; Sakabe, J.; Mosig, J.; Brouard, M.; Toki, F.; Shimokawa, M.; Kamiya, M.; Braschler, T.; Azzabi, F.; Lathion, S.D.; et al. Low temperature and mTOR inhibition favor stem cell maintenance in human keratinocyte cultures. EMBO Rep. 2023, 24, e55439. [Google Scholar] [CrossRef]
- Hemmings, B.A.; Restuccia, D.F. PI3K-PKB/Akt Pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011189, Corrected in Cold Spring Harb. Perspect. Biol. 2015, 7, a026609. [Google Scholar] [CrossRef] [PubMed]
- Alessi, D.R.; James, S.R.; Downes, C.; Holmes, A.B.; Gaffney, P.R.; Reese, C.B.; Cohen, P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Bα. Curr. Biol. 1997, 7, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Park, J.; Cron, P.; Hess, D.; Hemmings, B.A. Identification of a PKB/Akt Hydrophobic Motif Ser-473 Kinase as DNA-dependent Protein Kinase. J. Biol. Chem. 2004, 279, 41189–41196. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Dixon, J.E.; Cho, W. Membrane-binding and activation mechanism of PTEN. Proc. Natl. Acad. Sci. USA 2003, 100, 7491–7496. [Google Scholar] [CrossRef] [PubMed]
- Kotzampasi, D.M.; Premeti, K.; Papafotika, A.; Syropoulou, V.; Christoforidis, S.; Cournia, Z.; Leondaritis, G. The orchestrated signaling by PI3Kα and PTEN at the membrane interface. Comput. Struct. Biotec. 2022, 20, 5607–5621. [Google Scholar] [CrossRef]
- Masson, G.R.; Williams, R.L. Structural Mechanisms of PTEN Regulation. Cold Spring Harb. Perspect. Med. 2019, 10, a036152. [Google Scholar] [CrossRef] [PubMed]
- Al-Khouri, A.M.; Ma, Y.; Togo, S.H.; Williams, S.; Mustelin, T. Cooperative Phosphorylation of the Tumor Suppressor Phosphatase and Tensin Homologue (PTEN) by Casein Kinases and Glycogen Synthase Kinase 3β. J. Biol. Chem. 2005, 280, 35195–35202. [Google Scholar] [CrossRef]
- Torres, J.; Pulido, R. The Tumor Suppressor PTEN Is Phosphorylated by the Protein Kinase CK2 at Its C Terminus: Implications For Pten Stability To Proteasome-Mediated Degradation. J. Biol. Chem. 2001, 276, 993–998. [Google Scholar] [CrossRef]
- Vazquez, F.; Ramaswamy, S.; Nakamura, N.; Sellers, W.R. Phosphorylation of the PTEN Tail Regulates Protein Stability and Function. Mol. Cell. Biol. 2000, 20, 5010–5018. [Google Scholar] [CrossRef]
- Nakahata, S.; Ichikawa, T.; Maneesaay, P.; Saito, Y.; Nagai, K.; Tamura, T.; Manachai, N.; Yamakawa, N.; Hamasaki, M.; Kitabayashi, I.; et al. Loss of NDRG2 expression activates PI3K-AKT signalling via PTEN phosphorylation in ATLL and other cancers. Nat. Commun. 2014, 5, 3393. [Google Scholar] [CrossRef]
- Zhang, X.C.; Piccini, A.; Myers, M.P.; Van Aelst, L.; Tonks, N.K. Functional analysis of the protein phosphatase activity of PTEN. Biochem. J. 2012, 444, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Stambolic, V.; MacPherson, D.; Sas, D.; Lin, Y.; Snow, B.; Jang, Y.; Benchimol, S.; Mak, T. Regulation of PTEN Transcription by p53. Mol. Cell 2001, 8, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Martin, B.L.; Brautigan, D.L. Regulation of Protein Serine-Threonine Phosphatase Type-2A by Tyrosine Phosphorylation. Science 1992, 257, 1261–1264. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Damuni, Z. Autophosphorylation-activated protein kinase phosphorylates and inactivates protein phosphatase 2A. Proc. Natl. Acad. Sci. USA 1993, 90, 2500–2504. [Google Scholar] [CrossRef] [PubMed]
- Kuo, Y.-C.; Huang, K.-Y.; Yang, C.-H.; Yang, Y.-S.; Lee, W.-Y.; Chiang, C.-W. Regulation of Phosphorylation of Thr-308 of Akt, Cell Proliferation, and Survival by the B55α Regulatory Subunit Targeting of the Protein Phosphatase 2A Holoenzyme to Akt. J. Biol. Chem. 2008, 283, 1882–1892. [Google Scholar] [CrossRef]
- Lee, S.-R.; Yang, K.-S.; Kwon, J.; Lee, C.; Jeong, W.; Rhee, S.G. Reversible Inactivation of the Tumor Suppressor PTEN by H2O2. J. Biol. Chem. 2002, 277, 20336–20342. [Google Scholar] [CrossRef]
- Zhang, Y.; Park, J.; Han, S.-J.; Yang, S.Y.; Yoon, H.J.; Park, I.; Woo, H.A.; Lee, S.-R. Redox regulation of tumor suppressor PTEN in cell signaling. Redox Biol. 2020, 34, 101553. [Google Scholar] [CrossRef]
- Oatey, P.B.; Venkateswarlu, K.; Williams, A.G.; Fletcher, L.M.; Foulstone, E.J.; Cullen, P.J.; Tavaré, J.M. Confocal imaging of the subcellular distribution of phosphatidylinositol 3,4,5-trisphosphate in insulin- and PDGF-stimulated 3T3-L1 adipocytes. Biochem. J. 1999, 344 Pt 2, 511–518. [Google Scholar] [CrossRef]
- Leto, D.; Saltiel, A.R. Regulation of glucose transport by insulin: Traffic control of GLUT4. Nat. Rev. Mol. Cell Biol. 2012, 13, 383–396. [Google Scholar] [CrossRef]
- Cheng, Z.; Tseng, Y.; White, M.F. Insulin signaling meets mitochondria in metabolism. Trends Endocrinol. Metab. 2010, 21, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Salic, A.; Lee, E.; Mayer, L.; Kirschner, M.W. Control of β-Catenin Stability: Reconstitution of the Cytoplasmic Steps of the Wnt Pathway in Xenopus Egg Extracts. Mol. Cell 2000, 5, 523–532. [Google Scholar] [CrossRef]
- Ikeda, S.; Kishida, S.; Yamamoto, H.; Murai, H.; Koyama, S.; Kikuchi, A. Axin, a negative regulator of the Wnt signaling pathway, forms a complex with GSK-3β and β-catenin and promotes GSK-3β-dependent phosphorylation of β-catenin. EMBO J. 1998, 17, 1371–1384. [Google Scholar] [CrossRef] [PubMed]
- Behrens, J.; Von Kries, J.P.; Kühl, M.; Bruhn, L.; Wedlich, D.; Grosschedl, R.; Birchmeier, W. Functional interaction of β-catenin with the transcription factor LEF-1. Nature 1996, 382, 638–642. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, B.T.; Tamai, K.; He, X. Wnt/β-Catenin Signaling: Components, Mechanisms, and Diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef]
- Funato, Y.; Michiue, T.; Asashima, M.; Miki, H. The thioredoxin-related redox-regulating protein nucleoredoxin inhibits Wnt–β-catenin signalling through Dishevelled. Nature 2006, 8, 501–508. [Google Scholar] [CrossRef] [PubMed]
- He, T.-C.; Sparks, A.B.; Rago, C.; Hermeking, H.; Zawel, L.; da Costa, L.T.; Morin, P.J.; Vogelstein, B.; Kinzler, K.W. Identification of c-MYC as a Target of the APC Pathway. Science 1998, 281, 1509–1512. [Google Scholar] [CrossRef] [PubMed]
- Grandori, C.; Gomez-Roman, N.; Felton-Edkins, Z.A.; Ngouenet, C.; Galloway, D.A.; Eisenman, R.N.; White, R.J. c-Myc binds to human ribosomal DNA and stimulates transcription of rRNA genes by RNA polymerase I. Nat. Cell Biol. 2005, 7, 311–318. [Google Scholar] [CrossRef]
- Boon, K.; Caron, H.N.; van Asperen, R.; Valentijn, L.; Hermus, M.-C.; van Sluis, P.; Roobeek, I.; Weis, I.; Voûte, P.; Schwab, M.; et al. N-myc enhances the expression of a large set of genes functioning in ribosome biogenesis and protein synthesis. EMBO J. 2001, 20, 1383–1393. [Google Scholar] [CrossRef]
