

On the Chemical and Biological Characteristics of Multifunctional Compounds for the Treatment of Parkinson’s Disease




Abstract
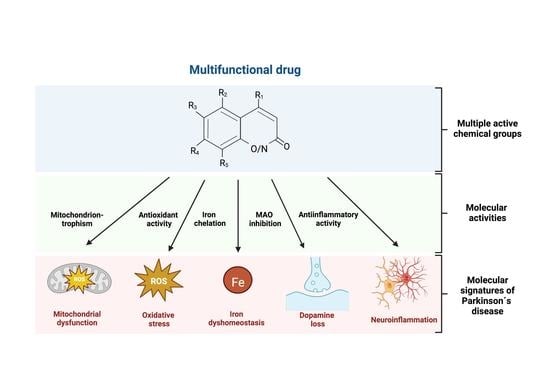
:
1. Introduction
2. Mitochondria-Associated Events in PD Neurodegeneration
2.1. Mitochondrial Dysfunction and ROS in PD
2.2. Mitochondrial Dysfunction and Iron Homeostasis in PD
2.3. Mitochondrial Dysfunction and Neuroinflammation in PD
2.4. Neuroinflammation and Iron Homeostasis in PD
3. Current Therapeutic Approaches to PD
3.1. Clinical Trials Targeting Oxidative Stress
3.2. Clinical Trials Using Iron Chelation Therapy
3.3. Clinical Trials Targeting a-Synuclein Aggregation
3.4. Clinical Trials Targeting Inflammation
4. Multifunctional Drugs for the Treatment of PD
4.1. Derivatives of Phenols
4.2. Derivatives of Polyphenols
4.3. Derivatives of Coumarins
4.4. Derivatives of Quinolines and Quinolones
4.5. Derivatives of Piperazine
4.6. Derivatives of Pyrazine and Pyridones
4.7. Derivatives of Terpenoids
4.8. Others Molecules
5. Drug-Likeness Prediction of the Multifunctional Compounds
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Fieblinger, T. Striatal Control of Movement: A Role for New Neuronal (Sub-) Populations? Front. Hum. Neurosci. 2021, 15, 697284. [Google Scholar] [CrossRef] [PubMed]
- Schneider, S.A.; Obeso, J.A. Clinical and pathological features of Parkinson’s disease. Curr. Top. Behav. Neurosci. 2015, 22, 205–220. [Google Scholar] [CrossRef] [PubMed]
- Collaborators, G.N. Global, regional, and national burden of neurological disorders, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 459–480. [Google Scholar] [CrossRef] [Green Version]
- Dorsey, E.R.; Sherer, T.; Okun, M.S.; Bloem, B.R. The Emerging Evidence of the Parkinson Pandemic. J. Park. Dis. 2018, 8, S3–S8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Hamilton, J.L.; Kopil, C.; Beck, J.C.; Tanner, C.M.; Albin, R.L.; Ray Dorsey, E.; Dahodwala, N.; Cintina, I.; Hogan, P.; et al. Current and projected future economic burden of Parkinson’s disease in the U.S. NPJ Park. Dis. 2020, 6, 15. [Google Scholar] [CrossRef]
- Klivenyi, P.; Vecsei, L. Novel therapeutic strategies in Parkinson’s disease. Eur. J. Clin. Pharmacol. 2010, 66, 119–125. [Google Scholar] [CrossRef] [Green Version]
- Schapira, A.H.; Emre, M.; Jenner, P.; Poewe, W. Levodopa in the treatment of Parkinson’s disease. Eur. J. Neurol. 2009, 16, 982–989. [Google Scholar] [CrossRef]
- Yanagisawa, N. [A prospect of treatment for Parkinson’s disease in the 21st century]. Nihon Rinsho 2000, 58, 1968–1974. [Google Scholar]
- Rane, P.; Sarmah, D.; Bhute, S.; Kaur, H.; Goswami, A.; Kalia, K.; Borah, A.; Dave, K.R.; Sharma, N.; Bhattacharya, P. Novel Targets for Parkinson’s Disease: Addressing Different Therapeutic Paradigms and Conundrums. ACS Chem. Neurosci. 2019, 10, 44–57. [Google Scholar] [CrossRef]
- Savelieff, M.G.; Nam, G.; Kang, J.; Lee, H.J.; Lee, M.; Lim, M.H. Development of Multifunctional Molecules as Potential Therapeutic Candidates for Alzheimer’s Disease, Parkinson’s Disease, and Amyotrophic Lateral Sclerosis in the Last Decade. Chem. Rev. 2019, 119, 1221–1322. [Google Scholar] [CrossRef]
- Cheong, S.L.; Federico, S.; Spalluto, G.; Klotz, K.N.; Pastorin, G. The current status of pharmacotherapy for the treatment of Parkinson’s disease: Transition from single-target to multitarget therapy. Drug Discov. Today 2019, 24, 1769–1783. [Google Scholar] [CrossRef] [PubMed]
- Rani, L.; Sahu, M.R.; Mondal, A.C. Age-related Mitochondrial Dysfunction in Parkinson’s Disease: New Insights Into the Disease Pathology. Neuroscience 2022, 499, 152–169. [Google Scholar] [CrossRef] [PubMed]
- Grünewald, A.; Kumar, K.R.; Sue, C.M. New insights into the complex role of mitochondria in Parkinson’s disease. Prog. Neurobiol. 2019, 177, 73–93. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, Y.; Carrasco, C.M.; Campos, J.D.; Aguirre, P.; Nuñez, M.T. Parkinson’s Disease: The Mitochondria-Iron Link. Park. Dis. 2016, 2016, 7049108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borsche, M.; Pereira, S.L.; Klein, C.; Grünewald, A. Mitochondria and Parkinson’s Disease: Clinical, Molecular, and Translational Aspects. J. Park. Dis. 2021, 11, 45–60. [Google Scholar] [CrossRef]
- Chaturvedi, R.K.; Beal, M.F. Mitochondrial approaches for neuroprotection. Ann. N. Y. Acad. Sci. 2008, 1147, 395–412. [Google Scholar] [CrossRef]
- Stamerra, C.A.; Di Giosia, P.; Giorgini, P.; Ferri, C.; Sukhorukov, V.N.; Sahebkar, A. Mitochondrial Dysfunction and Cardiovascular Disease: Pathophysiology and Emerging Therapies. Oxid. Med. Cell Longev. 2022, 2022, 9530007. [Google Scholar] [CrossRef]
- Langston, J.W.; Ballard, P.A., Jr. Parkinson’s disease in a chemist working with 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine. N. Engl. J. Med. 1983, 309, 310. [Google Scholar]
- Burns, R.S.; Markey, S.P.; Phillips, J.M.; Chiueh, C.C. The neurotoxicity of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine in the monkey and man. Can. J. Neurol. Sci. 1984, 11, 166–168. [Google Scholar] [CrossRef] [Green Version]
- Keeney, P.M.; Xie, J.; Capaldi, R.A.; Bennett, J.P., Jr. Parkinson’s disease brain mitochondrial complex I has oxidatively damaged subunits and is functionally impaired and misassembled. J. Neurosci. 2006, 26, 5256–5264. [Google Scholar] [CrossRef] [Green Version]
- Perier, C.; Bové, J.; Dehay, B.; Jackson-Lewis, V.; Rabinovitch, P.S.; Przedborski, S.; Vila, M. Apoptosis-inducing factor deficiency sensitizes dopaminergic neurons to parkinsonian neurotoxins. Ann. Neurol. 2010, 68, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Choi, W.S.; Kim, H.W.; Tronche, F.; Palmiter, R.D.; Storm, D.R.; Xia, Z. Conditional deletion of Ndufs4 in dopaminergic neurons promotes Parkinson’s disease-like non-motor symptoms without loss of dopamine neurons. Sci. Rep. 2017, 7, 44989. [Google Scholar] [CrossRef] [PubMed]
- Choi, W.S.; Kruse, S.E.; Palmiter, R.D.; Xia, Z. Mitochondrial complex I inhibition is not required for dopaminergic neuron death induced by rotenone, MPP+, or paraquat. Proc. Natl. Acad. Sci. USA 2008, 105, 15136–15141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, W.S.; Xia, Z. Maneb-induced dopaminergic neuronal death is not affected by loss of mitochondrial complex I activity: Results from primary mesencephalic dopaminergic neurons cultured from individual Ndufs4+/+ and Ndufs4−/− mouse embryos. Neuroreport 2014, 25, 1350–1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.W.; Choi, W.S.; Sorscher, N.; Park, H.J.; Tronche, F.; Palmiter, R.D.; Xia, Z. Genetic reduction of mitochondrial complex I function does not lead to loss of dopamine neurons in vivo. Neurobiol. Aging 2015, 36, 2617–2627. [Google Scholar] [CrossRef] [Green Version]
- Sterky, F.H.; Hoffman, A.F.; Milenkovic, D.; Bao, B.; Paganelli, A.; Edgar, D.; Wibom, R.; Lupica, C.R.; Olson, L.; Larsson, N.G. Altered dopamine metabolism and increased vulnerability to MPTP in mice with partial deficiency of mitochondrial complex I in dopamine neurons. Hum. Mol. Genet. 2012, 21, 1078–1089. [Google Scholar] [CrossRef] [Green Version]
- Calvaruso, M.A.; Willems, P.; van den Brand, M.; Valsecchi, F.; Kruse, S.; Palmiter, R.; Smeitink, J.; Nijtmans, L. Mitochondrial complex III stabilizes complex I in the absence of NDUFS4 to provide partial activity. Hum. Mol. Genet. 2012, 21, 115–120. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.S.; Saiki, S.; Kawajiri, S.; Sato, F.; et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 2010, 189, 211–221. [Google Scholar] [CrossRef]
- Meissner, C.; Lorenz, H.; Hehn, B.; Lemberg, M.K. Intramembrane protease PARL defines a negative regulator of PINK1- and PARK2/Parkin-dependent mitophagy. Autophagy 2015, 11, 1484–1498. [Google Scholar] [CrossRef] [Green Version]
- Vives-Bauza, C.; Zhou, C.; Huang, Y.; Cui, M.; de Vries, R.L.; Kim, J.; May, J.; Tocilescu, M.A.; Liu, W.; Ko, H.S.; et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc. Natl. Acad. Sci. USA 2010, 107, 378–383. [Google Scholar] [CrossRef] [Green Version]
