
Stimulation of Hepatic Ferritinophagy Mitigates Irp2 Depletion-Induced Anemia


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Tissue Collection
2.2. Drug Treatment
2.3. Routine Blood Examinations
2.4. Blood Biochemical Examinations
2.5. Western Blot Analysis
2.6. Quantitative Real-Time PCR
2.7. Histological Staining
2.8. Ferrozine Assays
2.9. ELISA
2.10. Ferritin Iron Staining in Gels
2.11. Statistical Analysis
3. Results
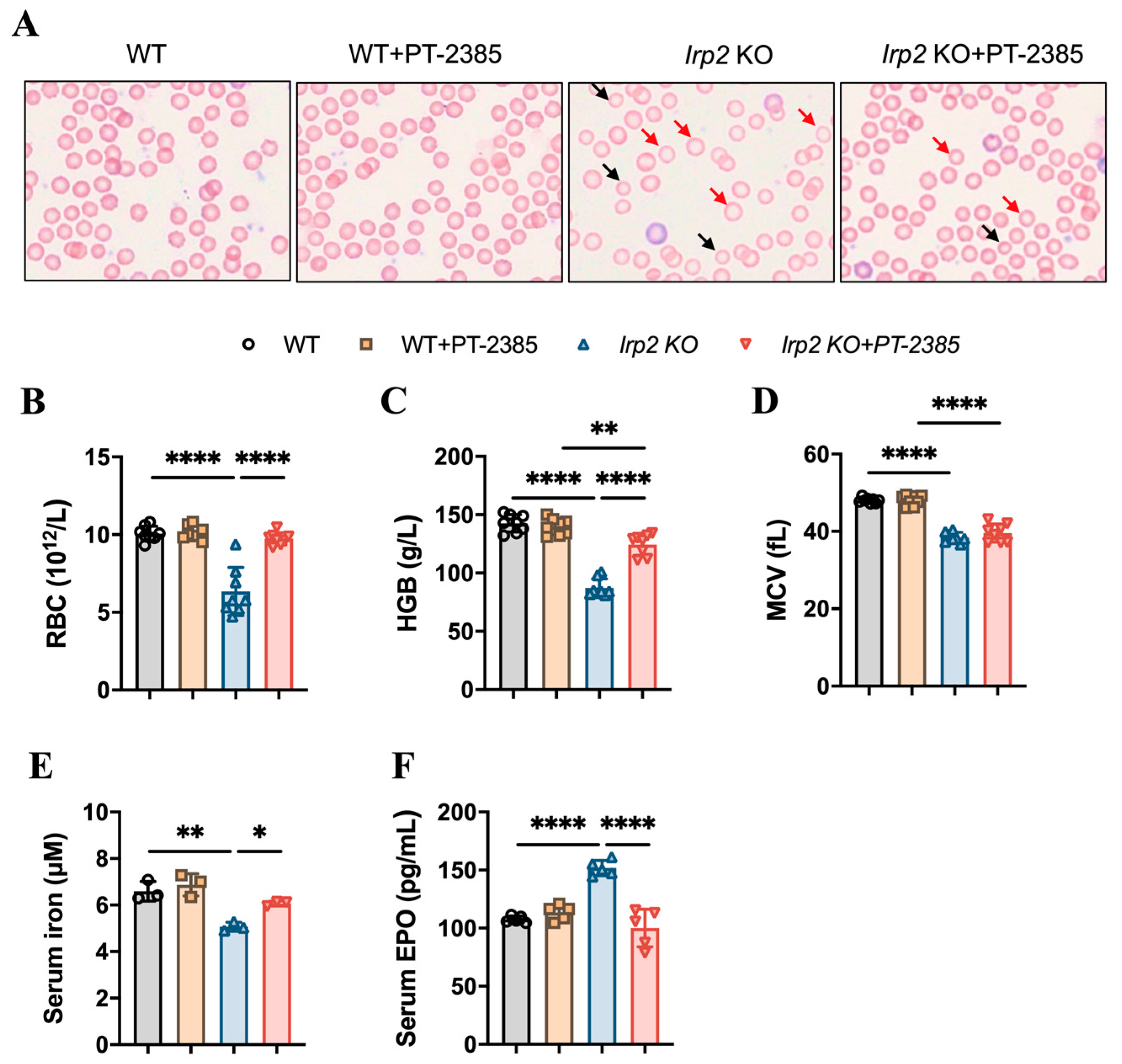
3.1. Hif2 Inhibition Ameliorates Anemia in Irp2 Knockout Mice
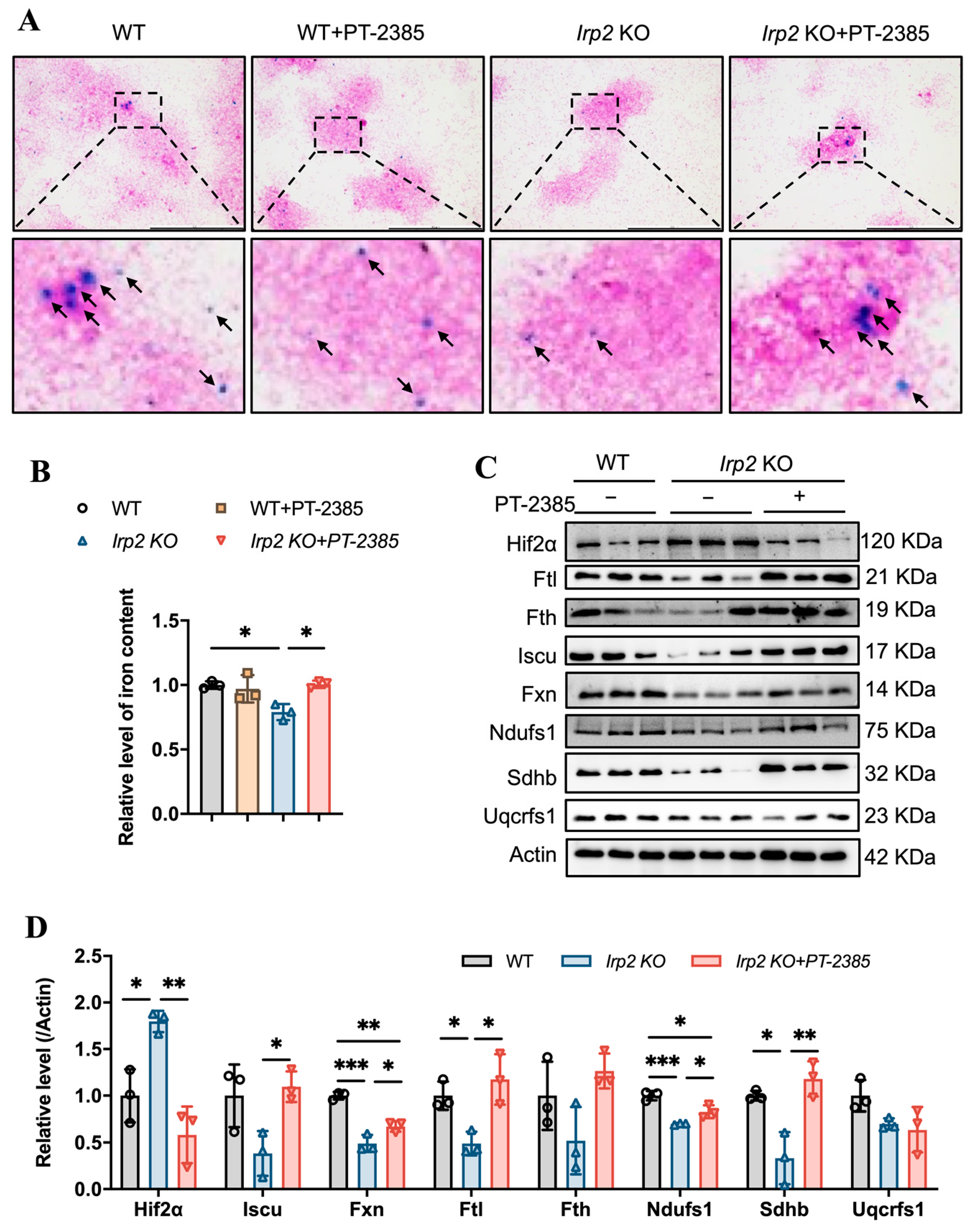
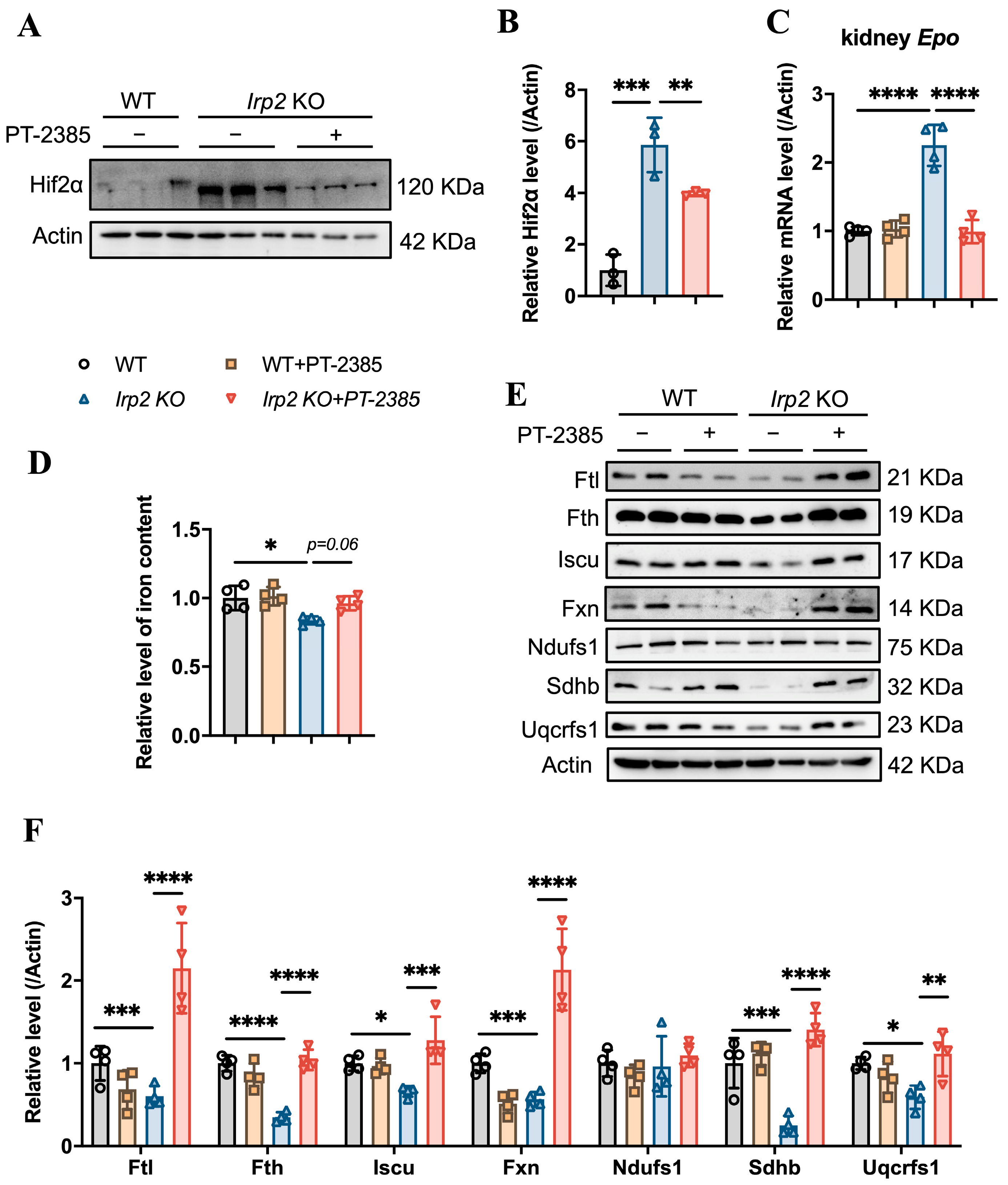
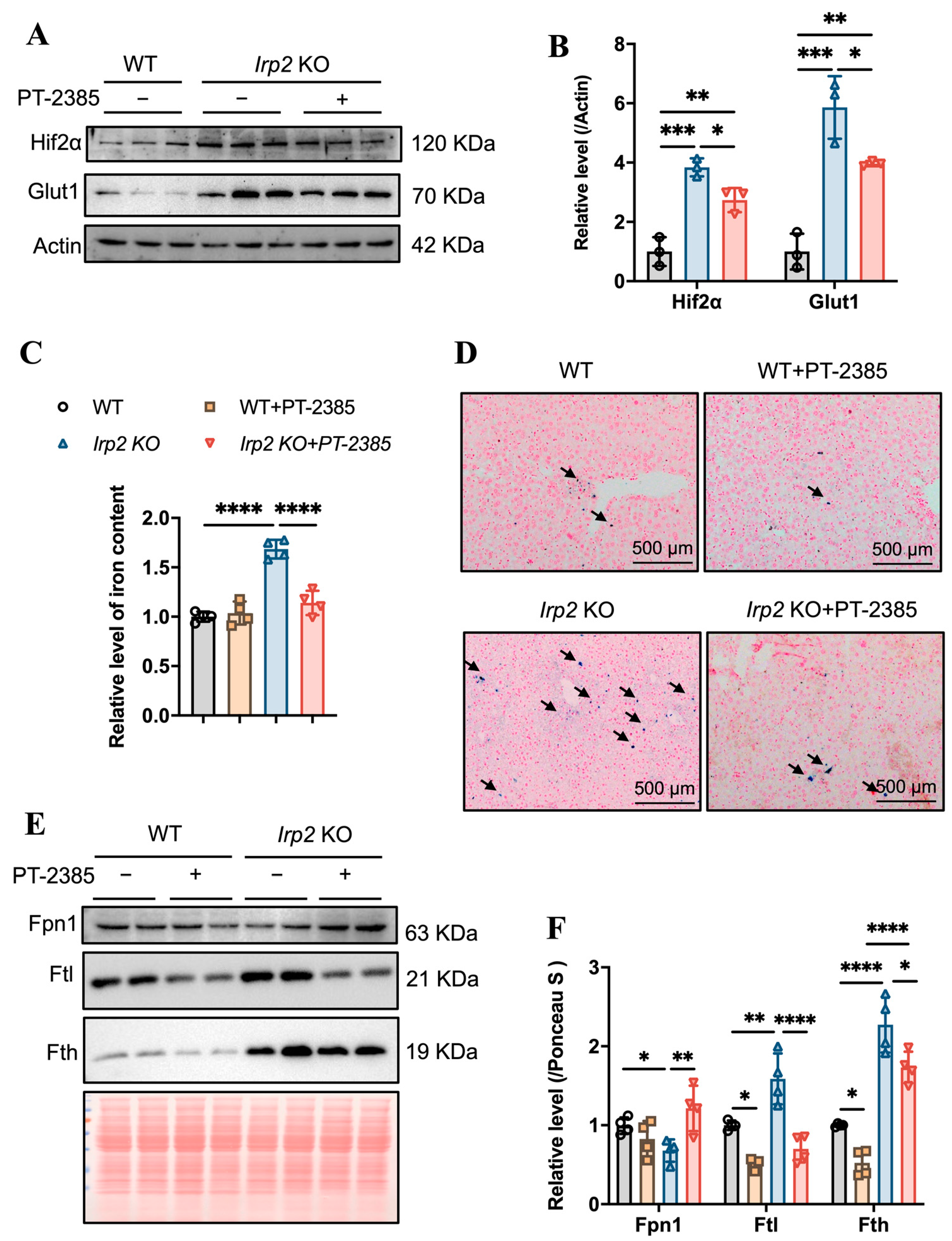
3.2. Enhanced Fe-S Cluster Biogenesis by Hif2 Inhibition Rescues Iron Deficiency to Recover Hematopoietic Function in Irp2 Knockout Mice
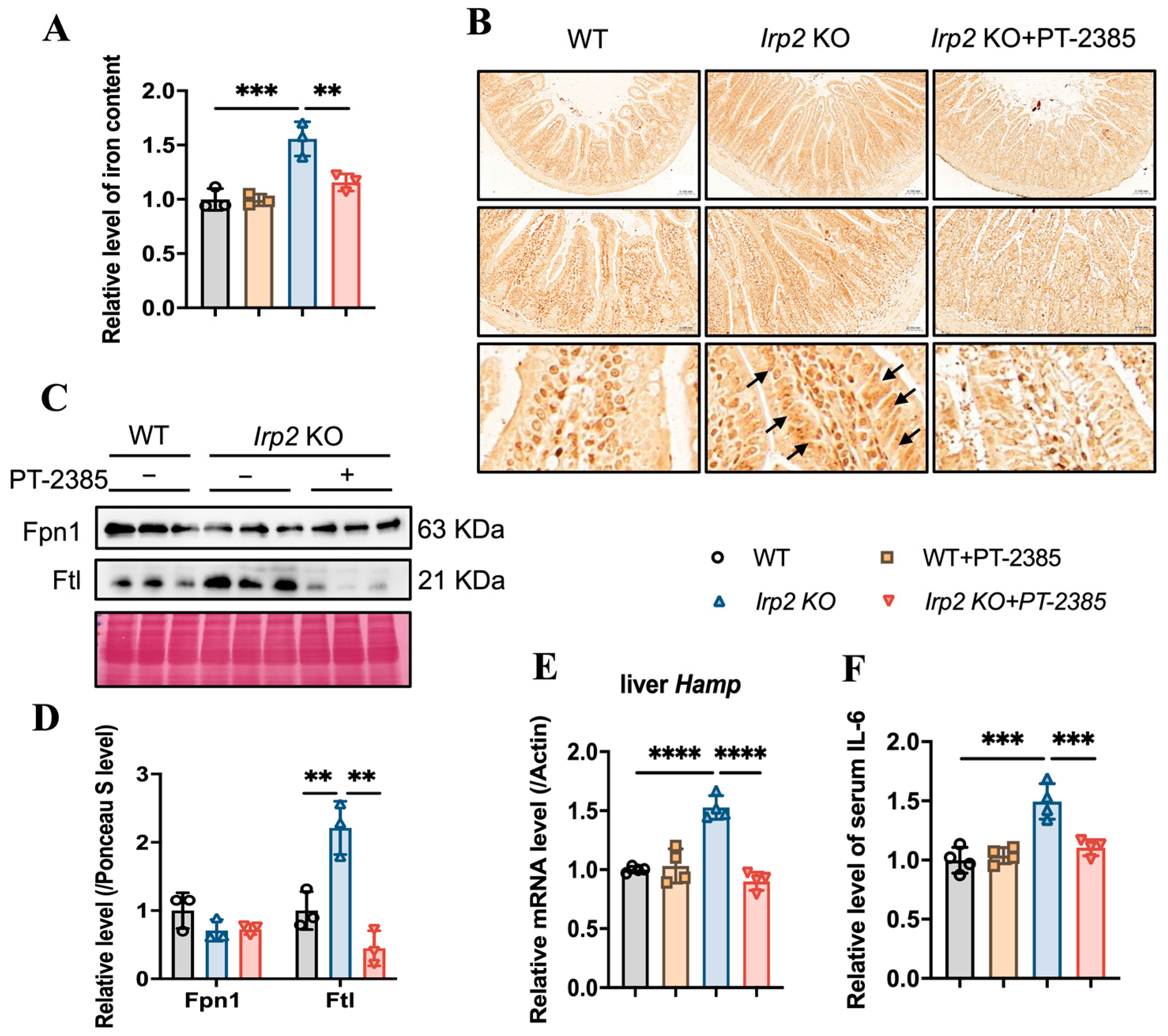
3.3. PT–2385 Treatment Modulated Intestinal Iron Absorption and Hepatic Iron Release in Irp2 KO Mice
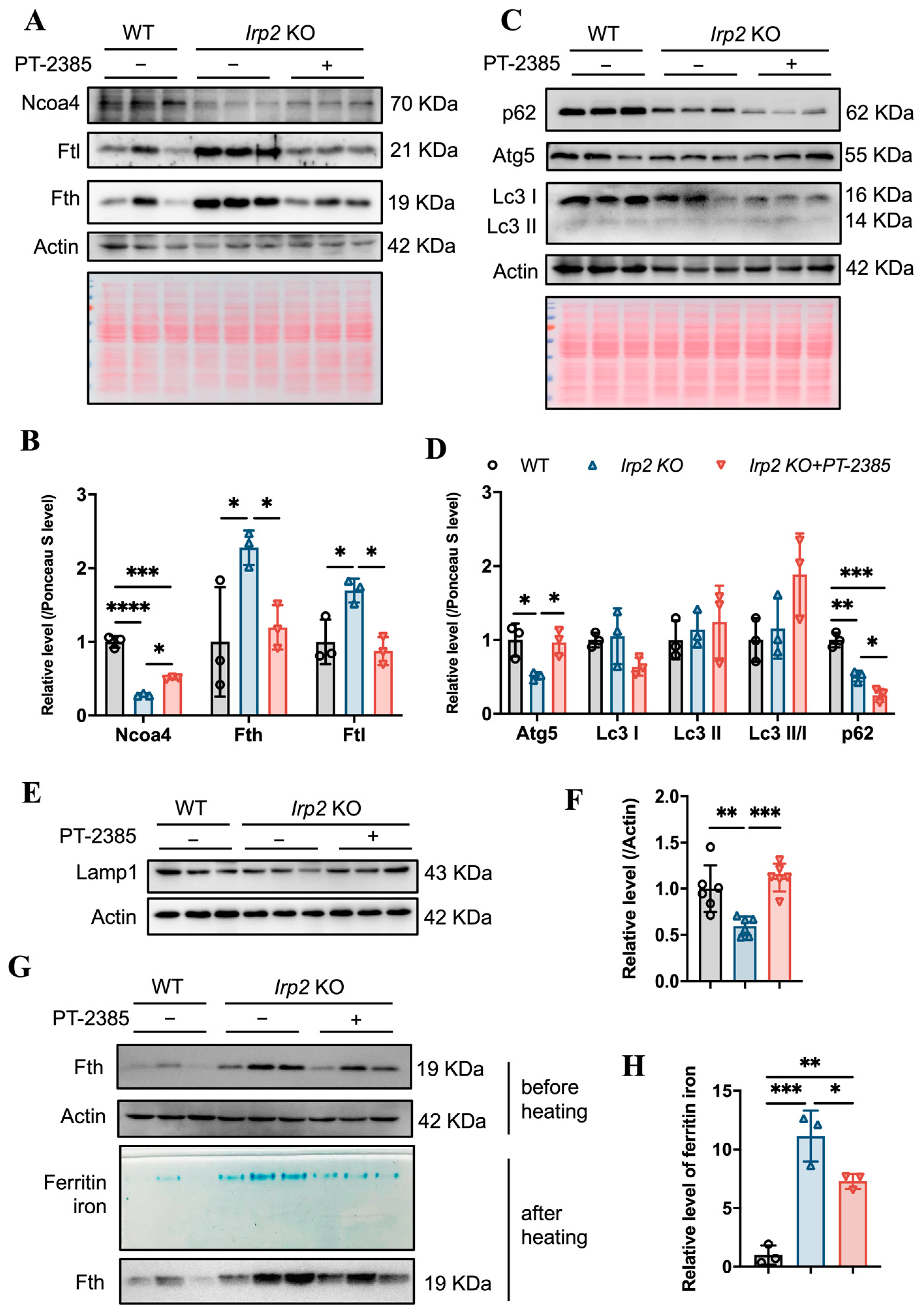
3.4. Hif2 Inhibition Reconstitutes Tissue Iron Homeostasis by Initiating Ncoa4-Mediated Ferritinophagy in Liver
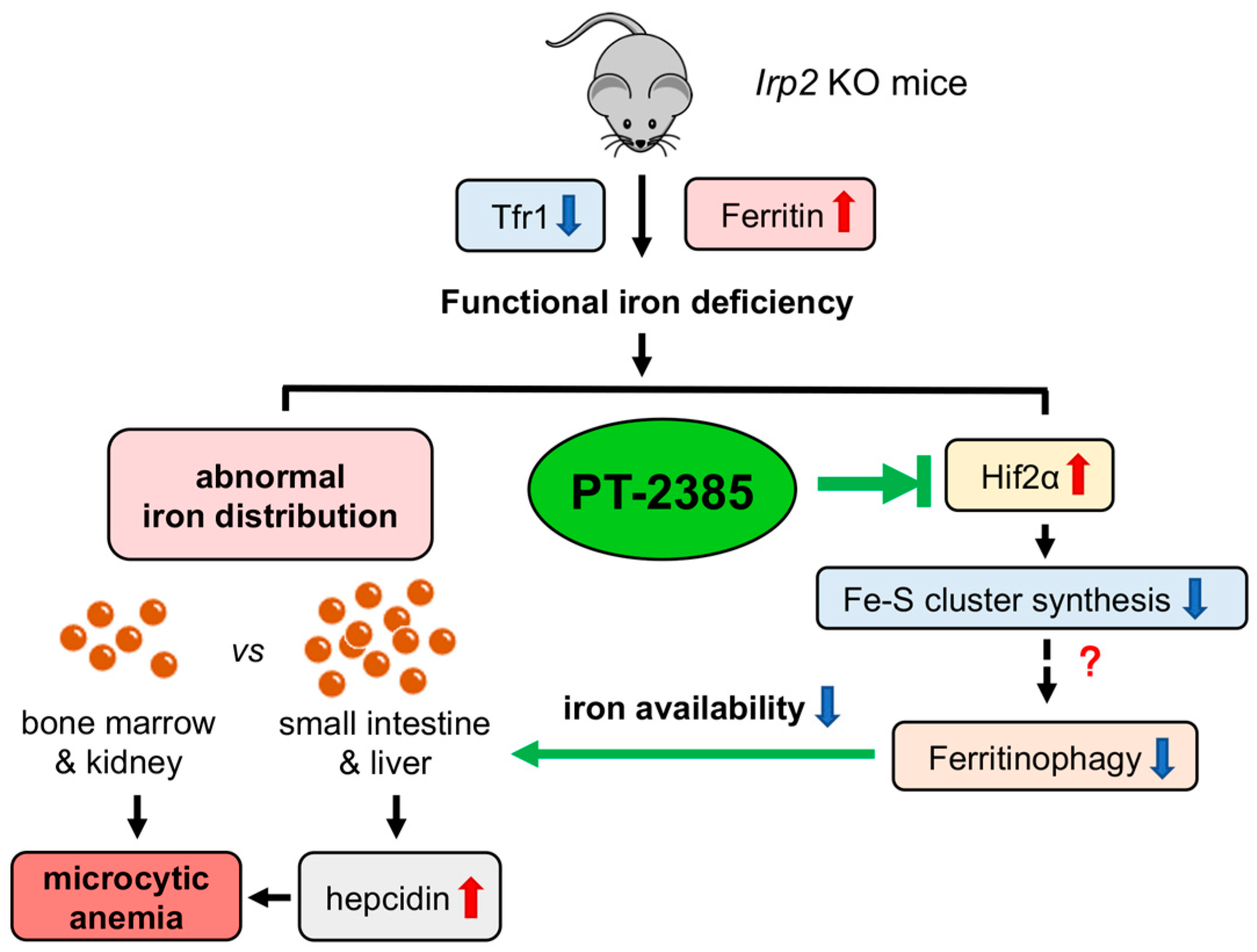
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Muckenthaler, M.U.; Rivella, S.; Hentze, M.W.; Galy, B. A Red Carpet for Iron Metabolism. Cell 2017, 168, 344–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maio, N.; Rouault, T.A. Outlining the Complex Pathway of Mammalian Fe-S Cluster Biogenesis. Trends Biochem. Sci. 2020, 45, 411–426. [Google Scholar] [CrossRef] [PubMed]