- Schmidt, E.V. The role of c-myc in regulation of translation initiation. Oncogene 2004, 23, 3217–3221. [Google Scholar] [CrossRef]
- Orford, K.; Orford, C.C.; Byers, S.W. Exogenous Expression of β-Catenin Regulates Contact Inhibition, Anchorage-Independent Growth, Anoikis, and Radiation-Induced Cell Cycle Arrest. J. Cell Biol. 1999, 146, 855–868. [Google Scholar] [CrossRef]
- Radisky, D.C.; Levy, D.D.; Littlepage, L.E.; Liu, H.; Nelson, C.M.; Fata, J.E.; Leake, D.; Godden, E.L.; Albertson, D.G.; Nieto, M.A.; et al. Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature 2005, 436, 123–127. [Google Scholar] [CrossRef]
- Dong, R.; Lu, J.-G.; Wang, Q.; He, X.-L.; Chu, Y.-K.; Ma, Q.-J. Stabilization of Snail by HuR in the process of hydrogen peroxide induced cell migration. Biochem. Biophys. Res. Commun. 2007, 356, 318–321. [Google Scholar] [CrossRef]
- Barnett, P.; Arnold, R.S.; Mezencev, R.; Chung, L.W.; Zayzafoon, M.; Odero-Marah, V. Snail-mediated regulation of reactive oxygen species in ARCaP human prostate cancer cells. Biochem. Biophys. Res. Commun. 2011, 404, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Giannoni, E.; Parri, M.; Chiarugi, P.; Di Francesco, A.M.; Toesca, A.; Cenciarelli, C.; Giordano, A.; Gasbarrini, A.; Puglisi, M.A.; Cirri, P.; et al. EMT and Oxidative Stress: A Bidirectional Interplay Affecting Tumor Malignancy. Antioxid. Redox Signal. 2012, 16, 1248–1263. [Google Scholar] [CrossRef] [PubMed]
- Andersson, E.R.; Sandberg, R.; Lendahl, U. Notch signaling: Simplicity in design, versatility in function. Development 2011, 138, 3593–3612. [Google Scholar] [CrossRef]
- Kovall, R.A.; Blacklow, S.C. Mechanistic Insights into Notch Receptor Signaling from Structural and Biochemical Studies. Curr. Top. Dev. Biol. 2010, 92, 31–71. [Google Scholar] [CrossRef]
- Palomero, T.; Lim, W.K.; Odom, D.T.; Sulis, M.L.; Real, P.J.; Margolin, A.; Barnes, K.C.; O’Neil, J.; Neuberg, D.; Weng, A.P.; et al. NOTCH1 directly regulates c-MYC and activates a feed-forward-loop transcriptional network promoting leukemic cell growth. Proc. Natl. Acad. Sci. USA 2006, 103, 18261–18266. [Google Scholar] [CrossRef] [PubMed]
- van Riggelen, J.; Yetil, A.; Felsher, D.W. MYC as a regulator of ribosome biogenesis and protein synthesis. Nat. Rev. Cancer 2010, 10, 301–309. [Google Scholar] [CrossRef]
- Aras, S.; Pak, O.; Sommer, N.; Finley, R., Jr.; Hüttemann, M.; Weissmann, N.; Grossman, L.I. Oxygen-dependent expression of cytochrome c oxidase subunit 4-2 gene expression is mediated by transcription factors RBPJ, CXXC5 and CHCHD2. Nucleic Acids Res. 2013, 41, 2255–2266. [Google Scholar] [CrossRef]
- Xu, J.; Chi, F.; Guo, T.; Punj, V.; Lee, W.P.; French, S.W.; Tsukamoto, H. NOTCH reprograms mitochondrial metabolism for proinflammatory macrophage activation. J. Clin. Investig. 2015, 125, 1579–1590. [Google Scholar] [CrossRef]
- Vujovic, F.; Hunter, N.; Farahani, R.M. Notch ankyrin domain: Evolutionary rise of a thermodynamic sensor. Cell Commun. Signal. 2022, 20, 1–18. [Google Scholar] [CrossRef]
- Foltz, D.R.; Santiago, M.C.; Berechid, B.E.; Nye, J.S. Glycogen Synthase Kinase-3β Modulates Notch Signaling and Stability. Curr. Biol. 2002, 12, 1006–1011. [Google Scholar] [CrossRef] [PubMed]
- Stambolic, V.; Woodgett, J.R. Mitogen inactivation of glycogen synthase kinase-3β in intact cells via serine 9 phosphorylation. Biochem. J. 1994, 303, 701–704. [Google Scholar] [CrossRef]
- Guarani, V.; Deflorian, G.; Franco, C.A.; Krüger, M.; Phng, L.-K.; Bentley, K.; Toussaint, L.; Dequiedt, F.; Mostoslavsky, R.; Schmidt, M.H.H.; et al. Acetylation-dependent regulation of endothelial Notch signalling by the SIRT1 deacetylase. Nature 2011, 473, 234–238. [Google Scholar] [CrossRef]
- Cardaci, S.; Filomeni, G.; Ciriolo, M.R. Redox implications of AMPK-mediated signal transduction beyond energetic clues. J. Cell Sci. 2012, 125, 2115–2125. [Google Scholar] [CrossRef] [PubMed]
- Cantó, C.; Gerhart-Hines, Z.; Feige, J.N.; Lagouge, M.; Noriega, L.; Milne, J.C.; Elliott, P.J.; Puigserver, P.; Auwerx, J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009, 458, 1056–1060. [Google Scholar] [CrossRef]
- Vujovic, F.; Rezaei-Lotfi, S.; Hunter, N.; Farahani, R.M. The fate of notch-1 transcript is linked to cell cycle dynamics by activity of a natural antisense transcript. Nucleic Acids Res. 2021, 49, 10419–10430. [Google Scholar] [CrossRef]
- Gardner, L.B. Hypoxic Inhibition of Nonsense-Mediated RNA Decay Regulates Gene Expression and the Integrated Stress Response. Mol. Cell. Biol. 2008, 28, 3729–3741. [Google Scholar] [CrossRef]
- Serra, H.; Chivite, I.; Angulo-Urarte, A.; Soler, A.; Sutherland, J.D.; Arruabarrena-Aristorena, A.; Ragab, A.; Lim, R.; Malumbres, M.; Fruttiger, M.; et al. PTEN mediates Notch-dependent stalk cell arrest in angiogenesis. Nat. Commun. 2015, 6, 7935. [Google Scholar] [CrossRef] [PubMed]
- Hayward, P.; Kalmar, T.; Arias, A.M. Wnt/Notch signalling and information processing during development. Development 2008, 135, 411–424. [Google Scholar] [CrossRef]
- Wang, G.L.; Jiang, B.-H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef]
- Jewell, U.R.; Kvietikova, I.; Scheid, A.; Bauer, C.; Wenger, R.H.; Gassmann, M. Induction of HIF–1α in response to hypoxia is instantaneous. FASEB J. 2001, 15, 1312–1314. [Google Scholar] [CrossRef]
- Jiang, B.H.; Semenza, G.L.; Bauer, C.; Marti, H.H. Hypoxia-inducible factor 1 levels vary exponentially over a physiologically relevant range of O2 tension. Am. J. Physiol. Cell Physiol. 1996, 271, C1172–C1180. [Google Scholar] [CrossRef]
- Kim, J.-W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S.; Maltepe, E.; Goldwasser, E.; Mathieu, C.E.; Simon, M.C.; Schumacker, P.T. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc. Natl. Acad. Sci. USA 1998, 95, 11715–11720. [Google Scholar] [CrossRef] [PubMed]
- Brunelle, J.K.; Bell, E.L.; Quesada, N.M.; Vercauteren, K.; Tiranti, V.; Zeviani, M.; Scarpulla, R.C.; Chandel, N.S. Oxygen sensing requires mitochondrial ROS but not oxidative phosphorylation. Cell Metab. 2005, 1, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Guzy, R.D.; Hoyos, B.; Robin, E.; Chen, H.; Liu, L.; Mansfield, K.D.; Simon, M.C.; Hammerling, U.; Schumacker, P.T. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 2005, 1, 401–408. [Google Scholar] [CrossRef]
- Kaewpila, S.; Venkataraman, S.; Buettner, G.R.; Oberley, L.W. Manganese Superoxide Dismutase Modulates Hypoxia-Inducible Factor-1α Induction via Superoxide. Cancer Res 2008, 68, 2781–2788. [Google Scholar] [CrossRef]
