
Therapeutic Dosage of Antipsychotic Drug Aripiprazole Induces Persistent Mitochondrial Hyperpolarisation, Moderate Oxidative Stress in Liver Cells, and Haemolysis
 , , and
, , and 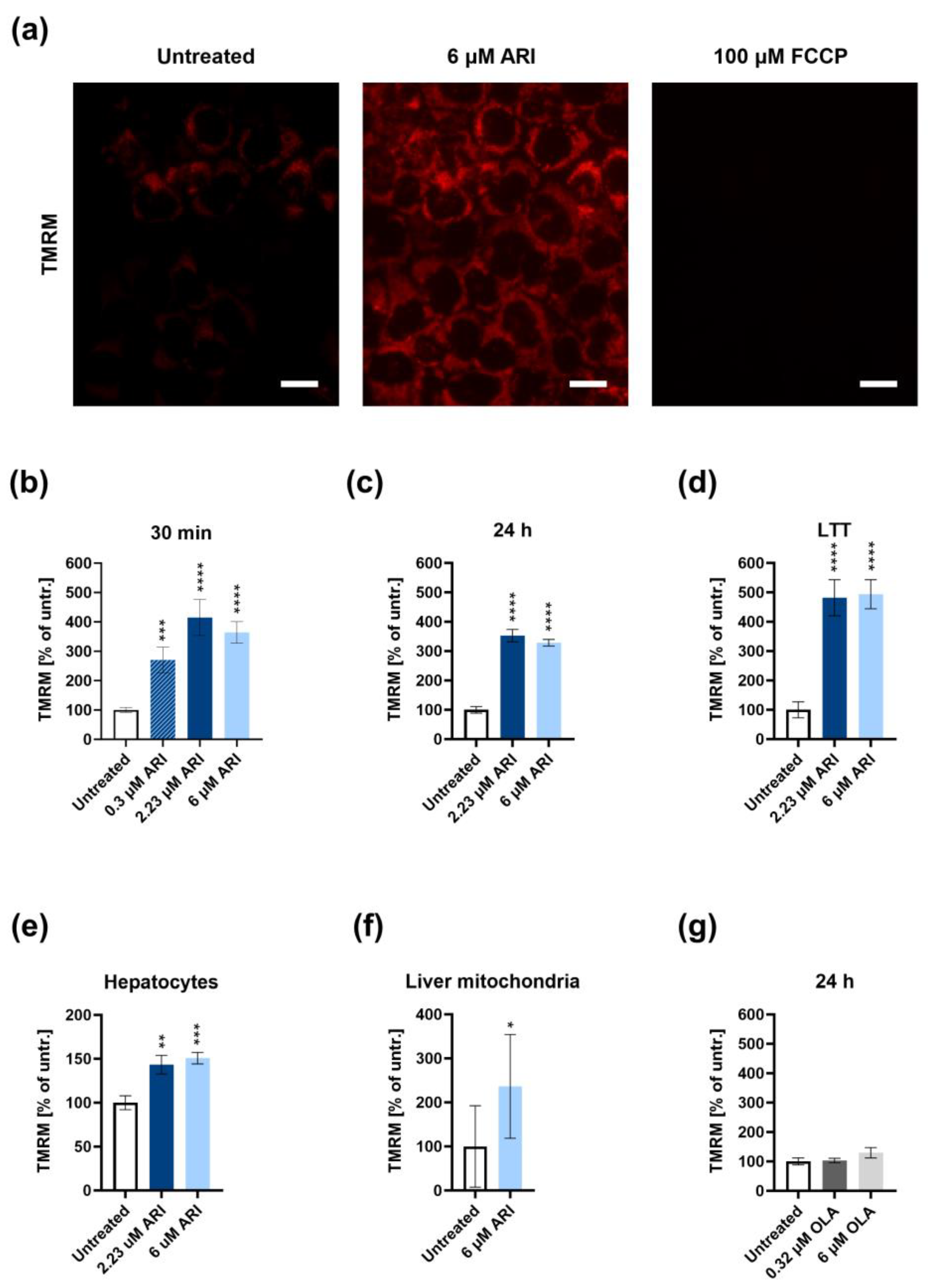{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Mitochondrial Isolation
2.3. Protein Content
2.4. Mitochondrial Membrane Potential Measurement in the Cells