- Iguchi, M.; Kujuro, Y.; Okatsu, K.; Koyano, F.; Kosako, H.; Kimura, M.; Suzuki, N.; Uchiyama, S.; Tanaka, K.; Matsuda, N. Parkin-catalyzed ubiquitin-ester transfer is triggered by PINK1-dependent phosphorylation. J. Biol. Chem. 2013, 288, 22019–22032. [Google Scholar] [CrossRef] [Green Version]
- Gautier, C.A.; Kitada, T.; Shen, J. Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. Proc. Natl. Acad. Sci. USA 2008, 105, 11364–11369. [Google Scholar] [CrossRef]
- Lopez-Fabuel, I.; Martin-Martin, L.; Resch-Beusher, M.; Azkona, G.; Sanchez-Pernaute, R.; Bolaños, J.P. Mitochondrial respiratory chain disorganization in Parkinson’s disease-relevant PINK1 and DJ1 mutants. Neurochem. Int. 2017, 109, 101–105. [Google Scholar] [CrossRef]
- Stauch, K.L.; Villeneuve, L.M.; Purnell, P.R.; Ottemann, B.M.; Emanuel, K.; Fox, H.S. Loss of Pink1 modulates synaptic mitochondrial bioenergetics in the rat striatum prior to motor symptoms: Concomitant complex I respiratory defects and increased complex II-mediated respiration. Proteom. Clin. Appl. 2016, 10, 1205–1217. [Google Scholar] [CrossRef] [Green Version]
- Villeneuve, L.M.; Purnell, P.R.; Boska, M.D.; Fox, H.S. Early Expression of Parkinson’s Disease-Related Mitochondrial Abnormalities in PINK1 Knockout Rats. Mol. Neurobiol. 2016, 53, 171–186. [Google Scholar] [CrossRef] [Green Version]
- Zhi, L.; Qin, Q.; Muqeem, T.; Seifert, E.L.; Liu, W.; Zheng, S.; Li, C.; Zhang, H. Loss of PINK1 causes age-dependent decrease of dopamine release and mitochondrial dysfunction. Neurobiol. Aging 2019, 75, 1–10. [Google Scholar] [CrossRef]
- Kuroda, Y.; Mitsui, T.; Kunishige, M.; Matsumoto, T. Parkin affects mitochondrial function and apoptosis in neuronal and myogenic cells. Biochem. Biophys. Res. Commun. 2006, 348, 787–793. [Google Scholar] [CrossRef]
- Bonifati, V.; Rizzu, P.; van Baren, M.J.; Schaap, O.; Breedveld, G.J.; Krieger, E.; Dekker, M.C.; Squitieri, F.; Ibanez, P.; Joosse, M.; et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 2003, 299, 256–259. [Google Scholar] [CrossRef] [Green Version]
- Kim, R.H.; Smith, P.D.; Aleyasin, H.; Hayley, S.; Mount, M.P.; Pownall, S.; Wakeham, A.; You-Ten, A.J.; Kalia, S.K.; Horne, P.; et al. Hypersensitivity of DJ-1-deficient mice to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyrindine (MPTP) and oxidative stress. Proc. Natl. Acad. Sci. USA 2005, 102, 5215–5220. [Google Scholar] [CrossRef] [Green Version]
- Biosa, A.; Sandrelli, F.; Beltramini, M.; Greggio, E.; Bubacco, L.; Bisaglia, M. Recent findings on the physiological function of DJ-1: Beyond Parkinson’s disease. Neurobiol. Dis. 2017, 108, 65–72. [Google Scholar] [CrossRef]
- Hao, L.Y.; Giasson, B.I.; Bonini, N.M. DJ-1 is critical for mitochondrial function and rescues PINK1 loss of function. Proc. Natl. Acad. Sci. USA 2010, 107, 9747–9752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malgieri, G.; Eliezer, D. Structural effects of Parkinson’s disease linked DJ-1 mutations. Protein Sci. 2008, 17, 855–868. [Google Scholar] [CrossRef] [PubMed]
- Larsen, N.J.; Ambrosi, G.; Mullett, S.J.; Berman, S.B.; Hinkle, D.A. DJ-1 knock-down impairs astrocyte mitochondrial function. Neuroscience 2011, 196, 251–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giaime, E.; Yamaguchi, H.; Gautier, C.A.; Kitada, T.; Shen, J. Loss of DJ-1 does not affect mitochondrial respiration but increases ROS production and mitochondrial permeability transition pore opening. PLoS ONE 2012, 7, e40501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Junn, E.; Jang, W.H.; Zhao, X.; Jeong, B.S.; Mouradian, M.M. Mitochondrial localization of DJ-1 leads to enhanced neuroprotection. J. Neurosci. Res. 2009, 87, 123–129. [Google Scholar] [CrossRef] [Green Version]
- Canet-Avilés, R.M.; Wilson, M.A.; Miller, D.W.; Ahmad, R.; McLendon, C.; Bandyopadhyay, S.; Baptista, M.J.; Ringe, D.; Petsko, G.A.; Cookson, M.R. The Parkinson’s disease protein DJ-1 is neuroprotective due to cysteine-sulfinic acid-driven mitochondrial localization. Proc. Natl. Acad. Sci. USA 2004, 101, 9103–9108. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.; Park, H.A.; Mnatsakanyan, N.; Niu, Y.; Licznerski, P.; Wu, J.; Miranda, P.; Graham, M.; Tang, J.; Boon, A.J.W.; et al. Parkinson’s disease protein DJ-1 regulates ATP synthase protein components to increase neuronal process outgrowth. Cell Death Dis. 2019, 10, 469. [Google Scholar] [CrossRef] [Green Version]
- Burre, J. The Synaptic Function of alpha-Synuclein. J. Park. Dis. 2015, 5, 699–713. [Google Scholar] [CrossRef] [Green Version]
- Burré, J.; Sharma, M.; Südhof, T.C. Cell Biology and Pathophysiology of α-Synuclein. Cold Spring Harb. Perspect. Med. 2018, 8, a024091. [Google Scholar] [CrossRef]
- Weinreb, P.H.; Zhen, W.; Poon, A.W.; Conway, K.A.; Lansbury, P.T., Jr. NACP, a protein implicated in Alzheimer’s disease and learning, is natively unfolded. Biochemistry 1996, 35, 13709–13715. [Google Scholar] [CrossRef]
- Shahmoradian, S.H.; Lewis, A.J.; Genoud, C.; Hench, J.; Moors, T.E.; Navarro, P.P.; Castano-Diez, D.; Schweighauser, G.; Graff-Meyer, A.; Goldie, K.N.; et al. Lewy pathology in Parkinson’s disease consists of crowded organelles and lipid membranes. Nat. Neurosci. 2019, 22, 1099–1109. [Google Scholar] [CrossRef] [Green Version]
- Beyer, K.; Domingo-Sàbat, M.; Ariza, A. Molecular pathology of Lewy body diseases. Int. J. Mol. Sci. 2009, 10, 724–745. [Google Scholar] [CrossRef]
- Ma, M.R.; Hu, Z.W.; Zhao, Y.F.; Chen, Y.X.; Li, Y.M. Phosphorylation induces distinct alpha-synuclein strain formation. Sci. Rep. 2016, 6, 37130. [Google Scholar] [CrossRef] [Green Version]
- Arsac, J.N.; Sedru, M.; Dartiguelongue, M.; Vulin, J.; Davoust, N.; Baron, T.; Mollereau, B. Chronic Exposure to Paraquat Induces Alpha-Synuclein Pathogenic Modifications in Drosophila. Int. J. Mol. Sci. 2021, 22, 1613. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, H. Environmental toxins and alpha-synuclein in Parkinson’s disease. Mol. Neurobiol. 2005, 31, 273–282. [Google Scholar] [CrossRef]
- Uversky, V.N.; Li, J.; Fink, A.L. Metal-triggered structural transformations, aggregation, and fibrillation of human alpha-synuclein. A possible molecular NK between Parkinson’s disease and heavy metal exposure. J. Biol. Chem. 2001, 276, 44284–44296. [Google Scholar] [CrossRef] [Green Version]
- Di Maio, R.; Barrett, P.J.; Hoffman, E.K.; Barrett, C.W.; Zharikov, A.; Borah, A.; Hu, X.; McCoy, J.; Chu, C.T.; Burton, E.A.; et al. α-Synuclein binds to TOM20 and inhibits mitochondrial protein import in Parkinson’s disease. Sci. Transl. Med. 2016, 8, 342ra378. [Google Scholar] [CrossRef] [Green Version]
- Reeve, A.K.; Ludtmann, M.H.; Angelova, P.R.; Simcox, E.M.; Horrocks, M.H.; Klenerman, D.; Gandhi, S.; Turnbull, D.M.; Abramov, A.Y. Aggregated alpha-synuclein and complex I deficiency: Exploration of their relationship in differentiated neurons. Cell Death Dis. 2015, 6, e1820. [Google Scholar] [CrossRef] [Green Version]
- Kamp, F.; Exner, N.; Lutz, A.K.; Wender, N.; Hegermann, J.; Brunner, B.; Nuscher, B.; Bartels, T.; Giese, A.; Beyer, K.; et al. Inhibition of mitochondrial fusion by α-synuclein is rescued by PINK1, Parkin and DJ-1. Embo J. 2010, 29, 3571–3589. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, K.; Nemani, V.M.; Azarbal, F.; Skibinski, G.; Levy, J.M.; Egami, K.; Munishkina, L.; Zhang, J.; Gardner, B.; Wakabayashi, J.; et al. Direct membrane association drives mitochondrial fission by the Parkinson disease-associated protein alpha-synuclein. J. Biol. Chem. 2011, 286, 20710–20726. [Google Scholar] [CrossRef] [Green Version]
- Pozo Devoto, V.M.; Dimopoulos, N.; Alloatti, M.; Pardi, M.B.; Saez, T.M.; Otero, M.G.; Cromberg, L.E.; Marín-Burgin, A.; Scassa, M.E.; Stokin, G.B.; et al. αSynuclein control of mitochondrial homeostasis in human-derived neurons is disrupted by mutations associated with Parkinson’s disease. Sci. Rep. 2017, 7, 5042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cookson, M.R. The role of leucine-rich repeat kinase 2 (LRRK2) in Parkinson’s disease. Nat. Rev. Neurosci. 2010, 11, 791–797. [Google Scholar] [CrossRef] [PubMed]