- Chua, Y.L.; Dufour, E.; Dassa, E.P.; Rustin, P.; Jacobs, H.T.; Taylor, C.T.; Hagen, T. Stabilization of Hypoxia-inducible Factor-1α Protein in Hypoxia Occurs Independently of Mitochondrial Reactive Oxygen Species Production*. J. Biol. Chem. 2010, 285, 31277–31284. [Google Scholar] [CrossRef]
- Schindler, C.; Plumlee, C. Inteferons pen the JAK–STAT pathway. Semin. Cell Dev. Biol. 2008, 19, 311–318. [Google Scholar] [CrossRef]
- Darnell, J.E., Jr.; Kerr, I.M.; Stark, G.R. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 1994, 264, 1415–1421. [Google Scholar] [CrossRef] [PubMed]
- Danial, N.N.; Rothman, P. JAK-STAT signaling activated by Abl oncogenes. Oncogene 2000, 19, 2523–2531. [Google Scholar] [CrossRef]
- Simon, A.R.; Rai, U.; Fanburg, B.L.; Cochran, B.H.; Natarajan, K.; Meganathan, V.; Mitchell, C.; Boggaram, V.; Bhavsar, S.K.; Hosseinzadeh, Z.; et al. Activation of the JAK-STAT pathway by reactive oxygen species. Am. J. Physiol. Physiol. 1998, 275, C1640–C1652. [Google Scholar] [CrossRef]
- Madamanchi, N.R.; Li, S.; Patterson, C.; Runge, M.S. Reactive Oxygen Species Regulate Heat-Shock Protein 70 via the JAK/STAT Pathway. Arter. Thromb. Vasc. Biol. 2001, 21, 321–326. [Google Scholar] [CrossRef]
- Oeckinghaus, A.; Ghosh, S. The NF-κB Family of Transcription Factors and Its Regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef]
- Espín-Palazón, R.; Traver, D. The NF-κB family: Key players during embryonic development and HSC emergence. Exp. Hematol. 2016, 44, 519–527. [Google Scholar] [CrossRef]
- Karin, M.; Delhase, M. The IκB kinase (IKK) and NF-κB: Key elements of proinflammatory signalling. Semin. Immunol. 2000, 12, 85–98. [Google Scholar] [CrossRef]
- Hayden, M.S.; Ghosh, S. Shared Principles in NF-κB Signaling. Cell 2008, 132, 344–362. [Google Scholar] [CrossRef] [PubMed]
- Xiao, G.; Harhaj, E.W.; Sun, S.-C. NF-κB-Inducing Kinase Regulates the Processing of NF-κB2 p100. Mol. Cell 2001, 7, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Senftleben, U.; Cao, Y.; Xiao, G.; Greten, F.R.; Krähn, G.; Bonizzi, G.; Chen, Y.; Hu, Y.; Fong, A.; Sun, S.-C.; et al. Activation by IKKα of a Second, Evolutionary Conserved, NF-κB Signaling Pathway. Science 2001, 293, 1495–1499. [Google Scholar] [CrossRef]
- Schreck, R.; Rieber, P.; Baeuerle, P. Reactive oxygen intermediates as apparently widely used messengers in the activation of the NF-kappa B transcription factor and HIV-1. EMBO J. 1991, 10, 2247–2258. [Google Scholar] [CrossRef]
- Manna, S.K.; Zhang, H.J.; Yan, T.; Oberley, L.W.; Aggarwal, B.B. Overexpression of Manganese Superoxide Dismutase Suppresses Tumor Necrosis Factor-induced Apoptosis and Activation of Nuclear Transcription Factor-κB and Activated Protein-1. J. Biol. Chem. 1998, 273, 13245–13254. [Google Scholar] [CrossRef]
- Kabe, Y.; Ando, K.; Hirao, S.; Yoshida, M.; Handa, H. Redox Regulation of NF-κB Activation: Distinct Redox Regulation between the Cytoplasm and the Nucleus. Antioxid. Redox Signal. 2005, 7, 395–403. [Google Scholar] [CrossRef]
- Toledano, M.B.; Leonard, W.J. Modulation of transcription factor NF-kappa B binding activity by oxidation-reduction in vitro. Proc. Natl. Acad. Sci. USA 1991, 88, 4328–4332. [Google Scholar] [CrossRef]
- Matthews, J.R.; Kaszubska, W.; Turcatti, G.; Wells, T.N.; Hay, R.T. Role of cysteine62in DNA recognition by the P50 subunit of NF-xB. Nucleic Acids Res. 1993, 21, 1727–1734. [Google Scholar] [CrossRef]
- Matthews, J.R.; Wakasugi, N.; Virelizier, J.-L.; Yodoi, J.; Hay, R.T. Thiordoxin regulates the DNA binding activity of NF-χB by reduction of a disulphid bond involving cysteine 62. Nucleic Acids Res. 1992, 20, 3821–3830. [Google Scholar] [CrossRef]
- Schoonbroodt, S.; Ferreira, V.; Best-Belpomme, M.; Boelaert, J.R.; Legrand-Poels, S.; Korner, M.; Piette, J. Crucial Role of the Amino-Terminal Tyrosine Residue 42 and the Carboxyl-Terminal PEST Domain of IκBα in NF-κB Activation by an Oxidative Stress. J. Immunol. 2000, 164, 4292–4300. [Google Scholar] [CrossRef]
- Takada, Y.; Mukhopadhyay, A.; Kundu, G.C.; Mahabeleshwar, G.H.; Singh, S.; Aggarwal, B.B. Hydrogen Peroxide Activates NF-κB through Tyrosine Phosphorylation of IκBα and Serine Phosphorylation of p65: Evidence for the involvement of IκBα kinase and Syk protein-tyrosine kinase. J. Biol. Chem. 2003, 278, 24233–24241. [Google Scholar] [CrossRef]
- Canty, T.G.; Boyle, E.M.; Farr, A.; Morgan, E.N.; Verrier, E.D.; Pohlman, T.H. Oxidative Stress Induces NF-κB Nuclear Translocation without Degradation of IκBα. Circulation 1999, 100, II-361–II-364. [Google Scholar] [CrossRef]
- Béraud, C.; Henzel, W.J.; Baeuerle, P.A. Involvement of regulatory and catalytic subunits of phosphoinositide 3-kinase in NF-κB activation. Proc. Natl. Acad. Sci. USA 1999, 96, 429–434. [Google Scholar] [CrossRef]
- Wu, M.; Bian, Q.; Liu, Y.; Fernandes, A.F.; Taylor, A.; Pereira, P.; Shang, F. Sustained oxidative stress inhibits NF-κB activation partially via inactivating the proteasome. Free. Radic. Biol. Med. 2009, 46, 62–69. [Google Scholar] [CrossRef]
- Jaspers, I.; Zhang, W.; Fraser, A.; Samet, J.M.; Reed, W. Hydrogen Peroxide Has Opposing Effects on IKK Activity and I κ B α Breakdown in Airway Epithelial Cells. Am. J. Respir. Cell Mol. Biol. 2001, 24, 769–777. [Google Scholar] [CrossRef]
- Reynaert, N.L.; van der Vliet, A.; Guala, A.S.; McGovern, T.; Hristova, M.; Pantano, C.; Heintz, N.H.; Heim, J.; Ho, Y.-S.; Matthews, D.E.; et al. Dynamic redox control of NF-κB through glutaredoxin-regulated S-glutathionylation of inhibitory κB kinase β. Proc. Natl. Acad. Sci. USA 2006, 103, 13086–13091. [Google Scholar] [CrossRef] [PubMed]
- Djavaheri-Mergny, M.; Javelaud, D.; Wietzerbin, J.; Besançon, F. NF-κB activation prevents apoptotic oxidative stress via an increase of both thioredoxin and MnSOD levels in TNFα-treated Ewing sarcoma cells. FEBS Lett. 2004, 578, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Pham, C.G.; Bubici, C.; Zazzeroni, F.; Papa, S.; Jones, J.; Alvarez, K.; Jayawardena, S.; De Smaele, E.; Cong, R.; Beaumont, C.; et al. Ferritin Heavy Chain Upregulation by NF-κB Inhibits TNFα-Induced Apoptosis by Suppressing Reactive Oxygen Species. Cell 2004, 119, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Kairisalo, M.; Korhonen, L.; Blomgren, K.; Lindholm, D. X-linked inhibitor of apoptosis protein increases mitochondrial antioxidants through NF-κB activation. Biochem. Biophys. Res. Commun. 2007, 364, 138–144. [Google Scholar] [CrossRef]
- Finley, D. Recognition and Processing of Ubiquitin-Protein Conjugates by the Proteasome. Annu. Rev. Biochem. 2009, 78, 477–513. [Google Scholar] [CrossRef]