- Paul, V.D.; Lill, R. Biogenesis of cytosolic and nuclear iron-sulfur proteins and their role in genome stability. Biochim. Biophys. Acta 2015, 1853, 1528–1539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrews, N.C.; Schmidt, P.J. Iron homeostasis. Annu. Rev. Physiol. 2007, 69, 69–85. [Google Scholar] [CrossRef] [PubMed]
- Rouault, T.A. The role of iron regulatory proteins in mammalian iron homeostasis and disease. Nat. Chem. Biol. 2006, 2, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Hentze, M.W.; Muckenthaler, M.U.; Galy, B.; Camaschella, C. Two to tango: Regulation of Mammalian iron metabolism. Cell 2010, 142, 24–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallander, M.L.; Leibold, E.A.; Eisenstein, R.S. Molecular control of vertebrate iron homeostasis by iron regulatory proteins. Biochim. Biophys. Acta 2006, 1763, 668–689. [Google Scholar] [CrossRef] [Green Version]
- Salahudeen, A.A.; Thompson, J.W.; Ruiz, J.C.; Ma, H.W.; Kinch, L.N.; Li, Q.; Grishin, N.V.; Bruick, R.K. An E3 ligase possessing an iron-responsive hemerythrin domain is a regulator of iron homeostasis. Science 2009, 326, 722–726. [Google Scholar] [CrossRef] [Green Version]
- Vashisht, A.A.; Zumbrennen, K.B.; Huang, X.; Powers, D.N.; Durazo, A.; Sun, D.; Bhaskaran, N.; Persson, A.; Uhlen, M.; Sangfelt, O.; et al. Control of iron homeostasis by an iron-regulated ubiquitin ligase. Science 2009, 326, 718–721. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Shi, H.; Rajan, M.; Canarie, E.R.; Hong, S.; Simoneschi, D.; Pagano, M.; Bush, M.F.; Stoll, S.; Leibold, E.A.; et al. FBXL5 Regulates IRP2 Stability in Iron Homeostasis via an Oxygen-Responsive [2Fe2S] Cluster. Mol. Cell 2020, 78, 31–41.e5. [Google Scholar] [CrossRef]