2.5. Mitochondrial Membrane Potential Measurement in Isolated Mitochondria
2.6. Mitochondrial Metabolic Function and Mitochondrial Complex Activity
2.7. ATP Content
2.8. Oxidative Stress (Superoxide Measurement)
2.9. H2O2 Production
2.10. Antioxidant Enzymes and Glutathione Measurements
2.11. Haemolysis Assay
2.12. Statistical Analysis
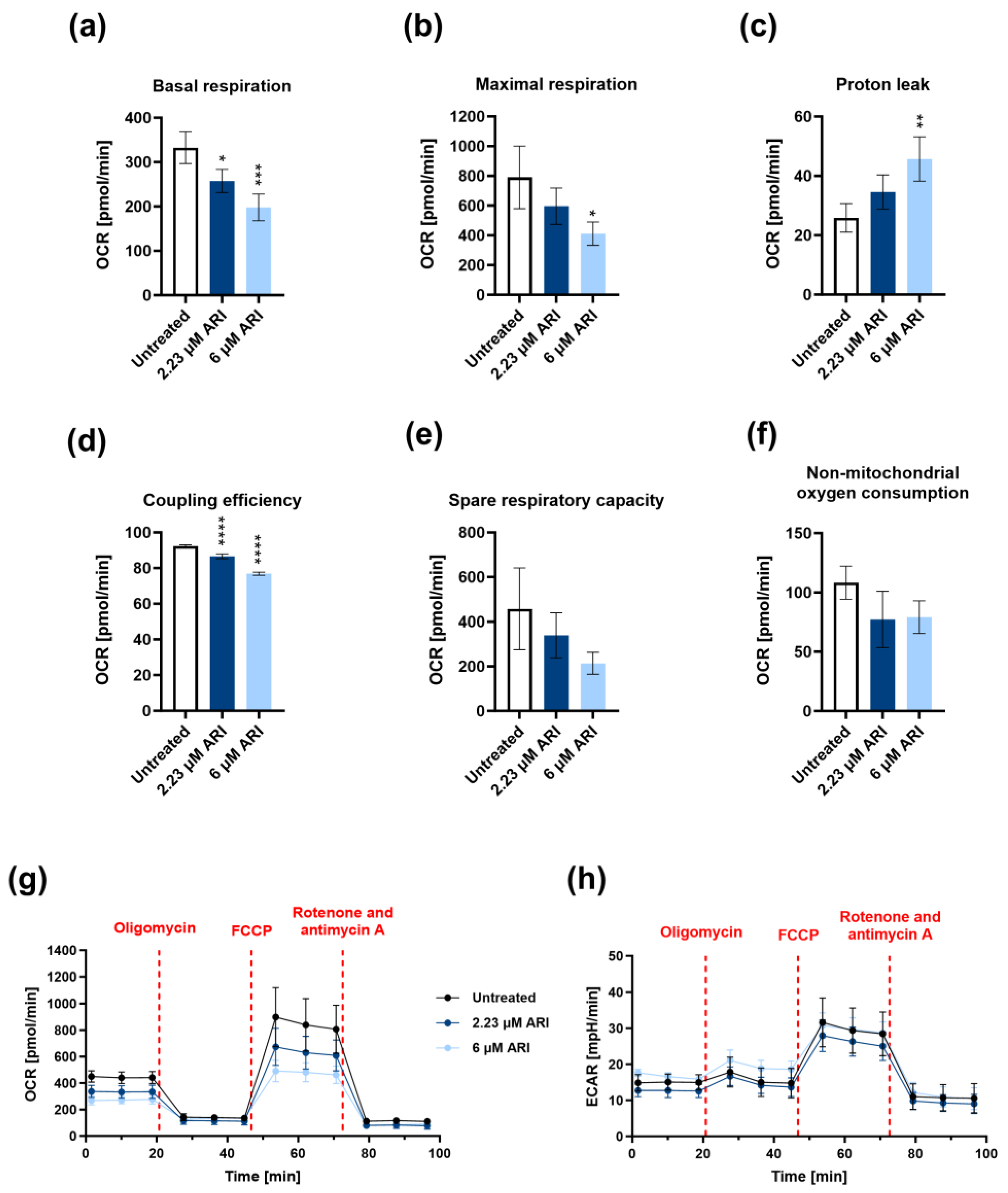
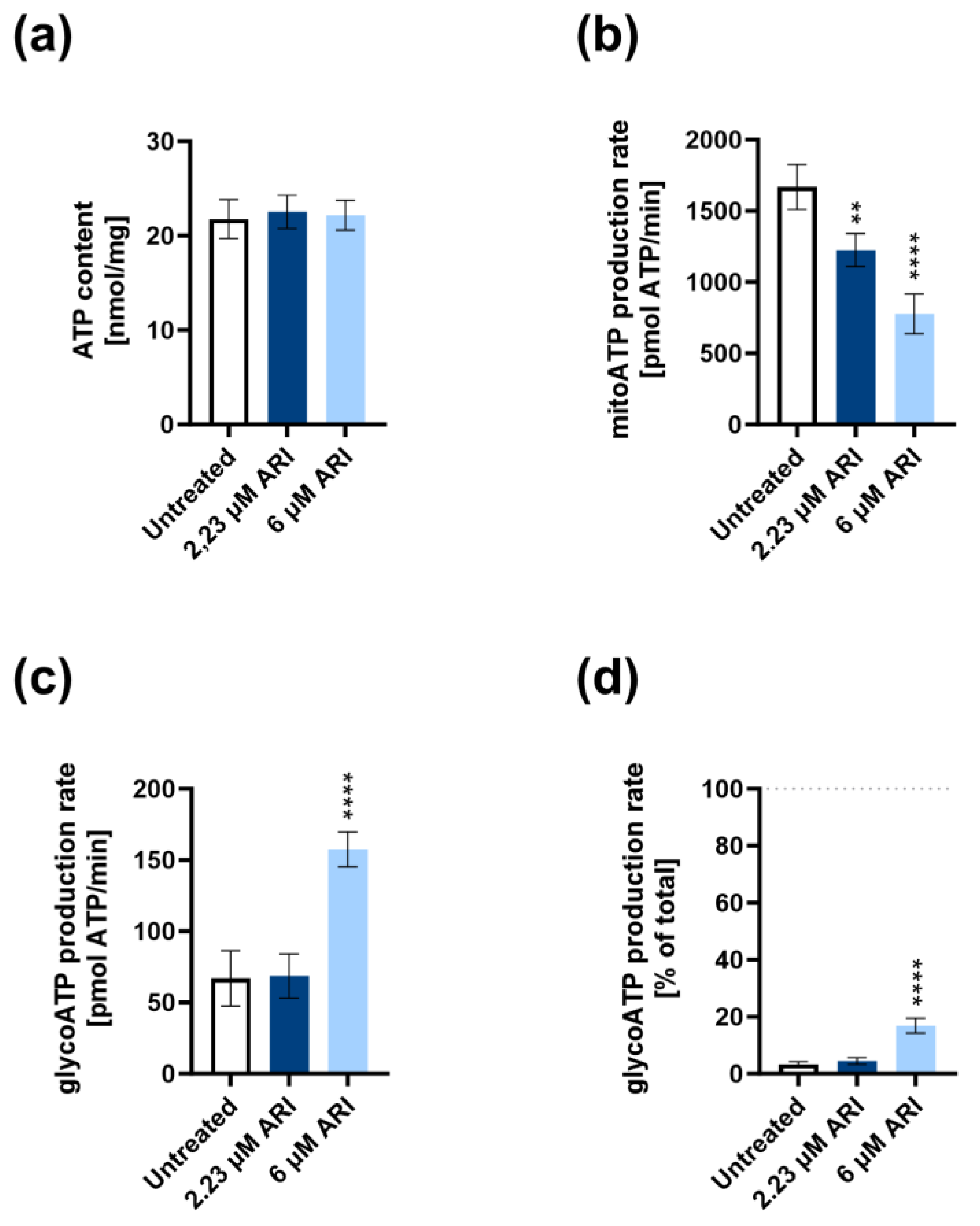