- Glickman, M.H.; Ciechanover, A. The Ubiquitin-Proteasome Proteolytic Pathway: Destruction for the Sake of Construction. Physiol. Rev. 2002, 82, 373–428. [Google Scholar] [CrossRef]
- Pickart, C.M. Mechanisms Underlying Ubiquitination. Annu. Rev. Biochem. 2001, 70, 503–533. [Google Scholar] [CrossRef]
- Komander, D. The emerging complexity of protein ubiquitination. Biochem. Soc. Trans. 2009, 37, 937–953. [Google Scholar] [CrossRef] [PubMed]
- Pacifici, R.E.; Salo, D.C.; Davies, K.J. Macroxyproteinase (M.O.P.): A 670 kDa Proteinase complex that degrades oxidatively denatured proteins in red blood cells. Free. Radic. Biol. Med. 1989, 7, 521–536. [Google Scholar] [CrossRef] [PubMed]
- Jahngen-Hodge, J.; Obin, M.S.; Gong, X.; Shang, F.; Nowell, T.R.; Gong, J.; Abasi, H.; Blumberg, J.; Taylor, A. Regulation of Ubiquitin-conjugating Enzymes by Glutathione Following Oxidative Stress. J. Biol. Chem. 1997, 272, 28218–28226. [Google Scholar] [CrossRef]
- Obin, M.; Shang, F.; Gong, X.; Handelman, G.; Blumberg, J.; Taylor, A. Redox regulation of ubiquitin-conjugating enzymes: Mechanistic insights using the thiol-specific oxidant diamide. FASEB J. 1998, 12, 561–569. [Google Scholar] [CrossRef]
- Sun, H.; Lesche, R.; Li, D.-M.; Liliental, J.; Zhang, H.; Gao, J.; Gavrilova, N.; Mueller, B.; Liu, X.; Wu, H. PTEN modulates cell cycle progression and cell survival by regulating phosphatidylinositol 3,4,5,-trisphosphate and Akt/protein kinase B signaling pathway. Proc. Natl. Acad. Sci. USA 1999, 96, 6199–6204. [Google Scholar] [CrossRef] [PubMed]
- Milne, S.B.; Ivanova, P.T.; DeCamp, D.; Hsueh, R.C.; Brown, H.A. A targeted mass spectrometric analysis of phosphatidylinositol phosphate species. J. Lipid Res. 2005, 46, 1796–1802. [Google Scholar] [CrossRef]
- Clark, J.; E Anderson, K.; Juvin, V.; Smith, T.S.; Karpe, F.; O Wakelam, M.J.; Stephens, L.R.; Hawkins, P.T. Quantification of PtdInsP3 molecular species in cells and tissues by mass spectrometry. Nat. Methods 2011, 8, 267–272. [Google Scholar] [CrossRef]
- Truebestein, L.; Hornegger, H.; Anrather, D.; Hartl, M.; Fleming, K.D.; Stariha, J.T.B.; Pardon, E.; Steyaert, J.; Burke, J.E.; Leonard, T.A. Structure of autoinhibited Akt1 reveals mechanism of PIP3-mediated activation. Proc. Natl. Acad. Sci. USA 2021, 118, e2101496118. [Google Scholar] [CrossRef]
- Sundaresan, M.; Yu, Z.-X.; Ferrans, V.J.; Irani, K.; Finkel, T. Requirement for Generation of H2O2 for Platelet-Derived Growth Factor Signal Transduction. Science 1995, 270, 296–299. [Google Scholar] [CrossRef]
- Bae, Y.S.; Kang, S.W.; Seo, M.S.; Baines, I.C.; Tekle, E.; Chock, P.B.; Rhee, S.G. Epidermal Growth Factor (EGF)-induced Generation of Hydrogen Peroxide. J. Biol. Chem. 1997, 272, 217–221. [Google Scholar] [CrossRef]
- Bos, J.L. All in the family? New insights and questions regarding interconnectivity of Ras, Rap1 and Ral. EMBO J. 1998, 17, 6776–6782. [Google Scholar] [CrossRef] [PubMed]
- Ohba, Y.; Ikuta, K.; Ogura, A.; Matsuda, J.; Mochizuki, N.; Nagashima, K.; Kurokawa, K.; Mayer, B.J.; Maki, K.; Miyazaki, J.; et al. Requirement for C3G-dependent Rap1 activation for cell adhesion and embryogenesis. EMBO J. 2001, 20, 3333–3341. [Google Scholar] [CrossRef] [PubMed]
- Kitayama, H.; Sugimoto, Y.; Matsuzaki, T.; Ikawa, Y.; Noda, M. A ras-related gene with transformation suppressor activity. Cell 1989, 56, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Mochizuki, N.; Yamashita, S.; Kurokawa, K.; Ohba, Y.; Nagai, T.; Miyawaki, A.; Matsuda, M. Spatio-temporal images of growth-factor-induced activation of Ras and Rap1. Nature 2001, 411, 1065–1068. [Google Scholar] [CrossRef]
- Keyes, J.; Ganesan, A.; Molinar-Inglis, O.; Hamidzadeh, A.; Zhang, J.; Ling, M.; Trejo, J.; Levchenko, A.; Zhang, J.; Department of Pharmacology; et al. Signaling diversity enabled by Rap1-regulated plasma membrane ERK with distinct temporal dynamics. eLife 2020, 9, e57410. [Google Scholar] [CrossRef]
- Hu, Y.; Lu, W.; Chen, G.; Wang, P.; Chen, Z.; Zhou, Y.; Ogasawara, M.; Trachootham, D.; Feng, L.; Pelicano, H.; et al. K-rasG12V transformation leads to mitochondrial dysfunction and a metabolic switch from oxidative phosphorylation to glycolysis. Cell Res. 2011, 22, 399–412. [Google Scholar] [CrossRef]
- Serasinghe, M.N.; Wieder, S.Y.; Renault, T.T.; Elkholi, R.; Asciolla, J.J.; Yao, J.L.; Jabado, O.; Hoehn, K.; Kageyama, Y.; Sesaki, H.; et al. Mitochondrial Division Is Requisite to RAS-Induced Transformation and Targeted by Oncogenic MAPK Pathway Inhibitors. Mol. Cell 2015, 57, 521–536. [Google Scholar] [CrossRef]
- Zhu, J.; Blenis, J.; Yuan, J. Activation of PI3K/Akt and MAPK pathways regulates Myc-mediated transcription by phosphorylating and promoting the degradation of Mad1. Proc. Natl. Acad. Sci. USA 2008, 105, 6584–6589. [Google Scholar] [CrossRef]
- Du, K.; Montminy, M. CREB Is a Regulatory Target for the Protein Kinase Akt/PKB. J. Biol. Chem. 1998, 273, 32377–32379. [Google Scholar] [CrossRef]
- Liu, J.; Park, E.; Gurney, A.; Roesler, W.; Hanson, R. Cyclic AMP induction of phosphoenolpyruvate carboxykinase (GTP) gene transcription is mediated by multiple promoter elements. J. Biol. Chem. 1991, 266, 19095–19102. [Google Scholar] [CrossRef]
- Quinn, P.G.; Granner, D.K. Cyclic AMP-dependent protein kinase regulates transcription of the phosphoenolpyruvate carboxykinase gene but not binding of nuclear factors to the cyclic AMP regulatory element. Mol. Cell. Biol. 1990, 10, 3357–3364. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishnan, L.; Scarpulla, R. Differential regulation of respiratory chain subunits by a CREB-dependent signal transduction pathway. Role of cyclic AMP in cytochrome c and COXIV gene expression. J. Biol. Chem. 1994, 269, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Herzig, R.P.; Scacco, S.; Scarpulla, R.C. Sequential Serum-dependent Activation of CREB and NRF-1 Leads to Enhanced Mitochondrial Respiration through the Induction of Cytochrome c. J. Biol. Chem. 2000, 275, 13134–13141. [Google Scholar] [CrossRef]
- Nandagopal, N.; Roux, P.P. Regulation of global and specific mRNA translation by the mTOR signaling pathway. Translation 2015, 3, e983402. [Google Scholar] [CrossRef] [PubMed]
- Rabanal-Ruiz, Y.; Otten, E.G.; Korolchuk, V.I. mTORC1 as the main gateway to autophagy. Essays Biochem. 2017, 61, 565–584. [Google Scholar] [CrossRef]
- Princiotta, M.F.; Finzi, D.; Qian, S.-B.; Gibbs, J.; Schuchmann, S.; Buttgereit, F.; Bennink, J.R.; Yewdell, J.W. Quantitating Protein Synthesis, Degradation, and Endogenous Antigen Processing. Immunity 2003, 18, 343–354. [Google Scholar] [CrossRef]
- Wagner, A. Energy Constraints on the Evolution of Gene Expression. Mol. Biol. Evol. 2005, 22, 1365–1374. [Google Scholar] [CrossRef]