- Cooperman, S.S.; Meyron-Holtz, E.G.; Olivierre-Wilson, H.; Ghosh, M.C.; McConnell, J.P.; Rouault, T.A. Microcytic anemia, erythropoietic protoporphyria, and neurodegeneration in mice with targeted deletion of iron-regulatory protein 2. Blood 2005, 106, 1084–1091. [Google Scholar] [CrossRef] [Green Version]
- LaVaute, T.; Smith, S.; Cooperman, S.; Iwai, K.; Land, W.; Meyron-Holtz, E.; Drake, S.K.; Miller, G.; Abu-Asab, M.; Tsokos, M.; et al. Targeted deletion of the gene encoding iron regulatory protein-2 causes misregulation of iron metabolism and neurodegenerative disease in mice. Nat. Genet. 2001, 27, 209–214. [Google Scholar] [CrossRef]
- Zhou, Y.; Wu, W.; Xu, Z.; Liu, Y.; Chang, H.; Yu, P.; Zhang, X.; Yang, Y.; Liu, K.; Chang, Y. Iron regulatory protein 2 deficiency may correlate with insulin resistance. Biochem. Biophys. Res. Commun. 2019, 510, 191–197. [Google Scholar] [CrossRef]
- Santos, M.; Anderson, C.P.; Neschen, S.; Zumbrennen-Bullough, K.B.; Romney, S.J.; Kahle-Stephan, M.; Rathkolb, B.; Gailus-Durner, V.; Fuchs, H.; Wolf, E.; et al. Irp2 regulates insulin production through iron-mediated Cdkal1-catalyzed tRNA modification. Nat. Commun. 2020, 11, 296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costain, G.; Ghosh, M.C.; Maio, N.; Carnevale, A.; Si, Y.C.; Rouault, T.A.; Yoon, G. Absence of iron-responsive element-binding protein 2 causes a novel neurodegenerative syndrome. Brain 2019, 142, 1195–1202. [Google Scholar] [CrossRef] [PubMed]