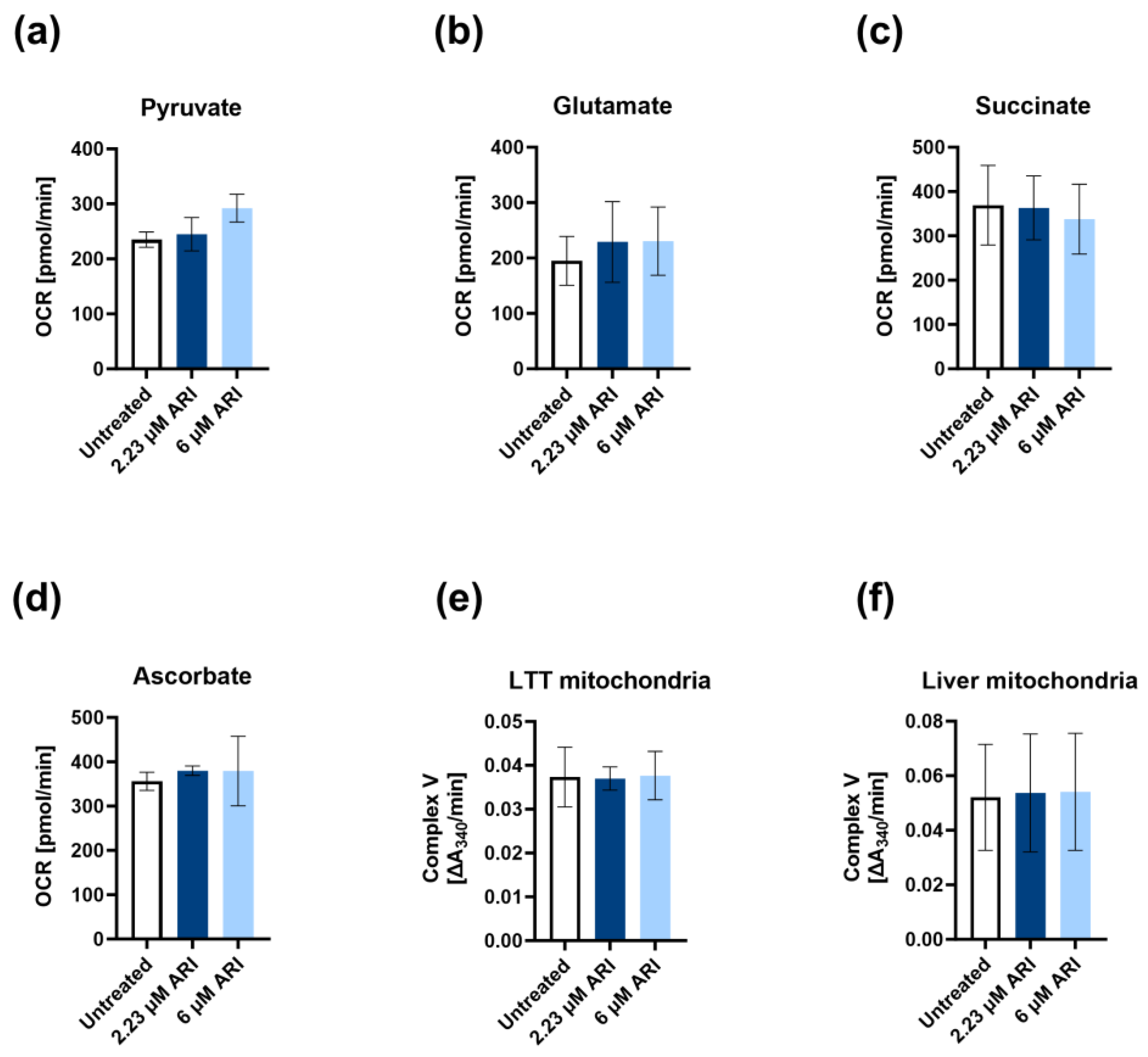
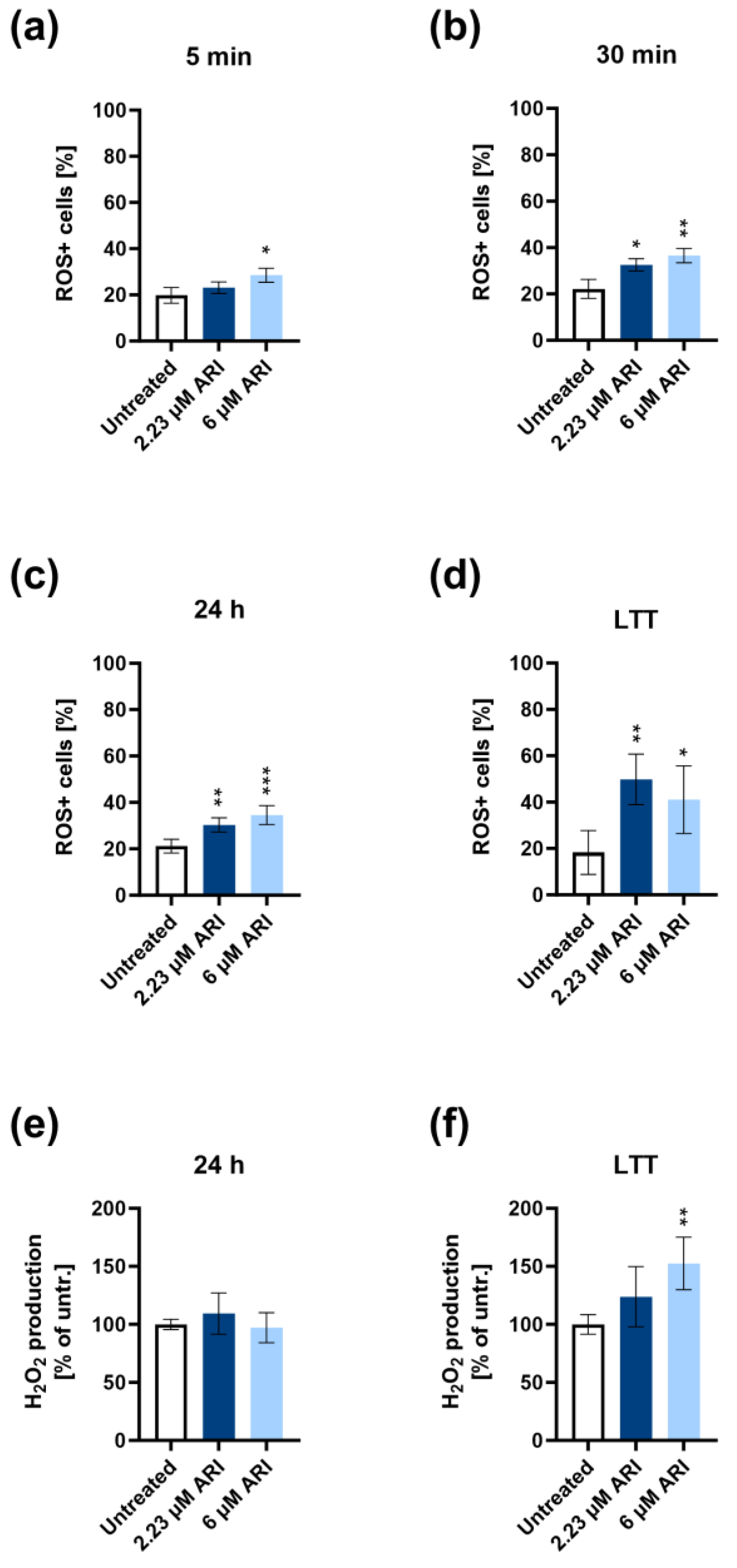
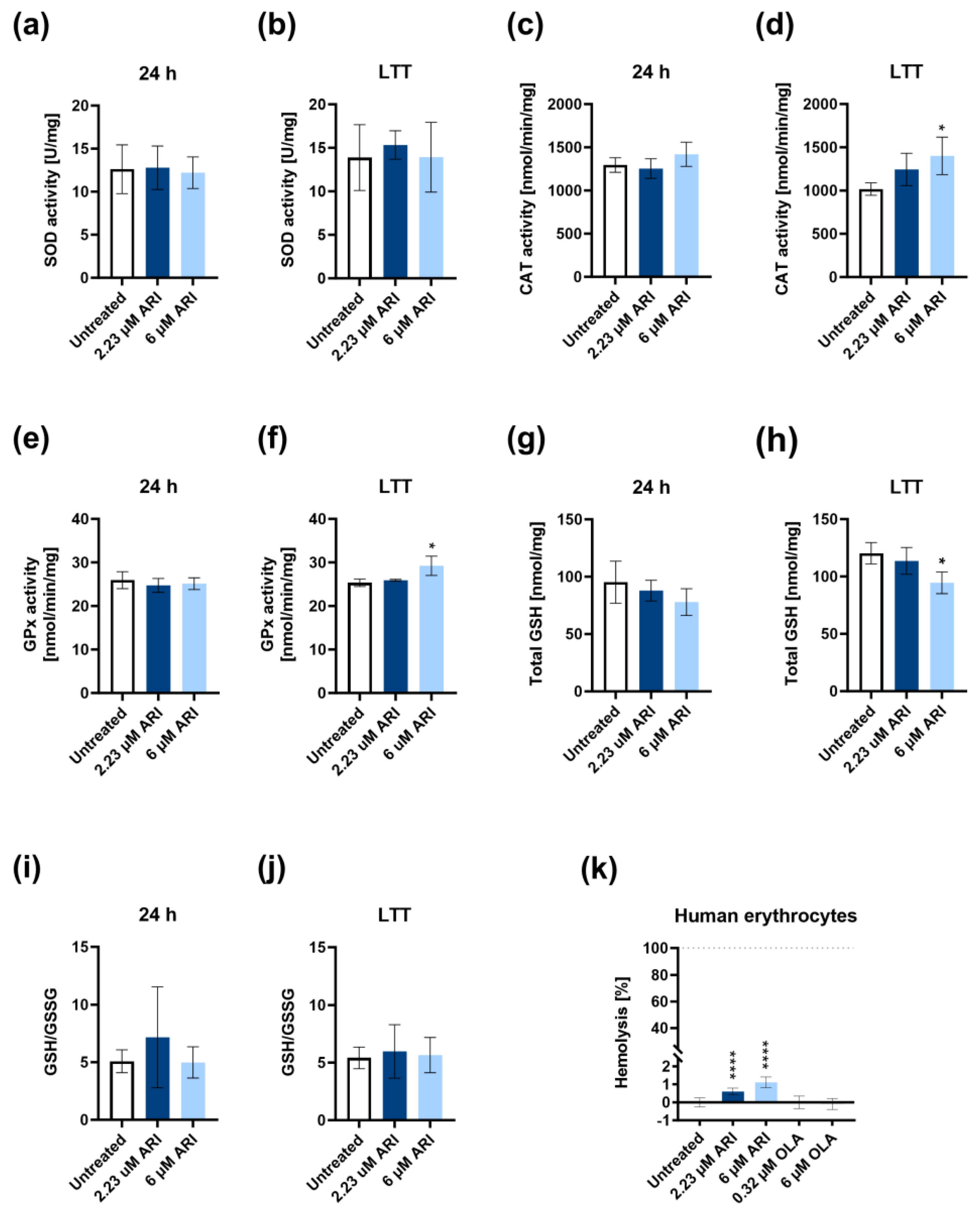
3. Results
Study Limitations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Christensen, A.P.; Boegevig, S.; Christensen, M.B.; Petersen, K.M.; Dalhoff, K.P.; Petersen, T.S. Overdoses with Aripiprazole: Signs, Symptoms and Outcome in 239 Exposures Reported to the Danish Poison Information Centre. Basic. Clin. Pharmacol. Toxicol. 2018, 122, 293–298. [Google Scholar] [CrossRef]
- FDA Warns about New Impulse-Control Problems Associated with Mental Health Drug Aripiprazole (Abilify, Abilify Maintena, Aristada). Available online: https://www.fda.gov/drugs/drug-safety-and-availability/fda-drug-safety-communication-fda-warns-about-new-impulse-control-problems-associated-mental-health (accessed on 30 May 2023).