- Xu, K.; Yin, N.; Peng, M.; Stamatiades, E.G.; Chhangawala, S.; Shyu, A.; Li, P.; Zhang, X.; Do, M.H.; Capistrano, K.J.; et al. Glycolytic ATP fuels phosphoinositide 3-kinase signaling to support effector T helper 17 cell responses. Immunity 2021, 54, 976–987.e7. [Google Scholar] [CrossRef]
- Liu, S.; Fu, S.; Wang, G.; Cao, Y.; Li, L.; Li, X.; Yang, J.; Li, N.; Shan, Y.; Cao, Y.; et al. Glycerol-3-phosphate biosynthesis regenerates cytosolic NAD+ to alleviate mitochondrial disease. Cell Metab. 2021, 33, 1974–1987.e9. [Google Scholar] [CrossRef]
- Miwa, S.; Brand, M.D. The topology of superoxide production by complex III and glycerol 3-phosphate dehydrogenase in Drosophila mitochondria. Biochim. Biophys. Acta 2005, 1709, 214–219. [Google Scholar] [CrossRef]
- Iwata, R.; Casimir, P.; Vanderhaeghen, P. Mitochondrial dynamics in postmitotic cells regulate neurogenesis. Science 2020, 369, 858–862. [Google Scholar] [CrossRef] [PubMed]
- Foo, M.X.R.; Ong, P.F.; Dreesen, O. Premature aging syndromes: From patients to mechanism. J. Dermatol. Sci. 2019, 96, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Sinha, J.K.; Ghosh, S.; Raghunath, M. Progeria: A rare genetic premature ageing disorder. Indian J. Med. Res. 2014, 139, 667–674. [Google Scholar] [PubMed]
- Rivera-Torres, J.; Acín-Perez, R.; Cabezas-Sánchez, P.; Osorio, F.G.; Gonzalez-Gómez, C.; Megias, D.; Cámara, C.; López-Otín, C.; Enríquez, J.A.; Luque-García, J.L.; et al. Identification of mitochondrial dysfunction in Hutchinson–Gilford progeria syndrome through use of stable isotope labeling with amino acids in cell culture. J. Proteom. 2013, 91, 466–477. [Google Scholar] [CrossRef]
- Mateos, J.; Landeira-Abia, A.; Fafián-Labora, J.A.; Fernández-Pernas, P.; Lesende-Rodríguez, I.; Fernández-Puente, P.; Fernández-Moreno, M.; Delmiro, A.; Martín, M.A.; Blanco, F.J.; et al. iTRAQ-based analysis of progerin expression reveals mitochondrial dysfunction, reactive oxygen species accumulation and altered proteostasis. Stem Cell Res. Ther. 2015, 6, 119. [Google Scholar] [CrossRef]
- Monterrubio-Ledezma, F.; Navarro-García, F.; Massieu, L.; Mondragón-Flores, R.; Soto-Ponce, L.A.; Magaña, J.J.; Cisneros, B. Rescue of Mitochondrial Function in Hutchinson-Gilford Progeria Syndrome by the Pharmacological Modulation of Exportin CRM1. Cells 2023, 12, 275. [Google Scholar] [CrossRef]
- Kuk, M.U.; Kim, J.W.; Lee, Y.-S.; A Cho, K.; Park, J.T.; Park, S.C. Alleviation of Senescence via ATM Inhibition in Accelerated Aging Models. Mol. Cells 2019, 42, 210–217. [Google Scholar] [CrossRef]
- Meng, F.; Henson, R.; Wehbe–Janek, H.; Ghoshal, K.; Jacob, S.T.; Patel, T. MicroRNA-21 Regulates Expression of the PTEN Tumor Suppressor Gene in Human Hepatocellular Cancer. Gastroenterology 2007, 133, 647–658. [Google Scholar] [CrossRef]
- Ghafouri-Fard, S.; Abak, A.; Shoorei, H.; Mohaqiq, M.; Majidpoor, J.; Sayad, A.; Taheri, M. Regulatory role of microRNAs on PTEN signaling. Biomed. Pharmacother. 2020, 133, 110986. [Google Scholar] [CrossRef]
- Farahani, R.M.; Rezaei-Lotfi, S.; Hunter, N. Genomic competition for noise reduction shaped evolutionary landscape of mir-4673. npj Syst. Biol. Appl. 2020, 6, 1–15. [Google Scholar] [CrossRef]
- Partin, A.C.; Ngo, T.D.; Herrell, E.; Jeong, B.-C.; Hon, G.; Nam, Y. Heme enables proper positioning of Drosha and DGCR8 on primary microRNAs. Nat. Commun. 2017, 8, 1737. [Google Scholar] [CrossRef]
- Quick-Cleveland, J.; Jacob, J.P.; Weitz, S.H.; Shoffner, G.; Senturia, R.; Guo, F. The DGCR8 RNA-Binding Heme Domain Recognizes Primary MicroRNAs by Clamping the Hairpin. Cell Rep. 2014, 7, 1994–2005. [Google Scholar] [CrossRef]
- McGeary, S.E.; Lin, K.S.; Shi, C.Y.; Pham, T.M.; Bisaria, N.; Kelley, G.M.; Bartel, D.P. The biochemical basis of microRNA targeting efficacy. Science 2019, 366, eaav1741. [Google Scholar] [CrossRef]
- Plascencia-Villa, G.; Perry, G. Roles of Oxidative Stress in Synaptic Dysfunction and Neuronal Cell Death in Alzheimer’s Disease. Antioxidants 2023, 12, 1628. [Google Scholar] [CrossRef] [PubMed]
- Roy, R.G.; Mandal, P.K.; Maroon, J.C. Oxidative Stress Occurs Prior to Amyloid Aβ Plaque Formation and Tau Phosphorylation in Alzheimer’s Disease: Role of Glutathione and Metal Ions. ACS Chem. Neurosci. 2023, 14, 2944–2954. [Google Scholar] [CrossRef] [PubMed]
- Mufson, E.; Chen, E.-Y.; Cochran, E.; Beckett, L.; Bennett, D.; Kordower, J. Entorhinal Cortex β-Amyloid Load in Individuals with Mild Cognitive Impairment. Exp. Neurol. 1999, 158, 469–490. [Google Scholar] [CrossRef] [PubMed]
- Markesbery, W.R.; Schmitt, F.A.; Kryscio, R.J.; Davis, D.G.; Smith, C.D.; Wekstein, D.R. Neuropathologic Substrate of Mild Cognitive Impairment. Arch. Neurol. 2006, 63, 38–46. [Google Scholar] [CrossRef]
- Price, J.L.; McKeel, D.W., Jr.; Buckles, V.D.; Roe, C.M.; Xiong, C.; Grundman, M.; Hansen, L.A.; Petersen, R.C.; Parisi, J.E.; Dickson, D.W.; et al. Neuropathology of nondemented aging: Presumptive evidence for preclinical Alzheimer disease. Neurobiol. Aging 2009, 30, 1026–1036. [Google Scholar] [CrossRef] [PubMed]
- Coyle, J.; Oster-Granite, M.; Gearhart, J. The neurobiologie consequences of down syndrome. Brain Res. Bull. 1986, 16, 773–787. [Google Scholar] [CrossRef]
- Pallardó, F.V.; Degan, P.; D’ischia, M.; Kelly, F.J.; Zatterale, A.; Calzone, R.; Castello, G.; Fernandez-Delgado, R.; Dunster, C.; Lloret, A.; et al. Multiple evidence for an early age pro-oxidant state in Down Syndrome patients. Biogerontology 2006, 7, 211–220. [Google Scholar] [CrossRef]
- Busciglio, J.; Yankner, B.A. Apoptosis and increased generation of reactive oxygen species in Down’s syndrome neurons in vitro. Nature 1995, 378, 776–779. [Google Scholar] [CrossRef] [PubMed]
- Yamane, T.; Ikari, Y.; Nishio, T.; Ishii, K.; Kato, T.; Ito, K.; Silverman, D.H.S.; Senda, M.; Asada, T.; Arai, H.; et al. Visual-Statistical Interpretation of 18F-FDG-PET Images for Characteristic Alzheimer Patterns in a Multicenter Study: Inter-Rater Concordance and Relationship to Automated Quantitative Evaluation. Am. J. Neuroradiol. 2013, 35, 244–249. [Google Scholar] [CrossRef] [PubMed]
- Shokouhi, S.; Claassen, D.; Kang, H.; Ding, Z.; Rogers, B.; Mishra, A.; Riddle, W.R.; Initiative, F.T.A.D.N. Longitudinal Progression of Cognitive Decline Correlates with Changes in the Spatial Pattern of Brain 18F-FDG PET. J. Nucl. Med. 2013, 54, 1564–1569. [Google Scholar] [CrossRef]
- Landau, S.M.; Harvey, D.; Madison, C.M.; Koeppe, R.A.; Reiman, E.M.; Foster, N.L.; Weiner, M.W.; Jagust, W.J.; The Alzheimer’s Disease Neuroimaging Initiative. Associations between cognitive, functional, and FDG-PET measures of decline in AD and MCI. Neurobiol. Aging 2011, 32, 1207–1218. [Google Scholar] [CrossRef]