- Galy, B.; Ferring, D.; Minana, B.; Bell, O.; Janser, H.G.; Muckenthaler, M.; Schumann, K.; Hentze, M.W. Altered body iron distribution and microcytosis in mice deficient in iron regulatory protein 2 (IRP2). Blood 2005, 106, 2580–2589. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Zhao, H.; Hao, S.; Shang, L.; Wu, J.; Song, C.; Meyron-Holtz, E.G.; Qiao, T.; Li, K. Iron regulatory protein deficiency compromises mitochondrial function in murine embryonic fibroblasts. Sci. Rep. 2018, 8, 5118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Liu, Y.; Shang, L.; Cai, J.; Wu, J.; Zhang, W.; Pu, X.; Dong, W.; Qiao, T.; Li, K. Iron regulatory protein 2 modulates the switch from aerobic glycolysis to oxidative phosphorylation in mouse embryonic fibroblasts. Proc. Natl. Acad. Sci. USA 2019, 116, 9871–9876. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.; Xu, L.; Li, Y.; Dong, W.; Cai, J.; Liu, Y.; Zhao, H.; Xu, T.; Holtz, E.M.; Chang, Y.; et al. Protective Effects of Hif2 Inhibitor PT-2385 on a Neurological Disorder Induced by Deficiency of Irp2. Front. Neurosci. 2021, 15, 715222. [Google Scholar] [CrossRef]
- Liu, G.; Sil, D.; Maio, N.; Tong, W.H.; Bollinger, J.M., Jr.; Krebs, C.; Rouault, T.A. Heme biosynthesis depends on previously unrecognized acquisition of iron-sulfur cofactors in human amino-levulinic acid dehydratase. Nat. Commun. 2020, 11, 6310. [Google Scholar] [CrossRef]
- Dailey, H.A.; Finnegan, M.G.; Johnson, M.K. Human ferrochelatase is an iron-sulfur protein. Biochemistry 1994, 33, 403–407. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Zhou, B. Ferritin is the key to dietary iron absorption and tissue iron detoxification in Drosophila melanogaster. FASEB J. 2013, 27, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Storti, F.; Santambrogio, S.; Crowther, L.M.; Otto, T.; Abreu-Rodriguez, I.; Kaufmann, M.; Hu, C.J.; Dame, C.; Fandrey, J.; Wenger, R.H.; et