- Reddymasu, S.; Bahta, E.; Levine, S.; Manas, K.; Slay, L.E. Elevated lipase and diabetic ketoacidosis associated with aripiprazole. JOP 2006, 7, 303–305. [Google Scholar] [PubMed]
- Zhu, X.; Hu, J.; Deng, S.; Qiu, C.; Shang, D.; Wen, Y. Rhabdomyolysis and elevated liver enzymes after rapid correction of hyponatremia due to pneumonia and concurrent use of aripiprazole: A case report. Aust. N. Z. J. Psychiatry 2018, 52, 206. [Google Scholar] [CrossRef] [PubMed]
- Seven, H.; Ayhan, M.G.; Kurkcu, A.; Ozbek, S.; Eren, I. Aripiprazole-induced Asymptomatic Hypertension: A Case Report. Psychopharmacol. Bull. 2017, 47, 53–56. [Google Scholar]
- Kornischka, J.; Cordes, J. Acute Drug-Induced Hepatitis during Aripiprazole Monotherapy: A Case Report. J. Pharmacovigil. 2016, 4, 201. [Google Scholar] [CrossRef]
- Castanheira, L.; Fernandes, E.; Levy, P.; Coentre, R. Aripiprazole-induced Hepatitis: A Case Report. Clin. Psychopharmacol. Neurosci. 2019, 17, 551–555. [Google Scholar] [CrossRef]
- Gonzalez-Munoz, M.; Monserrat Villatoro, J.; Marin-Serrano, E.; Stewart, S.; Bardon Rivera, B.; Marin, J.; Martinez de Soto, L.; Seco Meseguer, E.; Ramirez, E. A case report of a drug-induced liver injury (DILI) caused by multiple antidepressants with causality established by the updated Roussel Uclaf causality assessment method (RUCAM) and in vitro testing. Clin. Case Rep. 2020, 8, 3105–3109. [Google Scholar] [CrossRef]
- Pirc Marolt, T.; Kramar, B.; Bulc Rozman, K.; Suput, D.; Milisav, I. Aripiprazole reduces liver cell division. PLoS ONE 2020, 15, e0240754. [Google Scholar] [CrossRef]
- Baek, K.S.; Ahn, S.; Lee, J.; Kim, J.H.; Kim, H.G.; Kim, E.; Kim, J.H.; Sung, N.Y.; Yang, S.; Kim, M.S.; et al. Immunotoxicological Effects of Aripiprazole: In Vivo and In Vitro Studies. Korean J. Physiol. Pharmacol. 2015, 19, 365–372. [Google Scholar] [CrossRef]
- Aripiprazole. Available online: https://go.drugbank.com/drugs/DB01238 (accessed on 30 May 2023).
- Elmorsy, E.; Smith, P.A. Bioenergetic disruption of human micro-vascular endothelial cells by antipsychotics. Biochem. Biophys. Res. Commun. 2015, 460, 857–862. [Google Scholar] [CrossRef]
- Del Campo, A.; Bustos, C.; Mascayano, C.; Acuna-Castillo, C.; Troncoso, R.; Rojo, L.E. Metabolic Syndrome and Antipsychotics: The Role of Mitochondrial Fission/Fusion Imbalance. Front. Endocrinol. 2018, 9, 144. [Google Scholar] [CrossRef]
- Maurer, I.; Moller, H.J. Inhibition of complex I by neuroleptics in normal human brain cortex parallels the extrapyramidal toxicity of neuroleptics. Mol. Cell Biochem. 1997, 174, 255–259. [Google Scholar] [CrossRef]
- Prince, J.A.; Yassin, M.S.; Oreland, L. Neuroleptic-induced mitochondrial enzyme alterations in the rat brain. J. Pharmacol. Exp. Ther. 1997, 280, 261–267. [Google Scholar]
- Hroudova, J.; Fisar, Z. Connectivity between mitochondrial functions and psychiatric disorders. Psychiatry Clin. Neurosci. 2011, 65, 130–141. [Google Scholar] [CrossRef]
- Modica-Napolitano, J.S.; Lagace, C.J.; Brennan, W.A.; Aprille, J.R. Differential effects of typical and atypical neuroleptics on mitochondrial function in vitro. Arch. Pharm. Res. 2003, 26, 951–959. [Google Scholar] [CrossRef]
- Scaini, G.; Quevedo, J.; Velligan, D.; Roberts, D.L.; Raventos, H.; Walss-Bass, C. Second generation antipsychotic-induced mitochondrial alterations: Implications for increased risk of metabolic syndrome in patients with schizophrenia. Eur. Neuropsychopharmacol. 2018, 28, 369–380. [Google Scholar] [CrossRef]
- Cikankova, T.; Fisar, Z.; Bakhouche, Y.; Luptak, M.; Hroudova, J. In vitro effects of antipsychotics on mitochondrial respiration. Naunyn Schmiedebergs Arch. Pharmacol. 2019, 392, 1209–1223. [Google Scholar] [CrossRef]
- Takami, G.; Ota, M.; Nakashima, A.; Kaneko, Y.S.; Mori, K.; Nagatsu, T.; Ota, A. Effects of atypical antipsychotics and haloperidol on PC12 cells: Only aripiprazole phosphorylates AMP-activated protein kinase. J. Neural. Transm. 2010, 117, 1139–1153. [Google Scholar] [CrossRef]
- Racki, V.; Marcelic, M.; Stimac, I.; Petric, D.; Kucic, N. Effects of Haloperidol, Risperidone, and Aripiprazole on the Immunometabolic Properties of BV-2 Microglial Cells. Int. J. Mol. Sci. 2021, 22, 4399. [Google Scholar] [CrossRef]
- Ota, A.; Nakashima, A.; Kaneko, Y.S.; Mori, K.; Nagasaki, H.; Takayanagi, T.; Itoh, M.; Kondo, K.; Nagatsu, T.; Ota, M. Effects of aripiprazole and clozapine on the treatment of glycolytic carbon in PC12 cells. J. Neural. Transm. 2012, 119, 1327–1342. [Google Scholar] [CrossRef]
- Chen, J.; Chernatynskaya, A.V.; Li, J.W.; Kimbrell, M.R.; Cassidy, R.J.; Perry, D.J.; Muir, A.B.; Atkinson, M.A.; Brusko, T.M.; Mathews, C.E. T cells display mitochondria hyperpolarization in human type 1 diabetes. Sci. Rep. 2017, 7, 10835. [Google Scholar] [CrossRef]