- Paisán-Ruíz, C.; Jain, S.; Evans, E.W.; Gilks, W.P.; Simón, J.; van der Brug, M.; López de Munain, A.; Aparicio, S.; Gil, A.M.; Khan, N.; et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron 2004, 44, 595–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, E.E.S.; Ho, P.W.; Liu, H.F.; Pang, S.Y.; Leung, C.T.; Malki, Y.; Choi, Z.Y.; Ramsden, D.B.; Ho, S.L. LRRK2 mutant knock-in mouse models: Therapeutic relevance in Parkinson’s disease. Transl. Neurodegener. 2022, 11, 10. [Google Scholar] [CrossRef] [PubMed]
- Biskup, S.; Moore, D.J.; Celsi, F.; Higashi, S.; West, A.B.; Andrabi, S.A.; Kurkinen, K.; Yu, S.W.; Savitt, J.M.; Waldvogel, H.J.; et al. Localization of LRRK2 to membranous and vesicular structures in mammalian brain. Ann. Neurol. 2006, 60, 557–569. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yan, M.H.; Fujioka, H.; Liu, J.; Wilson-Delfosse, A.; Chen, S.G.; Perry, G.; Casadesus, G.; Zhu, X. LRRK2 regulates mitochondrial dynamics and function through direct interaction with DLP1. Hum. Mol. Genet. 2012, 21, 1931–1944. [Google Scholar] [CrossRef] [Green Version]
- Grünewald, A.; Arns, B.; Meier, B.; Brockmann, K.; Tadic, V.; Klein, C. Does uncoupling protein 2 expression qualify as marker of disease status in LRRK2-associated Parkinson’s disease? Antioxid. Redox Signal. 2014, 20, 1955–1960. [Google Scholar] [CrossRef] [Green Version]
- Cooper, O.; Seo, H.; Andrabi, S.; Guardia-Laguarta, C.; Graziotto, J.; Sundberg, M.; McLean, J.R.; Carrillo-Reid, L.; Xie, Z.; Osborn, T.; et al. Pharmacological rescue of mitochondrial deficits in iPSC-derived neural cells from patients with familial Parkinson’s disease. Sci. Transl. Med. 2012, 4, 141ra190. [Google Scholar] [CrossRef] [Green Version]
- Mendivil-Perez, M.; Velez-Pardo, C.; Jimenez-Del-Rio, M. Neuroprotective Effect of the LRRK2 Kinase Inhibitor PF-06447475 in Human Nerve-Like Differentiated Cells Exposed to Oxidative Stress Stimuli: Implications for Parkinson’s Disease. Neurochem. Res. 2016, 41, 2675–2692. [Google Scholar] [CrossRef]
- Howlett, E.H.; Jensen, N.; Belmonte, F.; Zafar, F.; Hu, X.; Kluss, J.; Schüle, B.; Kaufman, B.A.; Greenamyre, J.T.; Sanders, L.H. LRRK2 G2019S-induced mitochondrial DNA damage is LRRK2 kinase dependent and inhibition restores mtDNA integrity in Parkinson’s disease. Hum. Mol. Genet. 2017, 26, 4340–4351. [Google Scholar] [CrossRef] [Green Version]
- Mortiboys, H.; Johansen, K.K.; Aasly, J.O.; Bandmann, O. Mitochondrial impairment in patients with Parkinson disease with the G2019S mutation in LRRK2. Neurology 2010, 75, 2017–2020. [Google Scholar] [CrossRef]
- Stafa, K.; Tsika, E.; Moser, R.; Musso, A.; Glauser, L.; Jones, A.; Biskup, S.; Xiong, Y.; Bandopadhyay, R.; Dawson, V.L.; et al. Functional interaction of Parkinson’s disease-associated LRRK2 with members of the dynamin GTPase superfamily. Hum. Mol. Genet. 2014, 23, 2055–2077. [Google Scholar] [CrossRef]
- Su, Y.C.; Guo, X.; Qi, X. Threonine 56 phosphorylation of Bcl-2 is required for LRRK2 G2019S-induced mitochondrial depolarization and autophagy. Biochim. Biophys. Acta 2015, 1852, 12–21. [Google Scholar] [CrossRef] [Green Version]
- Smith, G.A.; Jansson, J.; Rocha, E.M.; Osborn, T.; Hallett, P.J.; Isacson, O. Fibroblast Biomarkers of Sporadic Parkinson’s Disease and LRRK2 Kinase Inhibition. Mol. Neurobiol. 2016, 53, 5161–5177. [Google Scholar] [CrossRef] [Green Version]
- Dehay, B.; Ramirez, A.; Martinez-Vicente, M.; Perier, C.; Canron, M.H.; Doudnikoff, E.; Vital, A.; Vila, M.; Klein, C.; Bezard, E. Loss of P-type ATPase ATP13A2/PARK9 function induces general lysosomal deficiency and leads to Parkinson disease neurodegeneration. Proc. Natl. Acad. Sci. USA 2012, 109, 9611–9616. [Google Scholar] [CrossRef] [Green Version]
- Kruer, M.C. The neuropathology of neurodegeneration with brain iron accumulation. Int. Rev. Neurobiol. 2013, 110, 165–194. [Google Scholar] [CrossRef] [Green Version]
- Kurz, T.; Gustafsson, B.; Brunk, U.T. Intralysosomal iron chelation protects against oxidative stress-induced cellular damage. Febs J. 2006, 273, 3106–3117. [Google Scholar] [CrossRef]
- Rajagopalan, S.; Rane, A.; Chinta, S.J.; Andersen, J.K. Regulation of ATP13A2 via PHD2-HIF1α Signaling Is Critical for Cellular Iron Homeostasis: Implications for Parkinson’s Disease. J. Neurosci. 2016, 36, 1086–1095. [Google Scholar] [CrossRef] [Green Version]
- Rinaldi, D.E.; Corradi, G.R.; Cuesta, L.M.; Adamo, H.P.; de Tezanos Pinto, F. The Parkinson-associated human P5B-ATPase ATP13A2 protects against the iron-induced cytotoxicity. Biochim. Biophys. Acta 2015, 1848, 1646–1655. [Google Scholar] [CrossRef] [Green Version]
- Park, J.S.; Koentjoro, B.; Veivers, D.; Mackay-Sim, A.; Sue, C.M. Parkinson’s disease-associated human ATP13A2 (PARK9) deficiency causes zinc dyshomeostasis and mitochondrial dysfunction. Hum. Mol. Genet. 2014, 23, 2802–2815. [Google Scholar] [CrossRef] [Green Version]
- Grünewald, A.; Arns, B.; Seibler, P.; Rakovic, A.; Münchau, A.; Ramirez, A.; Sue, C.M.; Klein, C. ATP13A2 mutations impair mitochondrial function in fibroblasts from patients with Kufor-Rakeb syndrome. Neurobiol. Aging 2012, 33, e1841–e1847. [Google Scholar] [CrossRef] [PubMed]
- Gusdon, A.M.; Zhu, J.; Van Houten, B.; Chu, C.T. ATP13A2 regulates mitochondrial bioenergetics through macroautophagy. Neurobiol. Dis. 2012, 45, 962–972. [Google Scholar] [CrossRef] [PubMed]
- Lill, R. Function and biogenesis of iron-sulphur proteins. Nature 2009, 460, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Lill, R.; Freibert, S.A. Mechanisms of Mitochondrial Iron-Sulfur Protein Biogenesis. Annu. Rev. Biochem. 2020, 89, 471–499. [Google Scholar] [CrossRef] [PubMed]
- Cadenas, E.; Davies, K.J. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic. Biol. Med. 2000, 29, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Núñez, M.T.; Urrutia, P.; Mena, N.; Aguirre, P.; Tapia, V.; Salazar, J. Iron toxicity in neurodegeneration. Biometals 2012, 25, 761–776. [Google Scholar] [CrossRef]
- Núñez, M.T.; Gaete, V.; Watkins, J.A.; Glass, J. Mobilization of iron from endocytic vesicles. The effects of acidification and reduction. J. Biol. Chem. 1990, 265, 6688–6692. [Google Scholar] [CrossRef]
- Radisky, D.C.; Kaplan, J. Iron in cytosolic ferritin can be recycled through lysosomal degradation in human fibroblasts. Biochem. J. 1998, 336 Pt 1, 201–205. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Mikhael, M.; Xu, D.; Li, Y.; Soe-Lin, S.; Ning, B.; Li, W.; Nie, G.; Zhao, Y.; Ponka, P. Lysosomal proteolysis is the primary degradation pathway for cytosolic ferritin and cytosolic ferritin degradation is necessary for iron exit. Antioxid. Redox Signal. 2010, 13, 999–1009. [Google Scholar] [CrossRef]
- Ashok, A.; Chaudhary, S.; Wise, A.S.; Rana, N.A.; McDonald, D.; Kritikos, A.E.; Lindner, E.; Singh, N. Release of Iron-Loaded Ferritin in Sodium Iodate-Induced Model of Age Related Macular Degeneration: An In-Vitro and In-Vivo Study. Antioxidants 2021, 10, 1253. [Google Scholar] [CrossRef]
- Hamdi, A.; Roshan, T.M.; Kahawita, T.M.; Mason, A.B.; Sheftel, A.D.; Ponka, P. Erythroid cell mitochondria receive endosomal iron by a “kiss-and-run” mechanism. Biochim. Biophys. Acta 2016, 1863, 2859–2867. [Google Scholar] [CrossRef]
- Barra, J.; Crosbourne, I.; Wang, L.; Bossardi-Ramos, R.; Jourd’heuil, F.; Nelson, I.; Adam, A.P.; Corr, D.T.; Jourd’heuil, D.; Barroso, M. DMT1 bridges endosomes and mitochondria to modulate mitochondrial iron translocation. bioRxiv 2022. [Google Scholar] [CrossRef]
- Shaw, G.C.; Cope, J.J.; Li, L.; Corson, K.; Hersey, C.; Ackermann, G.E.; Gwynn, B.; Lambert, A.J.; Wingert, R.A.; Traver, D.; et al. Mitoferrin is essential for erythroid iron assimilation. Nature 2006, 440, 96–100. [Google Scholar] [CrossRef]