al. A novel distal upstream hypoxia response element regulating oxygen-dependent erythropoietin gene expression. Haematologica 2014, 99, e45–e48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nemeth, E.; Tuttle, M.S.; Powelson, J.; Vaughn, M.B.; Donovan, A.; Ward, D.M.; Ganz, T.; Kaplan, J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004, 306, 2090–2093. [Google Scholar] [CrossRef] [Green Version]
- Nai, A.; Lidonnici, M.R.; Federico, G.; Pettinato, M.; Olivari, V.; Carrillo, F.; Geninatti Crich, S.; Ferrari, G.; Camaschella, C.; Silvestri, L.; et al. NCOA4-mediated ferritinophagy in macrophages is crucial to sustain erythropoiesis in mice. Haematologica 2021, 106, 795–805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mancias, J.D.; Wang, X.; Gygi, S.P.; Harper, J.W.; Kimmelman, A.C. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 2014, 509, 105–109. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Green, D.R. Autophagy-Independent Functions of the Autophagy Machinery. Cell 2019, 177, 1682–1699. [Google Scholar] [CrossRef]
- McKie, A.T.; Barrow, D.; Latunde-Dada, G.O.; Rolfs, A.; Sager, G.; Mudaly, E.; Mudaly, M.; Richardson, C.; Barlow, D.; Bomford, A.; et al. An iron-regulated ferric reductase associated with the absorption of dietary iron. Science 2001, 291, 1755–1759. [Google Scholar] [CrossRef]
- Gunshin, H.; Mackenzie, B.; Berger, U.V.; Gunshin, Y.; Romero, M.F.; Boron, W.F.; Nussberger, S.; Gollan, J.L.; Hediger, M.A. Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature 1997, 388, 482–488. [Google Scholar] [CrossRef]