- Johnson, L.V.; Walsh, M.L.; Bockus, B.J.; Chen, L.B. Monitoring of relative mitochondrial membrane potential in living cells by fluorescence microscopy. J. Cell Biol. 1981, 88, 526–535. [Google Scholar] [CrossRef]
- Banki, K.; Hutter, E.; Gonchoroff, N.J.; Perl, A. Elevation of mitochondrial transmembrane potential and reactive oxygen intermediate levels are early events and occur independently from activation of caspases in Fas signaling. J. Immunol. 1999, 162, 1466–1479. [Google Scholar] [CrossRef]
- Perl, A.; Gergely, P., Jr.; Nagy, G.; Koncz, A.; Banki, K. Mitochondrial hyperpolarization: A checkpoint of T-cell life, death and autoimmunity. Trends Immunol. 2004, 25, 360–367. [Google Scholar] [CrossRef]
- Sanchez-Alcazar, J.A.; Ault, J.G.; Khodjakov, A.; Schneider, E. Increased mitochondrial cytochrome c levels and mitochondrial hyperpolarization precede camptothecin-induced apoptosis in Jurkat cells. Cell Death Differ. 2000, 7, 1090–1100. [Google Scholar] [CrossRef]
- Gao, M.; Yi, J.; Zhu, J.; Minikes, A.M.; Monian, P.; Thompson, C.B.; Jiang, X. Role of Mitochondria in Ferroptosis. Mol. Cell 2019, 73, 354–363.e353. [Google Scholar] [CrossRef]
- Gergely, P., Jr.; Grossman, C.; Niland, B.; Puskas, F.; Neupane, H.; Allam, F.; Banki, K.; Phillips, P.E.; Perl, A. Mitochondrial hyperpolarization and ATP depletion in patients with systemic lupus erythematosus. Arthritis Rheum. 2002, 46, 175–190. [Google Scholar] [CrossRef]
- Perl, A.; Gergely, P., Jr.; Banki, K. Mitochondrial dysfunction in T cells of patients with systemic lupus erythematosus. Int. Rev. Immunol. 2004, 23, 293–313. [Google Scholar] [CrossRef]
- Chen, L.B. Mitochondrial membrane potential in living cells. Annu. Rev. Cell Biol. 1988, 4, 155–181. [Google Scholar] [CrossRef]
- Houston, M.A.; Augenlicht, L.H.; Heerdt, B.G. Stable differences in intrinsic mitochondrial membrane potential of tumor cell subpopulations reflect phenotypic heterogeneity. Int. J. Cell Biol. 2011, 2011, 978583. [Google Scholar] [CrossRef]
- Heerdt, B.G.; Houston, M.A.; Augenlicht, L.H. Growth properties of colonic tumor cells are a function of the intrinsic mitochondrial membrane potential. Cancer Res. 2006, 66, 1591–1596. [Google Scholar] [CrossRef] [PubMed]
- Korshunov, S.S.; Skulachev, V.P.; Starkov, A.A. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett. 1997, 416, 15–18. [Google Scholar] [CrossRef] [PubMed]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Lebiedzinska, M.; Karkucinska-Wieckowska, A.; Wojtala, A.; Suski, J.M.; Szabadkai, G.; Wilczynski, G.; Wlodarczyk, J.; Diogo, C.V.; Oliveira, P.J.; Tauber, J.; et al. Disrupted ATP synthase activity and mitochondrial hyperpolarisation-dependent oxidative stress is associated with p66Shc phosphorylation in fibroblasts of NARP patients. Int. J. Biochem. Cell Biol. 2013, 45, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Kuloglu, M.; Ustundag, B.; Atmaca, M.; Canatan, H.; Tezcan, A.E.; Cinkilinc, N. Lipid peroxidation and antioxidant enzyme levels in patients with schizophrenia and bipolar disorder. Cell Biochem. Funct. 2002, 20, 171–175. [Google Scholar] [CrossRef]
- Dietrich-Muszalska, A.; Kolodziejczyk-Czepas, J.; Nowak, P. Comparative Study of the Effects of Atypical Antipsychotic Drugs on Plasma and Urine Biomarkers of Oxidative Stress in Schizophrenic Patients. Neuropsychiatr. Dis. Treat. 2021, 17, 555–565. [Google Scholar] [CrossRef]
- Park, S.W.; Lee, C.H.; Lee, J.G.; Kim, L.W.; Shin, B.S.; Lee, B.J.; Kim, Y.H. Protective effects of atypical antipsychotic drugs against MPP(+)-induced oxidative stress in PC12 cells. Neurosci. Res. 2011, 69, 283–290. [Google Scholar] [CrossRef]
- Kramar, B.; Pirc Marolt, T.; Monsalve, M.; Suput, D.; Milisav, I. Antipsychotic Drug Aripiprazole Protects Liver Cells from Oxidative Stress. Int. J. Mol. Sci. 2022, 23, 8292. [Google Scholar] [CrossRef]
- Wei, Z.; Bai, O.; Richardson, J.S.; Mousseau, D.D.; Li, X.M. Olanzapine protects PC12 cells from oxidative stress induced by hydrogen peroxide. J. Neurosci. Res. 2003, 73, 364–368. [Google Scholar] [CrossRef]
- Bauman, J.N.; Frederick, K.S.; Sawant, A.; Walsky, R.L.; Cox, L.M.; Obach, R.S.; Kalgutkar, A.S. Comparison of the bioactivation potential of the antidepressant and hepatotoxin nefazodone with aripiprazole, a structural analog and marketed drug. Drug Metab. Dispos. 2008, 36, 1016–1029. [Google Scholar] [CrossRef]
- Miller, I.P.; Pavlovic, I.; Poljsak, B.; Suput, D.; Milisav, I. Beneficial Role of ROS in Cell Survival: Moderate Increases in H2O2 Production Induced by Hepatocyte Isolation Mediate Stress Adaptation and Enhanced Survival. Antioxidants 2019, 8, 434. [Google Scholar] [CrossRef] [PubMed]
- Lund, J.; Ouwens, D.M.; Wettergreen, M.; Bakke, S.S.; Thoresen, G.H.; Aas, V. Increased Glycolysis and Higher Lactate Production in Hyperglycemic Myotubes. Cells 2019, 8, 1101. [Google Scholar] [CrossRef]