- Liot, G.; Bossy, B.; Lubitz, S.; Kushnareva, Y.; Sejbuk, N.; Bossy-Wetzel, E. Complex II inhibition by 3-NP causes mitochondrial fragmentation and neuronal cell death via an NMDA- and ROS-dependent pathway. Cell Death Differ. 2009, 16, 899–909. [Google Scholar] [CrossRef]
- Wang, X.; Su, B.; Liu, W.; He, X.; Gao, Y.; Castellani, R.J.; Perry, G.; Smith, M.A.; Zhu, X. DLP1-dependent mitochondrial fragmentation mediates 1-methyl-4-phenylpyridinium toxicity in neurons: Implications for Parkinson’s disease. Aging Cell 2011, 10, 807–823. [Google Scholar] [CrossRef]
- Chandrasekaran, K.; Giordano, T.; Brady, D.R.; Stoll, J.; Martin, L.J.; Rapoport, S.I. Impairment in mitochondrial cytochrome oxidase gene expression in Alzheimer disease. Mol. Brain Res. 1994, 24, 336–340. [Google Scholar] [CrossRef] [PubMed]
- Cottrell, D.; Blakely, E.; Johnson, M.; Ince, P.; Turnbull, D. Mitochondrial enzyme-deficient hippocampal neurons and choroidal cells in AD. Neurology 2001, 57, 260–264. [Google Scholar] [CrossRef]
- Maurer, I. A selective defect of cytochrome c oxidase is present in brain of Alzheimer disease patients. Neurobiol. Aging 2000, 21, 455–462. [Google Scholar] [CrossRef]
- Parker, W.D.; Mahr, N.J.; Filley, C.M.; Parks, J.K.; Hughes, D.; Young, D.A.; Cullum, C.M. Reduced platelet cytochrome c oxidase activity in Alzheimer’s disease. Neurology 1994, 44, 1086. [Google Scholar] [CrossRef]
- Genova, M.L.; Ventura, B.; Giuliano, G.; Bovina, C.; Formiggini, G.; Castelli, G.P.; Lenaz, G. The site of production of superoxide radical in mitochondrial Complex I is not a bound ubisemiquinone but presumably iron–sulfur cluster N2. FEBS Lett. 2001, 505, 364–368. [Google Scholar] [CrossRef] [PubMed]
- Kushnareva, Y.; Murphy, A.N.; Andreyev, A. Complex I-mediated reactive oxygen species generation: Modulation by cytochrome c and NAD(P)+ oxidation–reduction state. Biochem. J. 2002, 368, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Niu, L.; Li, S.; Le, W. Pathological Impacts of Chronic Hypoxia on Alzheimer’s Disease. ACS Chem. Neurosci. 2018, 10, 902–909. [Google Scholar] [CrossRef]
- Guzy, R.D.; Schumacker, P.T. Oxygen sensing by mitochondria at complex III: The paradox of increased reactive oxygen species during hypoxia. Exp. Physiol. 2006, 91, 807–819. [Google Scholar] [CrossRef] [PubMed]
- Jaworski, J.; Spangler, S.; Seeburg, D.P.; Hoogenraad, C.C.; Sheng, M. Control of Dendritic Arborization by the Phosphoinositide-3′-Kinase–Akt–Mammalian Target of Rapamycin Pathway. J. Neurosci. 2005, 25, 11300–11312. [Google Scholar] [CrossRef] [PubMed]
- Cuesto, G.; Enriquez-Barreto, L.; Caramés, C.; Cantarero, M.; Gasull, X.; Sandi, C.; Ferrús, A.; Acebes, Á.; Morales, M. Phosphoinositide-3-Kinase Activation Controls Synaptogenesis and Spinogenesis in Hippocampal Neurons. J. Neurosci. 2011, 31, 2721–2733. [Google Scholar] [CrossRef]
- Akiyama, H.; Kamiguchi, H. Phosphatidylinositol 3-Kinase Facilitates Microtubule-dependent Membrane Transport for Neuronal Growth Cone Guidance. J. Biol. Chem. 2010, 285, 41740–41748. [Google Scholar] [CrossRef]
- Horwood, J.M.; Dufour, F.; Laroche, S.; Davis, S. Signalling mechanisms mediated by the phosphoinositide 3-kinase/Akt cascade in synaptic plasticity and memory in the rat. Eur. J. Neurosci. 2006, 23, 3375–3384. [Google Scholar] [CrossRef]
- Sui, L.; Wang, J.; Li, B.-M. Role of the phosphoinositide 3-kinase-Akt-mammalian target of the rapamycin signaling pathway in long-term potentiation and trace fear conditioning memory in rat medial prefrontal cortex. Learn. Mem. 2008, 15, 762–776. [Google Scholar] [CrossRef]
- Martin, T.F. Role of PI(4,5)P2 in Vesicle Exocytosis and Membrane Fusion. Phosphoinositides II Divers. Biol. Funct. 2012, 59, 111–130. [Google Scholar] [CrossRef]
- Wallroth, A.; Haucke, V. Phosphoinositide conversion in endocytosis and the endolysosomal system. J. Biol. Chem. 2018, 293, 1526–1535. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.F.; Trotman, L.C. PTEN: Bridging Endocytosis and Signaling. Cold Spring Harb. Perspect. Med. 2019, 10, a036103. [Google Scholar] [CrossRef]
- Hsu, J.-W.; Bai, M.; Li, K.; Yang, J.-S.; Chu, N.; Cole, P.A.; Eck, M.J.; Li, J.; Hsu, V.W. The protein kinase Akt acts as a coat adaptor in endocytic recycling. Nature 2020, 22, 927–933. [Google Scholar] [CrossRef]
- Fujioka, Y.; Tsuda, M.; Hattori, T.; Sasaki, J.; Sasaki, T.; Miyazaki, T.; Ohba, Y. The Ras–PI3K Signaling Pathway Is Involved in Clathrin-Independent Endocytosis and the Internalization of Influenza Viruses. PLoS ONE 2011, 6, e16324. [Google Scholar] [CrossRef]
- Fujioka, Y.; Satoh, A.O.; Horiuchi, K.; Fujioka, M.; Tsutsumi, K.; Sasaki, J.; Nepal, P.; Kashiwagi, S.; Paudel, S.; Nishide, S.; et al. A Peptide Derived from Phosphoinositide 3-kinase Inhibits Endocytosis and Influenza Virus Infection. Cell Struct. Funct. 2019, 44, 61–74. [Google Scholar] [CrossRef]
- Tsutsumi, K.; Fujioka, Y.; Tsuda, M.; Kawaguchi, H.; Ohba, Y. Visualization of Ras-PI3K interaction in the endosome using BiFC. Cell. Signal. 2009, 21, 1672–1679. [Google Scholar] [CrossRef] [PubMed]
- Merrifield, C.J.; Feldman, M.E.; Wan, L.; Almers, W. Imaging actin and dynamin recruitment during invagination of single clathrin-coated pits. Nature 2002, 4, 691–698. [Google Scholar] [CrossRef]
- Mooren, O.L.; Galletta, B.J.; Cooper, J.A. Roles for Actin Assembly in Endocytosis. Annu. Rev. Biochem. 2012, 81, 661–686. [Google Scholar] [CrossRef]
- Basquin, C.; Malardé, V.; Mellor, P.; Anderson, D.H.; Meas-Yedid, V.; Olivo-Marin, J.-C.; Dautry-Varsat, A.; Sauvonnet, N. The signalling factor PI3K is a specific regulator of the clathrin-independent dynamin-dependent endocytosis of IL-2 receptors. J. Cell Sci. 2013, 126, 1099–1108. [Google Scholar] [CrossRef]
- Innocenti, M.; Frittoli, E.; Ponzanelli, I.; Falck, J.R.; Brachmann, S.M.; Di Fiore, P.P.; Scita, G. Phosphoinositide 3-kinase activates Rac by entering in a complex with Eps8, Abi1, and Sos-1. J. Cell Biol. 2003, 160, 17–23. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, L.; Pei, L.; Ju, W.; Ahmadian, G.; Lu, J.; Wang, Y.; Liu, F.; Wang, Y.T. Control of Synaptic Strength, a Novel Function of Akt. Neuron 2003, 38, 915–928. [Google Scholar] [CrossRef]
- Satoh, A.O.; Fujioka, Y.; Kashiwagi, S.; Yoshida, A.; Fujioka, M.; Sasajima, H.; Nanbo, A.; Amano, M.; Ohba, Y. Interaction between PI3K and the VDAC2 channel tethers Ras-PI3K-positive endosomes to mitochondria and promotes endosome maturation. Cell Rep. 2023, 42, 112229. [Google Scholar] [CrossRef]