- Paradkar, P.N.; Zumbrennen, K.B.; Paw, B.H.; Ward, D.M.; Kaplan, J. Regulation of mitochondrial iron import through differential turnover of mitoferrin 1 and mitoferrin 2. Mol. Cell Biol. 2009, 29, 1007–1016. [Google Scholar] [CrossRef] [Green Version]
- Baldauf, L.; Endres, T.; Scholz, J.; Kirches, E.; Ward, D.M.; Lessmann, V.; Borucki, K.; Mawrin, C. Mitoferrin-1 is required for brain energy metabolism and hippocampus-dependent memory. Neurosci. Lett. 2019, 713, 134521. [Google Scholar] [CrossRef]
- Zhang, T.; Sun, L.; Hao, Y.; Suo, C.; Shen, S.; Wei, H.; Ma, W.; Zhang, P.; Wang, T.; Gu, X.; et al. ENO1 suppresses cancer cell ferroptosis by degrading the mRNA of iron regulatory protein 1. Nat. Cancer 2022, 3, 75–89. [Google Scholar] [CrossRef]
- Dexter, D.T.; Carter, C.J.; Wells, F.R.; Javoy-Agid, F.; Agid, Y.; Lees, A.; Jenner, P.; Marsden, C.D. Basal lipid peroxidation in substantia nigra is increased in Parkinson’s disease. J. Neurochem. 1989, 52, 381–389. [Google Scholar] [CrossRef]
- Hirsch, E.C.; Brandel, J.P.; Galle, P.; Javoy-Agid, F.; Agid, Y. Iron and aluminum increase in the substantia nigra of patients with Parkinson’s disease: An X-ray microanalysis. J. Neurochem. 1991, 56, 446–451. [Google Scholar] [CrossRef]
- Sofic, E.; Riederer, P.; Heinsen, H.; Beckmann, H.; Reynolds, G.P.; Hebenstreit, G.; Youdim, M.B. Increased iron (III) and total iron content in post mortem substantia nigra of parkinsonian brain. J. Neural Transm. 1988, 74, 199–205. [Google Scholar] [CrossRef]
- Bae, Y.J.; Kim, J.M.; Sohn, C.H.; Choi, J.H.; Choi, B.S.; Song, Y.S.; Nam, Y.; Cho, S.J.; Jeon, B.; Kim, J.H. Imaging the Substantia Nigra in Parkinson Disease and Other Parkinsonian Syndromes. Radiology 2021, 300, 260–278. [Google Scholar] [CrossRef]
- Abeyawardhane, D.L.; Fernández, R.D.; Murgas, C.J.; Heitger, D.R.; Forney, A.K.; Crozier, M.K.; Lucas, H.R. Iron Redox Chemistry Promotes Antiparallel Oligomerization of α-Synuclein. J. Am. Chem. Soc. 2018, 140, 5028–5032. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Wang, J.; Song, N.; Xie, J.; Jiang, H. Up-regulation of divalent metal transporter 1 is involved in 1-methyl-4-phenylpyridinium (MPP(+))-induced apoptosis in MES23.5 cells. Neurobiol. Aging 2009, 30, 1466–1476. [Google Scholar] [CrossRef] [PubMed]
- Urrutia, P.J.; Aguirre, P.; Tapia, V.; Carrasco, C.M.; Mena, N.P.; Nunez, M.T. Cell death induced by mitochondrial complex I inhibition is mediated by Iron Regulatory Protein 1. Biochim. Biophys. Acta 2017, 863, 2202–2209. [Google Scholar] [CrossRef] [PubMed]
- Mena, N.P.; Bulteau, A.L.; Salazar, J.; Hirsch, E.C.; Núñez, M.T. Effect of mitochondrial complex I inhibition on Fe-S cluster protein activity. Biochem. Biophys. Res. Commun. 2011, 409, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Grotemeyer, A.; McFleder, R.L.; Wu, J.; Wischhusen, J.; Ip, C.W. Neuroinflammation in Parkinson’s Disease-Putative Pathomechanisms and Targets for Disease-Modification. Front. Immunol. 2022, 13, 878771. [Google Scholar] [CrossRef]
- Tansey, M.G.; Wallings, R.L.; Houser, M.C.; Herrick, M.K.; Keating, C.E.; Joers, V. Inflammation and immune dysfunction in Parkinson disease. Nat. Rev. Immunol. 2022, 22, 657–673. [Google Scholar] [CrossRef]
- Hall, S.; Janelidze, S.; Surova, Y.; Widner, H.; Zetterberg, H.; Hansson, O. Cerebrospinal fluid concentrations of inflammatory markers in Parkinson’s disease and atypical parkinsonian disorders. Sci. Rep. 2018, 8, 13276. [Google Scholar] [CrossRef] [Green Version]
- Meredith, G.E.; Sonsalla, P.K.; Chesselet, M.F. Animal models of Parkinson’s disease progression. Acta Neuropathol. 2008, 115, 385–398. [Google Scholar] [CrossRef] [Green Version]
- Sliter, D.A.; Martinez, J.; Hao, L.; Chen, X.; Sun, N.; Fischer, T.D.; Burman, J.L.; Li, Y.; Zhang, Z.; Narendra, D.P.; et al. Parkin and PINK1 mitigate STING-induced inflammation. Nature 2018, 561, 258–262. [Google Scholar] [CrossRef]
- Jewell, S.; Herath, A.M.; Gordon, R. Inflammasome Activation in Parkinson’s Disease. J. Park. Dis. 2022, 12, S113–S128. [Google Scholar] [CrossRef]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef]
- Pérez-Treviño, P.; Velásquez, M.; García, N. Mechanisms of mitochondrial DNA escape and its relationship with different metabolic diseases. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165761. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef]
- Ding, Z.; Liu, S.; Wang, X.; Khaidakov, M.; Dai, Y.; Mehta, J.L. Oxidant stress in mitochondrial DNA damage, autophagy and inflammation in atherosclerosis. Sci. Rep. 2013, 3, 1077. [Google Scholar] [CrossRef] [Green Version]
- Zhong, Z.; Liang, S.; Sanchez-Lopez, E.; He, F.; Shalapour, S.; Lin, X.J.; Wong, J.; Ding, S.; Seki, E.; Schnabl, B.; et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature 2018, 560, 198–203. [Google Scholar] [CrossRef]
- Sarkar, S.; Malovic, E.; Harishchandra, D.S.; Ghaisas, S.; Panicker, N.; Charli, A.; Palanisamy, B.N.; Rokad, D.; Jin, H.; Anantharam, V.; et al. Mitochondrial impairment in microglia amplifies NLRP3 inflammasome proinflammatory signaling in cell culture and animal models of Parkinson’s disease. NPJ Park. Dis. 2017, 3, 30. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Gupta, R.; Blanco, L.P.; Yang, S.; Shteinfer-Kuzmine, A.; Wang, K.; Zhu, J.; Yoon, H.E.; Wang, X.; Kerkhofs, M.; et al. VDAC oligomers form mitochondrial pores to release mtDNA fragments and promote lupus-like disease. Science 2019, 366, 1531–1536. [Google Scholar] [CrossRef]
- West, A.P.; Khoury-Hanold, W.; Staron, M.; Tal, M.C.; Pineda, C.M.; Lang, S.M.; Bestwick, M.; Duguay, B.A.; Raimundo, N.; MacDuff, D.A.; et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature 2015, 520, 553–557. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Boyd, R.M.; Tree, M.O.; Samkari, F.; Zhao, L. Mitochondrial transcription factor A promotes DNA strand cleavage at abasic sites. Proc. Natl. Acad. Sci. USA 2019, 116, 17792–17799. [Google Scholar] [CrossRef] [Green Version]
- Garcia, A.; LaMontagne, K.; Reavis, D.; Stober-Grasser, U.; Lipsick, J.S. Determinants of sequence-specific DNA-binding by p48v-myb. Oncogene 1991, 6, 265–273. [Google Scholar] [PubMed]
- Grunewald, A.; Rygiel, K.A.; Hepplewhite, P.D.; Morris, C.M.; Picard, M.; Turnbull, D.M. Mitochondrial DNA Depletion in Respiratory Chain-Deficient Parkinson Disease Neurons. Ann. Neurol. 2016, 79, 366–378. [Google Scholar] [CrossRef] [PubMed]
- Ingrassia, R.; Lanzillotta, A.; Sarnico, I.; Benarese, M.; Blasi, F.; Borgese, L.; Bilo, F.; Depero, L.; Chiarugi, A.; Spano, P.F.; et al. 1B/(-)IRE DMT1 expression during brain ischemia contributes to cell death mediated by NF-κB/RelA acetylation at Lys310. PLoS ONE 2012, 7, e38019. [Google Scholar] [CrossRef] [PubMed]
- Paradkar, P.N.; Roth, J.A. Nitric oxide transcriptionally down-regulates specific isoforms of divalent metal transporter (DMT1) via NF-kappaB. J. Neurochem. 2006, 96, 1768–1777. [Google Scholar] [CrossRef] [PubMed]
- Shih, R.H.; Wang, C.Y.; Yang, C.M. NF-kappaB Signaling Pathways in Neurological Inflammation: A Mini Review. Front. Mol. Neurosci. 2015, 8, 77. [Google Scholar] [CrossRef] [Green Version]
- Mogi, M.; Togari, A.; Kondo, T.; Mizuno, Y.; Komure, O.; Kuno, S.; Ichinose, H.; Nagatsu, T. Caspase activities and tumor necrosis factor receptor R1 (p55) level are elevated in the substantia nigra from parkinsonian brain. J. Neural Transm. (Vienna) 2000, 107, 335–341. [Google Scholar] [CrossRef]
- Thomsen, M.S.; Andersen, M.V.; Christoffersen, P.R.; Jensen, M.D.; Lichota, J.; Moos, T. Neurodegeneration with inflammation is accompanied by accumulation of iron and ferritin in microglia and neurons. Neurobiol. Dis. 2015, 81, 108–118. [Google Scholar] [CrossRef]
- Urrutia, P.; Aguirre, P.; Esparza, A.; Tapia, V.; Mena, N.P.; Arredondo, M.; Gonzalez-Billault, C.; Nunez, M.T. Inflammation alters the expression of DMT1, FPN1 and hepcidin, and it causes iron accumulation in central nervous system cells. J. Neurochem. 2013, 126, 541–549. [Google Scholar] [CrossRef]