- Abboud, S.; Haile, D.J. A novel mammalian iron-regulated protein involved in intracellular iron metabolism. J. Biol. Chem. 2000, 275, 19906–19912. [Google Scholar] [CrossRef] [Green Version]
- Das, N.K.; Schwartz, A.J.; Barthel, G.; Inohara, N.; Liu, Q.; Sankar, A.; Hill, D.R.; Ma, X.; Lamberg, O.; Schnizlein, M.K.; et al. Microbial Metabolite Signaling Is Required for Systemic Iron Homeostasis. Cell Metab. 2020, 31, 115–130.e6. [Google Scholar] [CrossRef]
- Anderson, S.A.; Nizzi, C.P.; Chang, Y.I.; Deck, K.M.; Schmidt, P.J.; Galy, B.; Damnernsawad, A.; Broman, A.T.; Kendziorski, C.; Hentze, M.W.; et al. The IRP1-HIF-2alpha axis coordinates iron and oxygen sensing with erythropoiesis and iron absorption. Cell Metab. 2013, 17, 282–290. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, N.; Pantopoulos, K. IRP1 regulates erythropoiesis and systemic iron homeostasis by controlling HIF2alpha mRNA translation. Blood 2013, 122, 1658–1668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, M.C.; Zhang, D.L.; Jeong, S.Y.; Kovtunovych, G.; Ollivierre-Wilson, H.; Noguchi, A.; Tu, T.; Senecal, T.; Robinson, G.; Crooks, D.R.; et al. Deletion of iron regulatory protein 1 causes polycythemia and pulmonary hypertension in mice through translational derepression of HIF2alpha. Cell Metab. 2013, 17, 271–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nemeth, E.; Ganz, T. Hepcidin and Iron in Health and Disease. Annu. Rev. Med. 2022, 74, 261–277. [Google Scholar] [CrossRef] [PubMed]