- Accardi, M.V.; Daniels, B.A.; Brown, P.M.; Fritschy, J.-M.; Tyagarajan, S.K.; Bowie, D. Mitochondrial reactive oxygen species regulate the strength of inhibitory GABA-mediated synaptic transmission. Nat. Commun. 2014, 5, 3168. [Google Scholar] [CrossRef]
- Esparza-Moltó, P.B.; Romero-Carramiñana, I.; de Arenas, C.N.; Pereira, M.P.; Blanco, N.; Pardo, B.; Bates, G.R.; Sánchez-Castillo, C.; Artuch, R.; Murphy, M.P.; et al. Generation of mitochondrial reactive oxygen species is controlled by ATPase inhibitory factor 1 and regulates cognition. PLoS Biol. 2021, 19, e3001252. [Google Scholar] [CrossRef]
- González, A.N.B.; Pazos, M.I.L.; Calvo, D.J. Reactive Oxygen Species in the Regulation of the GABA Mediated Inhibitory Neurotransmission. Neuroscience 2019, 439, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Lam, A.D.; Deck, G.; Goldman, A.; Eskandar, E.N.; Noebels, J.; Cole, A.J. Silent hippocampal seizures and spikes identified by foramen ovale electrodes in Alzheimer’s disease. Nat. Med. 2017, 23, 678–680. [Google Scholar] [CrossRef]
- Vossel, K.A.; Beagle, A.J.; Rabinovici, G.D.; Shu, H.; Lee, S.E.; Naasan, G.; Hegde, M.; Cornes, S.B.; Henry, M.L.; Nelson, A.B.; et al. Seizures and Epileptiform Activity in the Early Stages of Alzheimer Disease. JAMA Neurol. 2013, 70, 1158–1166. [Google Scholar] [CrossRef] [PubMed]
- Palop, J.J.; Mucke, L. Epilepsy and Cognitive Impairments in Alzheimer Disease. Arch. Neurol. 2009, 66, 435–440. [Google Scholar] [CrossRef] [PubMed]
- Palop, J.J.; Mucke, L. Network abnormalities and interneuron dysfunction in Alzheimer disease. Nat. Rev. Neurosci. 2016, 17, 777–792. [Google Scholar] [CrossRef]
- Busche, M.A.; Eichhoff, G.; Adelsberger, H.; Abramowski, D.; Wiederhold, K.-H.; Haass, C.; Staufenbiel, M.; Konnerth, A.; Garaschuk, O. Clusters of Hyperactive Neurons Near Amyloid Plaques in a Mouse Model of Alzheimer’s Disease. Science 2008, 321, 1686–1689. [Google Scholar] [CrossRef] [PubMed]
- Verret, L.; Mann, E.O.; Hang, G.B.; Barth, A.M.; Cobos, I.; Ho, K.; Devidze, N.; Masliah, E.; Kreitzer, A.C.; Mody, I.; et al. Inhibitory Interneuron Deficit Links Altered Network Activity and Cognitive Dysfunction in Alzheimer Model. Cell 2012, 149, 708–721. [Google Scholar] [CrossRef]
- Limon, A.; Reyes-Ruiz, J.M.; Miledi, R. Loss of functional GABA A receptors in the Alzheimer diseased brain. Proc. Natl. Acad. Sci. USA 2012, 109, 10071–10076. [Google Scholar] [CrossRef]
- Griffin, R.J.; Moloney, A.; Kelliher, M.; A Johnston, J.; Ravid, R.; Dockery, P.; O’Connor, R.; O’Neill, C. Activation of Akt/PKB, increased phosphorylation of Akt substrates and loss and altered distribution of Akt and PTEN are features of Alzheimer’s disease pathology. J. Neurochem. 2005, 93, 105–117. [Google Scholar] [CrossRef]
- Pei, J.-J.; Khatoon, S.; An, W.-L.; Nordlinder, M.; Tanaka, T.; Braak, H.; Tsujio, I.; Takeda, M.; Alafuzoff, I.; Winblad, B.; et al. Role of protein kinase B in Alzheimer’s neurofibrillary pathology. Acta Neuropathol. 2002, 105, 381–392. [Google Scholar] [CrossRef]
- Kwak, Y.-D.; Ma, T.; Diao, S.; Zhang, X.; Chen, Y.; Hsu, J.; A Lipton, S.; Masliah, E.; Xu, H.; Liao, F.-F. NO signaling and S-nitrosylation regulate PTEN inhibition in neurodegeneration. Mol. Neurodegener. 2010, 5, 49. [Google Scholar] [CrossRef]
- Sonoda, Y.; Mukai, H.; Matsuo, K.; Takahashi, M.; Ono, Y.; Maeda, K.; Akiyama, H.; Kawamata, T. Accumulation of tumor-suppressor PTEN in Alzheimer neurofibrillary tangles. Neurosci. Lett. 2010, 471, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, F.; Bulloj, A.; Zhang, Y.-W.; Tong, G.; Zhang, Z.; Liao, F.-F.; Xu, H. Tumor suppressor PTEN affects tau phosphorylation, aggregation, and binding to microtubules. FASEB J. 2006, 20, 1272–1274. [Google Scholar] [CrossRef] [PubMed]
- Dickey, C.A.; Koren, J.; Zhang, Y.-J.; Xu, Y.-F.; Jinwal, U.K.; Birnbaum, M.J.; Monks, B.; Sun, M.; Cheng, J.Q.; Patterson, C.; et al. Akt and CHIP coregulate tau degradation through coordinated interactions. Proc. Natl. Acad. Sci. USA 2008, 105, 3622–3627. [Google Scholar] [CrossRef]
- Neill, C.O. PI3-kinase/Akt/mTOR signaling: Impaired on/off switches in aging, cognitive decline and Alzheimer’s disease. Exp. Gerontol. 2013, 48, 647–653. [Google Scholar] [CrossRef] [PubMed]
- Bonifati, V.; Rizzu, P.; van Baren, M.J.; Schaap, O.; Breedveld, G.J.; Krieger, E.; Dekker, M.C.J.; Squitieri, F.; Ibanez, P.; Joosse, M.; et al. Mutations in the DJ-1 Gene Associated with Autosomal Recessive Early-Onset Parkinsonism. Science 2003, 299, 256–259. [Google Scholar] [CrossRef] [PubMed]
- Kahle, P.J.; Waak, J.; Gasser, T. DJ-1 and prevention of oxidative stress in Parkinson’s disease and other age-related disorders. Free. Radic. Biol. Med. 2009, 47, 1354–1361. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.H.; Peters, M.; Jang, Y.; Shi, W.; Pintilie, M.; Fletcher, G.C.; DeLuca, C.; Liepa, J.; Zhou, L.; Snow, B.; et al. DJ-1, a novel regulator of the tumor suppressor PTEN. Cancer Cell 2005, 7, 263–273. [Google Scholar] [CrossRef]
- Unoki, M.; Nakamura, Y. Growth-suppressive effects of BPOZ and EGR2, two genes involved in the PTEN signaling pathway. Oncogene 2001, 20, 4457–4465. [Google Scholar] [CrossRef] [PubMed]
- Valente, E.M.; Salvi, S.; Ialongo, T.; Marongiu, R.; Elia, A.E.; Caputo, V.; Romito, L.; Albanese, A.; Dallapiccola, B.; Bentivoglio, A.R. PINK1 mutations are associated with sporadic early-onset parkinsonism. Ann. Neurol. 2004, 56, 336–341. [Google Scholar] [CrossRef] [PubMed]
- Lingappan, K. NF-κB in oxidative stress. Curr. Opin. Toxicol. 2018, 7, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-H.; Zhou, W.; Liu, S.; Deng, Y.; Cai, F.; Tone, M.; Tone, Y.; Tong, Y.; Song, W. Increased NF-κB signalling up-regulates BACE1 expression and its therapeutic potential in Alzheimer’s disease. Int. J. Neuropsychopharmacol. 2011, 15, 77–90. [Google Scholar] [CrossRef]
- Borghi, R.; Patriarca, S.; Traverso, N.; Piccini, A.; Storace, D.; Garuti, A.; Cirmena, G.; Odetti, P.; Tabaton, M. The increased activity of BACE1 correlates with oxidative stress in Alzheimer’s disease. Neurobiol. Aging 2007, 28, 1009–1014. [Google Scholar] [CrossRef] [PubMed]
- Tong, Y.; Zhou, W.; Fung, V.; Christensen, M.A.; Qing, H.; Sun, X.; Song, W. Oxidative stress potentiates BACE1 gene expression and A? generation. J. Neural Transm. 2004, 112, 455–469. [Google Scholar] [CrossRef]
- Moreno-Jiménez, E.P.; Flor-García, M.; Terreros-Roncal, J.; Rábano, A.; Cafini, F.; Pallas-Bazarra, N.; Ávila, J.; Llorens-Martín, M. Adult hippocampal neurogenesis is abundant in neurologically healthy subjects and drops sharply in patients with Alzheimer’s disease. Nat. Med. 2019, 25, 554–560. [Google Scholar] [CrossRef]