- Levi, S.; Yewdall, S.J.; Harrison, P.M.; Santambrogio, P.; Cozzi, A.; Rovida, E.; Albertini, A.; Arosio, P. Evidence of H- and L-chains have co-operative roles in the iron-uptake mechanism of human ferritin. Biochem. J. 1992, 288 Pt 2, 591–596. [Google Scholar] [CrossRef] [Green Version]
- Torti, F.M.; Torti, S.V. Regulation of ferritin genes and protein. Blood 2002, 99, 3505–3516. [Google Scholar] [CrossRef] [Green Version]
- Pham, C.G.; Bubici, C.; Zazzeroni, F.; Papa, S.; Jones, J.; Alvarez, K.; Jayawardena, S.; De Smaele, E.; Cong, R.; Beaumont, C.; et al. Ferritin heavy chain upregulation by NF-kappaB inhibits TNFalpha-induced apoptosis by suppressing reactive oxygen species. Cell 2004, 119, 529–542. [Google Scholar] [CrossRef] [Green Version]
- Kerins, M.J.; Ooi, A. The Roles of NRF2 in Modulating Cellular Iron Homeostasis. Antioxid. Redox Signal. 2018, 29, 1756–1773. [Google Scholar] [CrossRef]
- Dexter, D.T.; Carayon, A.; Javoy-Agid, F.; Agid, Y.; Wells, F.R.; Daniel, S.E.; Lees, A.J.; Jenner, P.; Marsden, C.D. Alterations in the levels of iron, ferritin and other trace metals in Parkinson’s disease and other neurodegenerative diseases affecting the basal ganglia. Brain 1991, 114 Pt 4, 1953–1975. [Google Scholar] [CrossRef]
- Galazka-Friedman, J.; Bauminger, E.R.; Koziorowski, D.; Friedman, A. Mössbauer spectroscopy and ELISA studies reveal differences between Parkinson’s disease and control substantia nigra. Biochim. Biophys. Acta 2004, 1688, 130–136. [Google Scholar] [CrossRef] [Green Version]
- Roth, M.P.; Meynard, D.; Coppin, H. Regulators of hepcidin expression. Vitam. Horm. 2019, 110, 101–129. [Google Scholar] [CrossRef]
- Zechel, S.; Huber-Wittmer, K.; von Bohlen und Halbach, O. Distribution of the iron-regulating protein hepcidin in the murine central nervous system. J. Neurosci. Res. 2006, 84, 790–800. [Google Scholar] [CrossRef]
- Wang, S.M.; Fu, L.J.; Duan, X.L.; Crooks, D.R.; Yu, P.; Qian, Z.M.; Di, X.J.; Li, J.; Rouault, T.A.; Chang, Y.Z. Role of hepcidin in murine brain iron metabolism. Cell Mol. Life Sci. 2010, 67, 123–133. [Google Scholar] [CrossRef] [Green Version]
- Hänninen, M.M.; Haapasalo, J.; Haapasalo, H.; Fleming, R.E.; Britton, R.S.; Bacon, B.R.; Parkkila, S. Expression of iron-related genes in human brain and brain tumors. BMC Neurosci. 2009, 10, 36. [Google Scholar] [CrossRef] [Green Version]
- Yanase, K.; Uemura, N.; Chiba, Y.; Murakami, R.; Fujihara, R.; Matsumoto, K.; Shirakami, G.; Araki, N.; Ueno, M. Immunoreactivities for hepcidin, ferroportin, and hephaestin in astrocytes and choroid plexus epithelium of human brains. Neuropathology 2020, 40, 75–83. [Google Scholar] [CrossRef]
- Xu, Q.; Kanthasamy, A.G.; Jin, H.; Reddy, M.B. Hepcidin Plays a Key Role in 6-OHDA Induced Iron Overload and Apoptotic Cell Death in a Cell Culture Model of Parkinson’s Disease. Park. Dis. 2016, 2016, 8684130. [Google Scholar] [CrossRef] [Green Version]
- You, L.; Yu, P.P.; Dong, T.; Guo, W.; Chang, S.; Zheng, B.; Ci, Y.; Wang, F.; Yu, P.; Gao, G.; et al. Astrocyte-derived hepcidin controls iron traffic at the blood-brain-barrier via regulating ferroportin 1 of microvascular endothelial cells. Cell Death Dis. 2022, 13, 667. [Google Scholar] [CrossRef] [PubMed]
- Du, F.; Qian, Z.M.; Luo, Q.; Yung, W.H.; Ke, Y. Hepcidin Suppresses Brain Iron Accumulation by Downregulating Iron Transport Proteins in Iron-Overloaded Rats. Mol. Neurobiol. 2015, 52, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Du, F.; Qian, Z.M.; Luo, Q.Q.; Sheng, Y.; Yung, W.H.; Xu, Y.X.; Ke, Y. Pre-treatment of rats with ad-hepcidin prevents iron-induced oxidative stress in the brain. Free Radic. Biol. Med. 2016, 90, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Liang, T.; Qian, Z.M.; Mu, M.D.; Yung, W.H.; Ke, Y. Brain Hepcidin Suppresses Major Pathologies in Experimental Parkinsonism. iScience 2020, 23, 101284. [Google Scholar] [CrossRef] [PubMed]
- Kwiatek-Majkusiak, J.; Geremek, M.; Koziorowski, D.; Tomasiuk, R.; Szlufik, S.; Friedman, A. Serum levels of hepcidin and interleukin 6 in Parkinson’s disease. Acta Neurobiol. Exp. (Wars) 2020, 80, 297–304. [Google Scholar] [CrossRef]
- Wang, J.; Song, N.; Jiang, H.; Wang, J.; Xie, J. Pro-inflammatory cytokines modulate iron regulatory protein 1 expression and iron transportation through reactive oxygen/nitrogen species production in ventral mesencephalic neurons. Biochim. Biophys. Acta 2013, 1832, 618–625. [Google Scholar] [CrossRef] [Green Version]
- Styś, A.; Galy, B.; Starzyński, R.R.; Smuda, E.; Drapier, J.C.; Lipiński, P.; Bouton, C. Iron regulatory protein 1 outcompetes iron regulatory protein 2 in regulating cellular iron homeostasis in response to nitric oxide. J. Biol. Chem. 2011, 286, 22846–22854. [Google Scholar] [CrossRef] [Green Version]
- Caltagirone, A.; Weiss, G.; Pantopoulos, K. Modulation of cellular iron metabolism by hydrogen peroxide. Effects of H2O2 on the expression and function of iron-responsive element-containing mRNAs in B6 fibroblasts. J. Biol. Chem. 2001, 276, 19738–19745. [Google Scholar] [CrossRef] [Green Version]
- Cairo, G.; Ronchi, R.; Recalcati, S.; Campanella, A.; Minotti, G. Nitric oxide and peroxynitrite activate the iron regulatory protein-1 of J774A.1 macrophages by direct disassembly of the Fe-S cluster of cytoplasmic aconitase. Biochemistry 2002, 41, 7435–7442. [Google Scholar] [CrossRef]
- Soum, E.; Drapier, J.C. Nitric oxide and peroxynitrite promote complete disruption of the [4Fe-4S] cluster of recombinant human iron regulatory protein 1. J. Biol. Inorg. Chem. 2003, 8, 226–232. [Google Scholar] [CrossRef]
- Lipinski, P.; Starzynski, R.R.; Drapier, J.C.; Bouton, C.; Bartlomiejczyk, T.; Sochanowicz, B.; Smuda, E.; Gajkowska, A.; Kruszewski, M. Induction of iron regulatory protein 1 RNA-binding activity by nitric oxide is associated with a concomitant increase in the labile iron pool: Implications for DNA damage. Biochem. Biophys. Res. Commun. 2005, 327, 349–355. [Google Scholar] [CrossRef]
- Crichton, R.R.; Dexter, D.T.; Ward, R.J. Brain iron metabolism and its perturbation in neurological diseases. J. Neural Transm. (Vienna) 2011, 118, 301–314. [Google Scholar] [CrossRef]
- McFarthing, K.; Rafaloff, G.; Baptista, M.; Mursaleen, L.; Fuest, R.; Wyse, R.K.; Stott, S.R.W. Parkinson’s Disease Drug Therapies in the Clinical Trial Pipeline: 2022 Update. J. Park. Dis. 2022, 12, 1073–1082. [Google Scholar] [CrossRef]
- Chandra, G.; Shenoi, R.A.; Anand, R.; Rajamma, U.; Mohanakumar, K.P. Reinforcing mitochondrial functions in aging brain: An insight into Parkinson’s disease therapeutics. J. Chem. Neuroanat. 2019, 95, 29–42. [Google Scholar] [CrossRef]
- Negida, A.; Menshawy, A.; El Ashal, G.; Elfouly, Y.; Hani, Y.; Hegazy, Y.; El Ghonimy, S.; Fouda, S.; Rashad, Y. Coenzyme Q10 for Patients with Parkinson’s Disease: A Systematic Review and Meta-Analysis. CNS Neurol. Disord. Drug Targets 2016, 15, 45–53. [Google Scholar] [CrossRef]
- Martin-Bastida, A.; Ward, R.J.; Newbould, R.; Piccini, P.; Sharp, D.; Kabba, C.; Patel, M.C.; Spino, M.; Connelly, J.; Tricta, F.; et al. Brain iron chelation by deferiprone in a phase 2 randomised double-blinded placebo controlled clinical trial in Parkinson’s disease. Sci. Rep. 2017, 7, 1398. [Google Scholar] [CrossRef] [Green Version]
- Devos, D.; Moreau, C.; Devedjian, J.C.; Kluza, J.; Petrault, M.; Laloux, C.; Jonneaux, A.; Ryckewaert, G.; Garçon, G.; Rouaix, N.; et al. Targeting chelatable iron as a therapeutic modality in Parkinson’s disease. Antioxid. Redox Signal. 2014, 21, 195–210. [Google Scholar] [CrossRef] [Green Version]
- Grolez, G.; Moreau, C.; Sablonniere, B.; Garcon, G.; Devedjian, J.C.; Meguig, S.; Gele, P.; Delmaire, C.; Bordet, R.; Defebvre, L.; et al. Ceruloplasmin activity and iron chelation treatment of patients with Parkinson’s disease. BMC Neurol. 2015, 15, 74. [Google Scholar] [CrossRef] [Green Version]
- Devos, D.; Labreuche, J.; Rascol, O.; Corvol, J.C.; Duhamel, A.; Guyon Delannoy, P.; Poewe, W.; Compta, Y.; Pavese, N.; Ruzicka, E.; et al. Trial of Deferiprone in Parkinson’s Disease. N. Engl. J. Med. 2022, 387, 2045–2055. [Google Scholar] [CrossRef]