- Kautz, L.; Jung, G.; Valore, E.V.; Rivella, S.; Nemeth, E.; Ganz, T. Identification of erythroferrone as an erythroid regulator of iron metabolism. Nat. Genet. 2014, 46, 678–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nemeth, E.; Rivera, S.; Gabayan, V.; Keller, C.; Taudorf, S.; Pedersen, B.K.; Ganz, T. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J. Clin. Investig. 2004, 113, 1271–1276. [Google Scholar] [CrossRef] [Green Version]
- Shanmugam, N.K.; Chen, K.; Cherayil, B.J. Commensal Bacteria-induced Interleukin 1beta (IL-1beta) Secreted by Macrophages Up-regulates Hepcidin Expression in Hepatocytes by Activating the Bone Morphogenetic Protein Signaling Pathway. J. Biol. Chem. 2015, 290, 30637–30647. [Google Scholar] [CrossRef] [Green Version]
- Pham, C.G.; Bubici, C.; Zazzeroni, F.; Papa, S.; Jones, J.; Alvarez, K.; Jayawardena, S.; De Smaele, E.; Cong, R.; Beaumont, C.; et al. Ferritin heavy chain upregulation by NF-kappaB inhibits TNFalpha-induced apoptosis by suppressing reactive oxygen species. Cell 2004, 119, 529–542. [Google Scholar] [CrossRef] [Green Version]
- Yanatori, I.; Richardson, D.R.; Dhekne, H.S.; Toyokuni, S.; Kishi, F. CD63 is regulated by iron via the IRE-IRP system and is important for ferritin secretion by extracellular vesicles. Blood 2021, 138, 1490–1503. [Google Scholar] [CrossRef]
- Kuno, S.; Fujita, H.; Tanaka, Y.K.; Ogra, Y.; Iwai, K. Iron-induced NCOA4 condensation regulates ferritin fate and iron homeostasis. EMBO Rep. 2022, 23, e54278. [Google Scholar] [CrossRef] [PubMed]
- Mancias, J.D.; Pontano Vaites, L.; Nissim, S.; Biancur, D.E.; Kim, A.J.; Wang, X.; Liu, Y.; Goessling, W.; Kimmelman, A.C.; Harper, J.W. Ferritinophagy via NCOA4 is required for erythropoiesis and is regulated by iron dependent HERC2-mediated proteolysis. Elife 2015, 4, e10308. [Google Scholar] [CrossRef]