- Hollands, C.; Tobin, M.K.; Hsu, M.; Musaraca, K.; Yu, T.-S.; Mishra, R.; Kernie, S.G.; Lazarov, O. Depletion of adult neurogenesis exacerbates cognitive deficits in Alzheimer’s disease by compromising hippocampal inhibition. Mol. Neurodegener. 2017, 12, 64. [Google Scholar] [CrossRef]
- Yan, P.; Liu, Z.; Song, M.; Wu, Z.; Xu, W.; Li, K.; Ji, Q.; Wang, S.; Liu, X.; Yan, K.; et al. Genome-wide R-loop Landscapes during Cell Differentiation and Reprogramming. Cell Rep. 2020, 32, 107870. [Google Scholar] [CrossRef]
- Rezaei-Lotfi, S.; Vujovic, F.; Simonian, M.; Hunter, N.; Farahani, R.M. Programmed genomic instability regulates neural transdifferentiation of human brain microvascular pericytes. Genome Biol. 2021, 22, 334. [Google Scholar] [CrossRef]
- A Watts, J.; Grunseich, C.; Rodriguez, Y.; Liu, Y.; Li, D.; Burdick, J.T.; Bruzel, A.; Crouch, R.J.; Mahley, R.W.; Wilson, S.H.; et al. A common transcriptional mechanism involving R-loop and RNA abasic site regulates an enhancer RNA of APOE. Nucleic Acids Res. 2022, 50, 12497–12514. [Google Scholar] [CrossRef]
- Crossley, M.P.; Bocek, M.; Cimprich, K.A. R-Loops as Cellular Regulators and Genomic Threats. Mol. Cell 2019, 73, 398–411. [Google Scholar] [CrossRef]
- de Murcia, G.; de Murcia, J.M. Poly(ADP-ribose) polymerase: A molecular nick-sensor. Trends Biochem. Sci. 1994, 19, 172–176. [Google Scholar] [CrossRef]
- Zhang, X.; He, X.; Li, Q.; Kong, X.; Ou, Z.; Zhang, L.; Gong, Z.; Long, D.; Li, J.; Zhang, M.; et al. PI3K/AKT/mTOR Signaling Mediates Valproic Acid-Induced Neuronal Differentiation of Neural Stem Cells through Epigenetic Modifications. Stem Cell Rep. 2017, 8, 1256–1269. [Google Scholar] [CrossRef]
- Chen, Y.L.; Monteith, N.; Law, P.-Y.; Loh, H.H. Dynamic association of p300 with the promoter of the G protein-coupled rat delta opioid receptor gene during NGF-induced neuronal differentiation. Biochem. Biophys. Res. Commun. 2010, 396, 294–298. [Google Scholar] [CrossRef]
- Chen, Y.L.; Law, P.-Y.; Loh, H.H. NGF/PI3K signaling-mediated epigenetic regulation of delta opioid receptor gene expression. Biochem. Biophys. Res. Commun. 2008, 368, 755–760. [Google Scholar] [CrossRef]
- Jacobs, S.; Lie, D.C.; DeCicco, K.L.; Shi, Y.; DeLuca, L.M.; Gage, F.H.; Evans, R.M. Retinoic acid is required early during adult neurogenesis in the dentate gyrus. Proc. Natl. Acad. Sci. USA 2006, 103, 3902–3907. [Google Scholar] [CrossRef]
- Janesick, A.; Wu, S.C.; Blumberg, B. Retinoic acid signaling and neuronal differentiation. Cell. Mol. Life Sci. 2015, 72, 1559–1576. [Google Scholar] [CrossRef]
- Katoh, M. Regulation of WNT signaling molecules by retinoic acid during neuronal differentiation in NT2 cells: Threshold model of WNT action (review). Int. J. Mol. Med. 2002, 10, 683–687. [Google Scholar] [CrossRef] [PubMed]
- McCaffery, P.J.; Adams, J.; Maden, M.; Rosa-Molinar, E. Too much of a good thing: Retinoic acid as an endogenous regulator of neural differentiation and exogenous teratogen. Eur. J. Neurosci. 2003, 18, 457–472. [Google Scholar] [CrossRef] [PubMed]
- Andrews, P.W. Retinoic acid induces neuronal differentiation of a cloned human embryonal carcinoma cell line in vitro. Dev. Biol. 1984, 103, 285–293. [Google Scholar] [CrossRef]
- López-Carballo, G.; Moreno, L.; Masiá, S.; Pérez, P.; Barettino, D. Activation of the Phosphatidylinositol 3-Kinase/Akt Signaling Pathway by Retinoic Acid Is Required for Neural Differentiation of SH-SY5Y Human Neuroblastoma Cells. J. Biol. Chem. 2002, 277, 25297–25304. [Google Scholar] [CrossRef] [PubMed]
- Dorigatti, A.O.; Riordan, R.; Yu, Z.; Ross, G.; Wang, R.; Reynolds-Lallement, N.; Magnusson, K.; Galvan, V.; Perez, V.I. Brain cellular senescence in mouse models of Alzheimer’s disease. GeroScience 2022, 44, 1157–1168. [Google Scholar] [CrossRef]
- Guerrero, A.; De Strooper, B.; Arancibia-Cárcamo, I.L. Cellular senescence at the crossroads of inflammation and Alzheimer’s disease. Trends Neurosci. 2021, 44, 714–727. [Google Scholar] [CrossRef]
- Liu, R.-M. Aging, Cellular Senescence, and Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 1989. [Google Scholar] [CrossRef]
- Masaldan, S.; Belaidi, A.A.; Ayton, S.; Bush, A.I. Cellular Senescence and Iron Dyshomeostasis in Alzheimer’s Disease. Pharmaceuticals 2019, 12, 93. [Google Scholar] [CrossRef]
- Saez-Atienzar, S.; Masliah, E. Cellular senescence and Alzheimer disease: The egg and the chicken scenario. Nat. Rev. Neurosci. 2020, 21, 433–444. [Google Scholar] [CrossRef]
- Jung, S.H.; Hwang, H.J.; Kang, D.; Park, H.A.; Lee, H.C.; Jeong, D.; Lee, K.; Park, H.J.; Ko, Y.-G.; Lee, J.-S. mTOR kinase leads to PTEN-loss-induced cellular senescence by phosphorylating p53. Oncogene 2018, 38, 1639–1650. [Google Scholar] [CrossRef]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic ras Provokes Premature Cell Senescence Associated with Accumulation of p53 and p16INK4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef]
- Minamino, T.; Yoshida, T.; Tateno, K.; Miyauchi, H.; Zou, Y.; Toko, H.; Komuro, I. Ras Induces Vascular Smooth Muscle Cell Senescence and Inflammation in Human Atherosclerosis. Circulation 2003, 108, 2264–2269. [Google Scholar] [CrossRef] [PubMed]
- Hardbower, D.M.; de Sablet, T.; Chaturvedi, R.; Wilson, K.T. Chronic inflammation and oxidative stress. Gut Microbes 2013, 4, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Kesika, P.; Suganthy, N.; Sivamaruthi, B.S.; Chaiyasut, C. Role of gut-brain axis, gut microbial composition, and probiotic intervention in Alzheimer’s disease. Life Sci. 2020, 264, 118627. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vujovic, F.; Shepherd, C.E.; Witting, P.K.; Hunter, N.; Farahani, R.M. Redox-Mediated Rewiring of Signalling Pathways: The Role of a Cellular Clock in Brain Health and Disease. Antioxidants 2023, 12, 1873. https://doi.org/10.3390/antiox12101873
Vujovic F, Shepherd CE, Witting PK, Hunter N, Farahani RM. Redox-Mediated Rewiring of Signalling Pathways: The Role of a Cellular Clock in Brain Health and Disease. Antioxidants. 2023; 12(10):1873. https://doi.org/10.3390/antiox12101873
Chicago/Turabian StyleVujovic, Filip, Claire E. Shepherd, Paul K. Witting, Neil Hunter, and Ramin M. Farahani. 2023. "Redox-Mediated Rewiring of Signalling Pathways: The Role of a Cellular Clock in Brain Health and Disease" Antioxidants 12, no. 10: 1873. https://doi.org/10.3390/antiox12101873
APA StyleVujovic, F., Shepherd, C. E., Witting, P. K., Hunter, N., & Farahani, R. M. (2023). Redox-Mediated Rewiring of Signalling Pathways: The Role of a Cellular Clock in Brain Health and Disease. Antioxidants, 12(10), 1873. https://doi.org/10.3390/antiox12101873