- Wu, B.J.; Yan, L.; Charlton, F.; Witting, P.; Barter, P.J.; Rye, K.A. Evidence that niacin inhibits acute vascular inflammation and improves endothelial dysfunction independent of changes in plasma lipids. Arter. Thromb. Vasc. Biol. 2010, 30, 968–975. [Google Scholar] [CrossRef] [Green Version]
- Wakade, C.; Chong, R.; Seamon, M.; Purohit, S.; Giri, B.; Morgan, J.C. Low-Dose Niacin Supplementation Improves Motor Function in US Veterans with Parkinson’s Disease: A Single-Center, Randomized, Placebo-Controlled Trial. Biomedicines 2021, 9, 1881. [Google Scholar] [CrossRef] [PubMed]
- Aoun, M.; Tiranti, V. Mitochondria: A crossroads for lipid metabolism defect in neurodegeneration with brain iron accumulation diseases. Int. J. Biochem. Cell Biol. 2015, 63, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Benek, O.; Korabecny, J.; Soukup, O. A Perspective on Multi-target Drugs for Alzheimer’s Disease. Trends Pharmacol. Sci. 2020, 41, 434–445. [Google Scholar] [CrossRef] [PubMed]
- Miguel, C.A.; Noya-Riobó, M.V.; Mazzone, G.L.; Villar, M.J.; Coronel, M.F. Antioxidant, anti-inflammatory and neuroprotective actions of resveratrol after experimental nervous system insults. Special focus on the molecular mechanisms involved. Neurochem. Int. 2021, 150, 105188. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Chen, Z.; Huang, X.; Shi, W.; Zhang, R.; Chen, M.; Huang, H.; Wu, L. Insights on the Multifunctional Activities of Magnolol. Biomed. Res. Int. 2019, 2019, 1847130. [Google Scholar] [CrossRef] [Green Version]
- Ortiz, C.J.C.; Damasio, C.M.; Pruccoli, L.; Nadur, N.F.; de Azevedo, L.L.; Guedes, I.A.; Dardenne, L.E.; Kümmerle, A.E.; Tarozzi, A.; Viegas, C., Jr. Cinnamoyl-N-Acylhydrazone-Donepezil Hybrids: Synthesis and Evaluation of Novel Multifunctional Ligands Against Neurodegenerative Diseases. Neurochem. Res. 2020, 45, 3003–3020. [Google Scholar] [CrossRef]
- Xie, S.; Chen, J.; Li, X.; Su, T.; Wang, Y.; Wang, Z.; Huang, L.; Li, X. Synthesis and evaluation of selegiline derivatives as monoamine oxidase inhibitor, antioxidant and metal chelator against Alzheimer’s disease. Bioorg. Med. Chem. 2015, 23, 3722–3729. [Google Scholar] [CrossRef]
- Chen, Y.; Hao, Y.; Qian, Y.; Chengguo, L.; Wu, B.; Liu, Y.; Zhang, Z.; Tian, C.; Ning, X.; Guo, Y.; et al. Design, Synthesis and Biological Evaluation of Novel (E)-Hydroxystyryl Aralkyl Sulfones as Neuroprotective Agents. ChemistrySelect 2020, 5, 6268–6273. [Google Scholar] [CrossRef]
- Wang, Y.; Plastina, P.; Vincken, J.P.; Jansen, R.; Balvers, M.; Ten Klooster, J.P.; Gruppen, H.; Witkamp, R.; Meijerink, J. N-Docosahexaenoyl Dopamine, an Endocannabinoid-like Conjugate of Dopamine and the n-3 Fatty Acid Docosahexaenoic Acid, Attenuates Lipopolysaccharide-Induced Activation of Microglia and Macrophages via COX-2. ACS Chem. Neurosci. 2017, 8, 548–557. [Google Scholar] [CrossRef]
- Wang, Y.Q.; Wang, M.Y.; Fu, X.R.; Peng, Y.; Gao, G.F.; Fan, Y.M.; Duan, X.L.; Zhao, B.L.; Chang, Y.Z.; Shi, Z.H. Neuroprotective effects of ginkgetin against neuroinjury in Parkinson’s disease model induced by MPTP via chelating iron. Free Radic. Res. 2015, 49, 1069–1080. [Google Scholar] [CrossRef]
- Sarfraz, A.; Javeed, M.; Shah, M.A.; Hussain, G.; Shafiq, N.; Sarfraz, I.; Riaz, A.; Sadiqa, A.; Zara, R.; Zafar, S.; et al. Biochanin A: A novel bioactive multifunctional compound from nature. Sci. Total Environ. 2020, 722, 137907. [Google Scholar] [CrossRef]
- Chen, M.; Wang, T.; Yue, F.; Li, X.; Wang, P.; Li, Y.; Chan, P.; Yu, S. Tea polyphenols alleviate motor impairments, dopaminergic neuronal injury, and cerebral α-synuclein aggregation in MPTP-intoxicated parkinsonian monkeys. Neuroscience 2015, 286, 383–392. [Google Scholar] [CrossRef]
- Aguirre, P.; Garcia-Beltran, O.; Tapia, V.; Munoz, Y.; Cassels, B.K.; Nunez, M.T. Neuroprotective effect of a new 7,8-dihydroxycoumarin-based Fe2+/Cu2+ chelator in cell and animal models of Parkinson’s disease. ACS Chem. Neurosci. 2017, 12, 178–185. [Google Scholar] [CrossRef]
- Garcia-Beltran, O.; Mena, N.P.; Aguirre, P.; Barriga-Gonzalez, G.; Galdamez, A.; Nagles, E.; Adasme, T.; Hidalgo, C.; Nunez, M.T. Development of an iron-selective antioxidant probe with protective effects on neuronal function. PLoS ONE 2017, 12, e0189043. [Google Scholar] [CrossRef] [Green Version]
- Tao, D.; Wang, Y.; Bao, X.Q.; Yang, B.B.; Gao, F.; Wang, L.; Zhang, D.; Li, L. Discovery of coumarin Mannich base derivatives as multifunctional agents against monoamine oxidase B and neuroinflammation for the treatment of Parkinson’s disease. Eur. J. Med. Chem. 2019, 173, 203–212. [Google Scholar] [CrossRef]
- Youdim, M.B.H. Monoamine oxidase inhibitors, and iron chelators in depressive illness and neurodegenerative diseases. J. Neural Transm. (Vienna) 2018, 125, 1719–1733. [Google Scholar] [CrossRef]
- Zheng, H.; Weiner, L.M.; Bar-Am, O.; Epsztejn, S.; Cabantchik, Z.I.; Warshawsky, A.; Youdim, M.B.; Fridkin, M. Design, synthesis, and evaluation of novel bifunctional iron-chelators as potential agents for neuroprotection in Alzheimer’s, Parkinson’s, and other neurodegenerative diseases. Bioorg. Med. Chem. 2005, 13, 773–783. [Google Scholar] [CrossRef]
- Selvaraj, B.; Woon Kim, D.; Park, J.S.; Cheol Kwon, H.; Lee, H.; Yoo, K.Y.; Wook Lee, J. Neuroprotective effects of 2-heptyl-3-hydroxy-4-quinolone in HT22 mouse hippocampal neuronal cells. Bioorg. Med. Chem. Lett. 2021, 49, 128312. [Google Scholar] [CrossRef]
- Das, B.; Rajagopalan, S.; Joshi, G.S.; Xu, L.; Luo, D.; Andersen, J.K.; Todi, S.V.; Dutta, A.K. A novel iron (II) preferring dopamine agonist chelator D-607 significantly suppresses alpha-syn- and MPTP-induced toxicities in vivo. Neuropharmacology 2017, 123, 88–99. [Google Scholar] [CrossRef]
- Yedlapudi, D.; Xu, L.; Luo, D.; Marsh, G.B.; Todi, S.V.; Dutta, A.K. Targeting alpha synuclein and amyloid beta by a multifunctional, brain-penetrant dopamine D2/D3 agonist D-520: Potential therapeutic application in Parkinson’s disease with dementia. Sci. Rep. 2019, 9, 19648. [Google Scholar] [CrossRef] [Green Version]
- Elmabruk, A.; Das, B.; Yedlapudi, D.; Xu, L.; Antonio, T.; Reith, M.E.A.; Dutta, A.K. Design, Synthesis, and Pharmacological Characterization of Carbazole Based Dopamine Agonists as Potential Symptomatic and Neuroprotective Therapeutic Agents for Parkinson’s Disease. ACS Chem. Neurosci. 2019, 10, 396–411. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, B.; Antonio, T.; Reith, M.E.; Dutta, A.K. Discovery of 4-(4-(2-((5-Hydroxy-1,2,3,4-tetrahydronaphthalen-2-yl)(propyl)amino)ethyl)piperazin-1-yl)quinolin-8-ol and its analogues as highly potent dopamine D2/D3 agonists and as iron chelator: In vivo activity indicates potential application in symptomatic and neuroprotective therapy for Parkinson’s disease. J. Med. Chem. 2010, 53, 2114–2125. [Google Scholar] [CrossRef] [PubMed]
- Lewis, F.W.; Fairooz, S.; Elson, J.L.; Hubscher-Bruder, V.; Brandel, J.; Soundararajan, M.; Smith, D.; Dexter, D.T.; Tétard, D.; Pienaar, I.S. Novel 1-hydroxypyridin-2-one metal chelators prevent and rescue ubiquitin proteasomal-related neuronal injury in an in vitro model of Parkinson’s disease. Arch. Toxicol. 2020, 94, 813–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lunić, T.; Lađarević, J.; Mandić, M.; Veruševski, V.; Nedeljković, B.B.; Mijin, D.; Božić, B. Antioxidant and neuroprotective activities of selected 2-pyridones: In vitro and in silico study. J. Mol. Struct. 2022, 1256, 132546. [Google Scholar] [CrossRef]
- Zhou, H.; Shao, M.; Yang, X.; Li, C.; Cui, G.; Gao, C.; Di, L.; Zhong, H.; Wang, Y.; Zhang, Z.; et al. Tetramethylpyrazine Analogue T-006 Exerts Neuroprotective Effects against 6-Hydroxydopamine-Induced Parkinson’s Disease In Vitro and In Vivo. Oxid. Med. Cell Longev. 2019, 2019, 8169125. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Zhao, M.; Wang, B.; Su, Z.; Guo, B.; Qin, L.; Zhang, W.; Zheng, R. The Nrf2-NLRP3-caspase-1 axis mediates the neuroprotective effects of Celastrol in Parkinson’s disease. Redox Biol. 2021, 47, 102134. [Google Scholar] [CrossRef]