- Li, X.; Lozovatsky, L.; Sukumaran, A.; Gonzalez, L.; Jain, A.; Liu, D.; Ayala-Lopez, N.; Finberg, K.E. NCOA4 is regulated by HIF and mediates mobilization of murine hepatic iron stores after blood loss. Blood 2020, 136, 2691–2702. [Google Scholar] [CrossRef] [PubMed]
- Das, N.K.; Jain, C.; Sankar, A.; Schwartz, A.J.; Santana-Codina, N.; Solanki, S.; Zhang, Z.; Ma, X.; Parimi, S.; Rui, L.; et al. Modulation of the HIF2alpha-NCOA4 axis in enterocytes attenuates iron loading in a mouse model of hemochromatosis. Blood 2022, 139, 2547–2552. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Li, Y.; Yang, L.; Shen, J.; Zhao, H.; Dong, W.; Chang, Y.; Qiao, T.; Li, K. Stimulation of Hepatic Ferritinophagy Mitigates Irp2 Depletion-Induced Anemia. Antioxidants 2023, 12, 566. https://doi.org/10.3390/antiox12030566
Liu Y, Li Y, Yang L, Shen J, Zhao H, Dong W, Chang Y, Qiao T, Li K. Stimulation of Hepatic Ferritinophagy Mitigates Irp2 Depletion-Induced Anemia. Antioxidants. 2023; 12(3):566. https://doi.org/10.3390/antiox12030566
Chicago/Turabian StyleLiu, Yutong, Yuxuan Li, Liu Yang, Jiaqi Shen, Hongting Zhao, Weichen Dong, Yanzhong Chang, Tong Qiao, and Kuanyu Li. 2023. "Stimulation of Hepatic Ferritinophagy Mitigates Irp2 Depletion-Induced Anemia" Antioxidants 12, no. 3: 566. https://doi.org/10.3390/antiox12030566
APA StyleLiu, Y., Li, Y., Yang, L., Shen, J., Zhao, H., Dong, W., Chang, Y., Qiao, T., & Li, K. (2023). Stimulation of Hepatic Ferritinophagy Mitigates Irp2 Depletion-Induced Anemia. Antioxidants, 12(3), 566. https://doi.org/10.3390/antiox12030566