- Romero, N.; Rogers, G.; Neilson, A.; Dranka, B.P. Quantifying Cellular ATP Production Rate Using Agilent Seahorse XF Technology. Available online: https://www.agilent.com/cs/library/whitepaper/public/whitepaper-quantify-atp-production-rate-cell-analysis-5991-9303en-agilent.pdf (accessed on 6 June 2023).
- Evans, B.C.; Nelson, C.E.; Yu, S.S.; Beavers, K.R.; Kim, A.J.; Li, H.; Nelson, H.M.; Giorgio, T.D.; Duvall, C.L. Ex vivo red blood cell hemolysis assay for the evaluation of pH-responsive endosomolytic agents for cytosolic delivery of biomacromolecular drugs. J. Vis. Exp. 2013, 73, e50166. [Google Scholar] [CrossRef]
- Hiemke, C.; Bergemann, N.; Clement, H.W.; Conca, A.; Deckert, J.; Domschke, K.; Eckermann, G.; Egberts, K.; Gerlach, M.; Greiner, C.; et al. Consensus Guidelines for Therapeutic Drug Monitoring in Neuropsychopharmacology: Update 2017. Pharmacopsychiatry 2018, 51, e1. [Google Scholar] [CrossRef] [PubMed]
- Riccardi, K.; Lin, J.; Li, Z.; Niosi, M.; Ryu, S.; Hua, W.; Atkinson, K.; Kosa, R.E.; Litchfield, J.; Di, L. Novel Method to Predict In Vivo Liver-to-Plasma K(puu) for OATP Substrates Using Suspension Hepatocytes. Drug Metab. Dispos. 2017, 45, 576–580. [Google Scholar] [CrossRef]
- Beresford, T.P.; Clapp, L.; Martin, B.; Wiberg, J.L.; Alfers, J.; Beresford, H.F. Aripiprazole in schizophrenia with cocaine dependence: A pilot study. J. Clin. Psychopharmacol. 2005, 25, 363–366. [Google Scholar] [CrossRef]
- Scarino, M.L.; Howell, K.E. The Fao Cell. A tissue culture model for lipoprotein synthesis and secretion. Exp. Cell Res. 1987, 170, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Bayly, A.C.; French, N.J.; Dive, C.; Roberts, R.A. Nongenotoxic hepatocarcinogenesis in vitro: The FaO hepatoma line responds to peroxisome proliferators and retains the ability to undergo apoptosis. J. Cell Sci. 1993, 104 Pt 2, 307–315. [Google Scholar] [CrossRef]
- Monsalve, M.; Prieto, I.; de Bem, A.F.; Olmos, Y. Methodological Approach for the Evaluation of FOXO as a Positive Regulator of Antioxidant Genes. Methods Mol. Biol. 2019, 1890, 61–76. [Google Scholar] [CrossRef]
- Pillinger, T.; McCutcheon, R.A.; Vano, L.; Mizuno, Y.; Arumuham, A.; Hindley, G.; Beck, K.; Natesan, S.; Efthimiou, O.; Cipriani, A.; et al. Comparative effects of 18 antipsychotics on metabolic function in patients with schizophrenia, predictors of metabolic dysregulation, and association with psychopathology: A systematic review and network meta-analysis. Lancet Psychiatry 2020, 7, 64–77. [Google Scholar] [CrossRef]
- Burger, K.N.J.; Beulens, J.W.J.; van der Schouw, Y.T.; Sluijs, I.; Spijkerman, A.M.W.; Sluik, D.; Boeing, H.; Kaaks, R.; Teucher, B.; Dethlefsen, C.; et al. Dietary Fiber, Carbohydrate Quality and Quantity, and Mortality Risk of Individuals with Diabetes Mellitus. PLoS ONE 2012, 7, e43127. [Google Scholar] [CrossRef] [PubMed]
- Nagy, G.; Koncz, A.; Perl, A. T cell activation-induced mitochondrial hyperpolarization is mediated by Ca2+- and redox-dependent production of nitric oxide. J. Immunol. 2003, 171, 5188–5197. [Google Scholar] [CrossRef] [PubMed]
- Badran, A.; Tul-Wahab, A.; Zafar, H.; Mohammad, N.; Imad, R.; Ashfaq Khan, M.; Baydoun, E.; Choudhary, M.I. Antipsychotics drug aripiprazole as a lead against breast cancer cell line (MCF-7) in vitro. PLoS ONE 2020, 15, e0235676. [Google Scholar] [CrossRef] [PubMed]
- Locasale, J.W.; Cantley, L.C. Metabolic flux and the regulation of mammalian cell growth. Cell Metab. 2011, 14, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J. Energy metabolism of cancer: Glycolysis versus oxidative phosphorylation (Review). Oncol. Lett. 2012, 4, 1151–1157. [Google Scholar] [CrossRef]
- Esteras, N.; Rohrer, J.D.; Hardy, J.; Wray, S.; Abramov, A.Y. Mitochondrial hyperpolarization in iPSC-derived neurons from patients of FTDP-17 with 10+16 MAPT mutation leads to oxidative stress and neurodegeneration. Redox Biol. 2017, 12, 410–422. [Google Scholar] [CrossRef]
- Divakaruni, A.S.; Rogers, G.W.; Murphy, A.N. Measuring Mitochondrial Function in Permeabilized Cells Using the Seahorse XF Analyzer or a Clark-Type Oxygen Electrode. Curr. Protoc. Toxicol. 2014, 60, 25.2.1–25.2.16. [Google Scholar] [CrossRef]
- Affourtit, C.; Brand, M.D. Measuring mitochondrial bioenergetics in INS-1E insulinoma cells. Methods Enzymol. 2009, 457, 405–424. [Google Scholar] [CrossRef]
- Zhao, R.Z.; Jiang, S.; Zhang, L.; Yu, Z.B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef]
- Using PMP to Measure Substrate Specific ETC/OxPhos Activity in Permeabilized Cells. Available online: https://www.agilent.com/cs/library/technicaloverviews/public/xf%20pmp%20substrate%20oxidation.pdf (accessed on 30 May 2023).