- Wang, X.; Cao, G.; Ding, D.; Li, F.; Zhao, X.; Wang, J.; Yang, Y. Ferruginol prevents degeneration of dopaminergic neurons by enhancing clearance of α-synuclein in neuronal cells. Fitoterapia 2022, 156, 105066. [Google Scholar] [CrossRef]
- Miao, Q.; Chai, Z.; Song, L.J.; Wang, Q.; Song, G.B.; Wang, J.; Yu, J.Z.; Xiao, B.G.; Ma, C.G. The neuroprotective effects and transdifferentiation of astrocytes into dopaminergic neurons of Ginkgolide K on Parkinson’ disease mice. J. Neuroimmunol. 2022, 364, 577806. [Google Scholar] [CrossRef]
- Moyano, P.; Vicente-Zurdo, D.; Blázquez-Barbadillo, C.; Menéndez, J.C.; González, J.F.; Rosales-Conrado, N.; Del Pino, J. Neuroprotective Action of Multitarget 7-Aminophenanthridin-6(5H)-one Derivatives against Metal-Induced Cell Death and Oxidative Stress in SN56 Cells. ACS Chem. Neurosci. 2021, 12, 3358–3372. [Google Scholar] [CrossRef]
- Liu, Z.; Cai, W.; Lang, M.; Yan, R.; Li, Z.; Zhang, G.; Yu, P.; Wang, Y.; Sun, Y.; Zhang, Z. Neuroprotective Effects and Mechanisms of Action of Multifunctional Agents Targeting Free Radicals, Monoamine Oxidase B and Cholinesterase in Parkinson’s Disease Model. J. Mol. Neurosci. 2017, 61, 498–510. [Google Scholar] [CrossRef]
- Xia, N.; Daiber, A.; Förstermann, U.; Li, H. Antioxidant effects of resveratrol in the cardiovascular system. Br. J. Pharmacol. 2017, 174, 1633–1646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griñán-Ferré, C.; Bellver-Sanchis, A.; Izquierdo, V.; Corpas, R.; Roig-Soriano, J.; Chillón, M.; Andres-Lacueva, C.; Somogyvári, M.; Sőti, C.; Sanfeliu, C.; et al. The pleiotropic neuroprotective effects of resveratrol in cognitive decline and Alzheimer’s disease pathology: From antioxidant to epigenetic therapy. Ageing Res. Rev. 2021, 67, 101271. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.D.; Luo, M.; Huang, S.Y.; Saimaiti, A.; Shang, A.; Gan, R.Y.; Li, H.B. Effects and Mechanisms of Resveratrol on Aging and Age-Related Diseases. Oxid. Med. Cell Longev. 2021, 2021, 9932218. [Google Scholar] [CrossRef] [PubMed]
- Moussa, C.; Hebron, M.; Huang, X.; Ahn, J.; Rissman, R.A.; Aisen, P.S.; Turner, R.S. Resveratrol regulates neuro-inflammation and induces adaptive immunity in Alzheimer’s disease. J. Neuroinflamm. 2017, 14, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Huang, N.; Xu, S.; Luo, Y.; Li, Y.; Jin, H.; Yu, C.; Shi, J.; Jin, F. Signaling mechanisms underlying inhibition of neuroinflammation by resveratrol in neurodegenerative diseases. J. Nutr. Biochem. 2021, 88, 108552. [Google Scholar] [CrossRef]
- Renaud, J.; Martinoli, M.G. Resveratrol as a protective molecule for neuroinflammation: A review of mechanisms. Curr. Pharm. Biotechnol. 2014, 15, 318–329. [Google Scholar] [CrossRef]
- Wang, P.; Jiang, L.; Zhou, N.; Zhou, H.; Liu, H.; Zhao, W.; Zhang, H.; Zhang, X.; Hu, Z. Resveratrol ameliorates autophagic flux to promote functional recovery in rats after spinal cord injury. Oncotarget 2018, 9, 8427–8440. [Google Scholar] [CrossRef] [Green Version]
- Molaei, A.; Molaei, E.; Sadeghnia, H.; Hayes, A.W.; Karimi, G. LKB1: An emerging therapeutic target for cardiovascular diseases. Life Sci. 2022, 306, 120844. [Google Scholar] [CrossRef]
- Lin, H.Y.; Tang, H.Y.; Davis, F.B.; Davis, P.J. Resveratrol and apoptosis. Ann. N. Y. Acad. Sci. 2011, 1215, 79–88. [Google Scholar] [CrossRef]
- Repossi, G.; Das, U.N.; Eynard, A.R. Molecular Basis of the Beneficial Actions of Resveratrol. Arch. Med. Res. 2020, 51, 105–114. [Google Scholar] [CrossRef]
- Surkov, S.A.; Mingazov, E.R.; Blokhin, V.E.; Sturova, A.I.; Gretskaya, N.M.; Zinchenko, G.N.; Bezuglov, V.V.; Ugrumov, M.V. The Neuroprotective Effect of N-Docosahexaenoyldopamine on Degenerating Dopaminergic Neurons of the Mesencephalon. Biol. Bull. 2020, 47, 466–473. [Google Scholar] [CrossRef]
- Beg, M.S.; Ahmad, S.; Jan, K.; Bashir, K. Status, supply chain and processing of cocoa—A review. Trends Food Sci. Technol. 2017, 66, 108–116. [Google Scholar] [CrossRef]
- Yuan, S.; Li, X.; Jin, Y.; Lu, J. Chocolate Consumption and Risk of Coronary Heart Disease, Stroke, and Diabetes: A Meta-Analysis of Prospective Studies. Nutrients 2017, 9, 688. [Google Scholar] [CrossRef] [Green Version]
- De Araujo, Q.R.; Gattward, J.N.; Almoosawi, S.; Silva, M.; Dantas, P.A.; De Araujo Júnior, Q.R. Cocoa and Human Health: From Head to Foot—A Review. Crit. Rev. Food Sci. Nutr. 2016, 56, 1–12. [Google Scholar] [CrossRef]
- Liang, F.; Cao, W.; Huang, Y.; Fang, Y.; Cheng, Y.; Pan, S.; Xu, X. Isoflavone biochanin A, a novel nuclear factor erythroid 2-related factor 2 (Nrf2)-antioxidant response element activator, protects against oxidative damage in HepG2 cells. BioFactors 2019, 45, 563–574. [Google Scholar] [CrossRef]
- Jalaludeen, A.M.; Ha, W.T.; Lee, R.; Kim, J.H.; Do, J.T.; Park, C.; Heo, Y.T.; Lee, W.Y.; Song, H. Biochanin A Ameliorates Arsenic-Induced Hepato- and Hematotoxicity in Rats. Molecules 2016, 21, 69. [Google Scholar] [CrossRef] [Green Version]
- Kupershmidt, L.; Weinreb, O.; Amit, T.; Mandel, S.; Bar-Am, O.; Youdim, M.B. Novel molecular targets of the neuroprotective/neurorescue multimodal iron chelating drug M30 in the mouse brain. Neuroscience 2011, 189, 345–358. [Google Scholar] [CrossRef]
- Bar-Am, O.; Amit, T.; Kupershmidt, L.; Aluf, Y.; Mechlovich, D.; Kabha, H.; Danovitch, L.; Zurawski, V.R.; Youdim, M.B.; Weinreb, O. Neuroprotective and neurorestorative activities of a novel iron chelator-brain selective monoamine oxidase-A/monoamine oxidase-B inhibitor in animal models of Parkinson’s disease and aging. Neurobiol. Aging 2015, 36, 1529–1542. [Google Scholar] [CrossRef]
- Kupershmidt, L.; Amit, T.; Bar-Am, O.; Youdim, M.B.; Weinreb, O. Neuroprotection by the multitarget iron chelator M30 on age-related alterations in mice. Mech. Ageing Dev. 2012, 133, 267–274. [Google Scholar] [CrossRef]
- Mechlovich, D.; Amit, T.; Bar-Am, O.; Mandel, S.; Youdim, M.B.; Weinreb, O. The novel multi-target iron chelator, M30 modulates HIF-1α-related glycolytic genes and insulin signaling pathway in the frontal cortex of APP/PS1 Alzheimer’s disease mice. Curr. Alzheimer. Res. 2014, 11, 119–127. [Google Scholar] [CrossRef]
- Mechlovich, D.; Amit, T.; Bar-Am, O.; Weinreb, O.; Youdim, M.B. Molecular targets of the multifunctional iron-chelating drug, M30, in the brains of mouse models of type 2 diabetes mellitus. Br. J. Pharmacol. 2014, 171, 5636–5649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Youdim, M.B.; Buccafusco, J.J. Multi-functional drugs for various CNS targets in the treatment of neurodegenerative disorders. Trends Pharmacol. Sci. 2005, 26, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Mandel, S.; Weinreb, O.; Amit, T.; Youdim, M.B. Mechanism of neuroprotective action of the anti-Parkinson drug rasagiline and its derivatives. Brain Res. Brain Res. Rev. 2005, 48, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Youdim, M.B.; Fridkin, M.; Zheng, H. Bifunctional drug derivatives of MAO-B inhibitor rasagiline and iron chelator VK-28 as a more effective approach to treatment of brain ageing and ageing neurodegenerative diseases. Mech. Ageing Dev. 2005, 126, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Das, B.; Kandegedara, A.; Xu, L.; Antonio, T.; Stemmler, T.; Reith, M.E.A.; Dutta, A.K. A Novel Iron(II) Preferring Dopamine Agonist Chelator as Potential Symptomatic and Neuroprotective Therapeutic Agent for Parkinson’s Disease. ACS Chem. Neurosci. 2017, 8, 723–730. [Google Scholar] [CrossRef]
- Frogley, B.J.; Hill, A.F. Metal coordination to bipyridyl carbynes. Dalton Trans. 2020, 49, 3272–3283. [Google Scholar] [CrossRef]
- Hales, N.J.; Beattie, J.F. Novel inhibitors of prolyl 4-hydroxylase. 5. The intriguing structure-activity relationships seen with 2,2’-bipyridine and its 5,5’-dicarboxylic acid derivatives. J. Med. Chem. 1993, 36, 3853–3858. [Google Scholar] [CrossRef]
- Lindenbach, D.; Das, B.; Conti, M.M.; Meadows, S.M.; Dutta, A.K.; Bishop, C. D-512, a novel dopamine D2/3 receptor agonist, demonstrates greater anti-Parkinsonian efficacy than ropinirole in Parkinsonian rats. Br. J. Pharmacol. 2017, 174, 3058–3071. [Google Scholar] [CrossRef] [Green Version]
- Hider, R.C.; Hoffbrand, A.V. The Role of Deferiprone in Iron Chelation. N. Engl. J. Med. 2018, 379, 2140–2150. [Google Scholar] [CrossRef] [Green Version]
- Gyparaki, M.; Porter, J.B.; Hirani, S.; Streater, M.; Hider, R.C.; Huehns, E.R. In vivo evaluation of hydroxypyridone iron chelators in a mouse model. Acta Haematol. 1987, 78, 217–221. [Google Scholar] [CrossRef]
- Lewis, F.W.; Bird, K.; Navarro, J.P.; El Fallah, R.; Brandel, J.; Hubscher-Bruder, V.; Tsatsanis, A.; Duce, J.A.; Tétard, D.; Bourne, S.; et al. Synthesis, physicochemical characterization and neuroprotective evaluation of novel 1-hydroxypyrazin-2(1H)-one iron chelators in an in vitro cell model of Parkinson’s disease. Dalton Trans. 2022, 51, 3590–3603. [Google Scholar] [CrossRef]
- Chen, H.; Cao, J.; Zha, L.; Wang, P.; Liu, Z.; Guo, B.; Zhang, G.; Sun, Y.; Zhang, Z.; Wang, Y. Neuroprotective and neurogenic effects of novel tetramethylpyrazine derivative T-006 in Parkinson’s disease models through activating the MEF2-PGC1α and BDNF/CREB pathways. Aging 2020, 12, 14897–14917. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Daina, A.; Zoete, V. A BOILED-Egg To Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules. ChemMedChem 2016, 11, 1117–1121. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Active Chemical Groups | Metal Chelation | ROS Neutralization | Dopamine Metabolism # | Mitochondria Tropism | Anti-Inflammation | In Vivo Effectiveness | Reference |
|---|---|---|---|---|---|---|---|---|
| Resveratrol | Phenol | Not tested | Yes | Not tested | Not tested | Yes | Yes | [164] |
| Magnolol | Phenol | Not tested | Yes | No | No | Yes | Yes | [165] |
| Cinnamoyl-N-Acylhydrazone-Donepezil Hybrids (PQM263) | Catechol and | No tested | Yes | Not tested | Not tested | Not tested | Not tested | [166] |
| Clioquinol-selegiline hybrids (compound 8a) | amino-hydroxyl | Cu2+; Fe2+; Zn2+ | Yes | Yes | Not tested | Not tested | Yes | [167] |
| (E)-Hydroxystyryl Aralkyl Sulfones | Catechol | Not tested | Yes | Yes | Not tested | Yes | Not tested | [168] |
| N-Docosahexaenoyl Dopamine | Cathecol | Not tested | Yes | Yes | Not tested | Yes | Not tested | [169] |
| Ginkgetin | Enol and phenol | Fe2+ | Yes | Not tested | Not tested | Yes | Yes | [170] |
| Biochanin A | Phenol | Not tested | Yes | Yes | Not tested | Yes | Yes | [171] |
| Epigallocatechin gallate (EGCG) | Catechol | Cu2+; Fe3+; Al3+; Mn2+ | Yes | No | Not tested | Not tested | Yes | [172] |
| 7,8-Dihydroxycoumarin derivative (DHC12) | Catechol | Cu2+∼ Fe2+ > Zn2+ > Fe3+ | Yes | Yes | Yes | Not tested | Yes | [173] |
| Coumarin–tris hybrid (CT51) | 2-amino-2-(hydroxymethyl)propane-1,3-diol (TRIS) and carbonyls | Fe2+ > Fe3+ | Yes | Not tested | Yes | Not tested | Not tested | [174] |
| Coumarin Mannich base derivatives | No Present | Not tested | No | Yes | Not tested | Yes | Yes | [175] |
| Hydroxyquinoline-propargyl hybrids (M30) | quinolin-8-ol | Fe3+ > Cu2+ > Zn2+(19b) | No | Yes | Not tested | Not tested | Yes | [176] |
| Hydroxyquinoline-propargyl piperazine hybrids (HLA20) | quinolin-8-ol | Fe3+ > Cu2+ | Yes | Yes | Not tested | Not tested | Yes | [177] |
| 2-heptyl-3-hydroxy-4-quinolone | 3-hydroxy-4-quinolone | Not tested | Yes | Not tested | Not tested | Not tested | Not tested | [178] |
| Dipyridyl-D2R/D3R agonist hybrids (D-607) | 2,2′-Bipyridyl | Fe2+ >>> Fe3+ | No | Yes | Not tested | Not tested | Yes | [179] |
| Cathecol-D2/D3 agonist (D-520) | Catechol | No tested | No | Yes | Not tested | Not tested | Yes | [180] |
| Carbazole- D2/D3 agonist hybrids (D-653) | No present | No tested | Yes | Yes | Not tested | Not tested | Yes | [181] |
| Piperazine–8-OH-quinolone hybrids (19b) | quinolin-8-ol | Fe2+; Fe3+ | No | No | Not tested | Not tested | Yes | [182] |
| 1-hydroxypyrazin-2(1H)-one (1,2-HOPY) | N-hydroxy-N-methylacetamide | Fe3+ | Not tested | Yes | Not tested | Not tested | Not tested | [183] |
| 2-pyridones derivatives | 2-pyridones | Not tested | Yes | Not tested | Not tested | Yes | Not tested | [184] |
| Tetramethylpyrazine (T-006) | hydrazineylidene-pyrazine | Not tested | Yes | Yes | Not tested | Not tested | Yes | [185] |
| Celastrol | 2-hydroxycyclohexanone | Not tested | Yes | Yes | Not tested | Yes | Yes | [186] |
| Ferruginol | Phenol | Not tested | Not tested | Yes | Not tested | Not tested | Yes | [187] |
| Ginkgolide K | Ascorbic Acid Derivatives | Not tested | Yes | Yes | Not tested | Yes | Yes | [188] |
| 7-Aminophenanthridin- 6(5H)-one (APH-4) | 8-aminoisoquinolin-1(2H)-one | Fe2+, Cu2+ | Yes | Not tested | Not tested | Not tested | Not tested | [189] |
| Ladostigil derivative (MT-20R) | No | Not tested | No | Yes | Not tested | Not tested | Yes | [190] |
| Compound | MW | RB | HBA | HBD | MR | TPSA | Log P | Lipinski Violations | BBB Permeability | |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Resveratrol | 221.21 | 2 | 5 | 3 | 59.10 | 82.70 | 0.80 | 0 | Yes |
| 2 | Magnolol | 281.35 | 3 | 4 | 1 | 92.05 | 39.60 | 1.90 | 0 | Yes |
| 3 | PM263 | 310.75 | 4 | 5 | 1 | 82.84 | 70.75 | 1.23 | 0 | Yes |
| 4 | Compound 8a | 286.45 | 1 | 1 | 1 | 91.63 | 20.23 | 5.26 | 1 | No |
| 5 | Compound 5h | 367.44 | 10 | 5 | 2 | 107.96 | 74.16 | 2.84 | 0 | Yes |
| 6 | N-Docosahexa-enoyl dopamine | 373.49 | 9 | 4 | 0 | 112.18 | 61.53 | 2.95 | 0 | Yes |
| 7 | Ginkgetin | 448.64 | 7 | 4 | 1 | 143.84 | 42.84 | 4.08 | 0 | Yes |
| 8 | Biochanin A | 266.33 | 5 | 2 | 2 | 84.14 | 40.46 | 4.25 | 0 | Yes |
| 9 | EGCG | 428.6 | 2 | 4 | 3 | 126.90 | 66.76 | 5.29 | 0 | No |
| 10 | DHC12 | 382.41 | 4 | 3 | 2 | 115.81 | 67.26 | 4.43 | 0 | No |
| 11 | CT51 | 406.38 | 1 | 9 | 2 | 91.62 | 128.59 | 0.58 | 0 | No |
| 12 | Coumarin 24 | 323.3 | 7 | 7 | 5 | 80.03 | 140.23 | -0.31 | 0 | Yes |
| 13 | M30 | 482.66 | 7 | 3 | 2 | 157.97 | 45.74 | 4.89 | 0 | Yes |
| 14 | HLA20 | 228.24 | 2 | 3 | 3 | 67.88 | 60.69 | 2.48 | 0 | Yes |
| 15 | 2-heptyl-3-hydroxyquinolin-4(1H)-one | 318.39 | 6 | 4 | 2 | 88.45 | 82.98 | 3.00 | 0 | No |
| 16 | D-607 | 501.66 | 8 | 5 | 3 | 158.09 | 70.41 | 4.29 | 1 | Yes |
| 17 | D-520 | 449.62 | 17 | 3 | 3 | 141.37 | 69.56 | 4.29 | 1 | No |
| 18 | D-653 | 566.51 | 5 | 10 | 4 | 155.91 | 159.80 | 4.34 | 1 | No |
| 19 | 19b | 364.36 | 5 | 7 | 0 | 94.44 | 58.45 | 3.94 | 0 | Yes |
| 20 | 1,2-HOPY | 284.26 | 2 | 5 | 2 | 78.46 | 79.90 | 2.44 | 0 | Yes |
| 21 | 2-Pyridones derivatives | 150.13 | 0 | 3 | 2 | 38.77 | 76.88 | 0.45 | 0 | No |
| 22 | T-006 | 477.67 | 8 | 5 | 1 | 147.69 | 102.65 | 3.16 | 0 | No |
| 23 | Celastrol | 259.34 | 6 | 2 | 2 | 80.40 | 53.09 | 3.70 | 0 | Yes |
| 24 | Ferruginol | 458.37 | 4 | 11 | 8 | 112.06 | 197.37 | 1.01 | 1 | No |
| 25 | Ginkgolide K | 112.09 | 0 | 3 | 1 | 26.04 | 55.12 | -0.35 | 0 | No |
| 26 | APH-4 | 308.42 | 7 | 2 | 2 | 97.12 | 35.50 | 3.58 | 0 | Yes |
| 27 | MT-20R | 226.27 | 3 | 3 | 1 | 69.02 | 36.36 | 2.07 | 0 | No |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-Beltrán, O.; Urrutia, P.J.; Núñez, M.T. On the Chemical and Biological Characteristics of Multifunctional Compounds for the Treatment of Parkinson’s Disease. Antioxidants 2023, 12, 214. https://doi.org/10.3390/antiox12020214
García-Beltrán O, Urrutia PJ, Núñez MT. On the Chemical and Biological Characteristics of Multifunctional Compounds for the Treatment of Parkinson’s Disease. Antioxidants. 2023; 12(2):214. https://doi.org/10.3390/antiox12020214
Chicago/Turabian StyleGarcía-Beltrán, Olimpo, Pamela J. Urrutia, and Marco T. Núñez. 2023. "On the Chemical and Biological Characteristics of Multifunctional Compounds for the Treatment of Parkinson’s Disease" Antioxidants 12, no. 2: 214. https://doi.org/10.3390/antiox12020214
APA StyleGarcía-Beltrán, O., Urrutia, P. J., & Núñez, M. T. (2023). On the Chemical and Biological Characteristics of Multifunctional Compounds for the Treatment of Parkinson’s Disease. Antioxidants, 12(2), 214. https://doi.org/10.3390/antiox12020214