- Garcia, J.J.; Ogilvie, I.; Robinson, B.H.; Capaldi, R.A. Structure, functioning, and assembly of the ATP synthase in cells from patients with the T8993G mitochondrial DNA mutation. Comparison with the enzyme in Rho(0) cells completely lacking mtdna. J. Biol. Chem. 2000, 275, 11075–11081. [Google Scholar] [CrossRef]
- Kim, E.H.; Koh, E.H.; Park, J.Y.; Lee, K.U. Adenine nucleotide translocator as a regulator of mitochondrial function: Implication in the pathogenesis of metabolic syndrome. Korean Diabetes J. 2010, 34, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, D.G. Mitochondrial membrane potential and aging. Aging Cell 2004, 3, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Ursini, F.; Maiorino, M.; Gregolin, C. The selenoenzyme phospholipid hydroperoxide glutathione peroxidase. Biochim. Et Biophys. Acta 1985, 839, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Franco, R.; Schoneveld, O.J.; Pappa, A.; Panayiotidis, M.I. The central role of glutathione in the pathophysiology of human diseases. Arch. Physiol. Biochem. 2007, 113, 234–258. [Google Scholar] [CrossRef]
- Gong, A.; Guillory, C.; Creel, L.; Livingtson, J.E.; McKee-Garrett, T.M.; Fortunov, R. A Multicenter Initiative for Critical Congenital Heart Disease Newborn Screening in Texas Neonatal Intensive Care Units. Am. J. Perinatol. 2017, 34, 839–844. [Google Scholar] [CrossRef]
- Dausset, J.; Contu, L. Drug-induced hemolysis. Annu. Rev. Med. 1967, 18, 55–70. [Google Scholar] [CrossRef]
- Leppien, E.; Bystrak, T.; Doughty, B. Antipsychotic agents. Side Eff. Drugs Annu. 2019, 41, 51–63. [Google Scholar] [CrossRef]
- Ozaslan, M.S.; Balci, N.; Demir, Y.; Gurbuz, M.; Kufrevioglu, O.I. Inhibition effects of some antidepressant drugs on pentose phosphate pathway enzymes. Environ. Toxicol. Pharmacol. 2019, 72, 103244. [Google Scholar] [CrossRef]
- Benito, A.; Polat, I.H.; Noe, V.; Ciudad, C.J.; Marin, S.; Cascante, M. Glucose-6-phosphate dehydrogenase and transketolase modulate breast cancer cell metabolic reprogramming and correlate with poor patient outcome. Oncotarget 2017, 8, 106693–106706. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pirc Marolt, T.; Kramar, B.; Vovk, A.; Podgornik, H.; Šuput, D.; Milisav, I. Therapeutic Dosage of Antipsychotic Drug Aripiprazole Induces Persistent Mitochondrial Hyperpolarisation, Moderate Oxidative Stress in Liver Cells, and Haemolysis. Antioxidants 2023, 12, 1930. https://doi.org/10.3390/antiox12111930
Pirc Marolt T, Kramar B, Vovk A, Podgornik H, Šuput D, Milisav I. Therapeutic Dosage of Antipsychotic Drug Aripiprazole Induces Persistent Mitochondrial Hyperpolarisation, Moderate Oxidative Stress in Liver Cells, and Haemolysis. Antioxidants. 2023; 12(11):1930. https://doi.org/10.3390/antiox12111930
Chicago/Turabian StylePirc Marolt, Tinkara, Barbara Kramar, Andrej Vovk, Helena Podgornik, Dušan Šuput, and Irina Milisav. 2023. "Therapeutic Dosage of Antipsychotic Drug Aripiprazole Induces Persistent Mitochondrial Hyperpolarisation, Moderate Oxidative Stress in Liver Cells, and Haemolysis" Antioxidants 12, no. 11: 1930. https://doi.org/10.3390/antiox12111930
APA StylePirc Marolt, T., Kramar, B., Vovk, A., Podgornik, H., Šuput, D., & Milisav, I. (2023). Therapeutic Dosage of Antipsychotic Drug Aripiprazole Induces Persistent Mitochondrial Hyperpolarisation, Moderate Oxidative Stress in Liver Cells, and Haemolysis. Antioxidants, 12(11), 1930. https://doi.org/10.3390/antiox12111930