
Dysregulated Iron Homeostasis as Common Disease Etiology and Promising Therapeutic Target



Abstract
:1. Introduction
1.1. Iron as Biological Catalyst
1.2. Excess Iron Toxicity
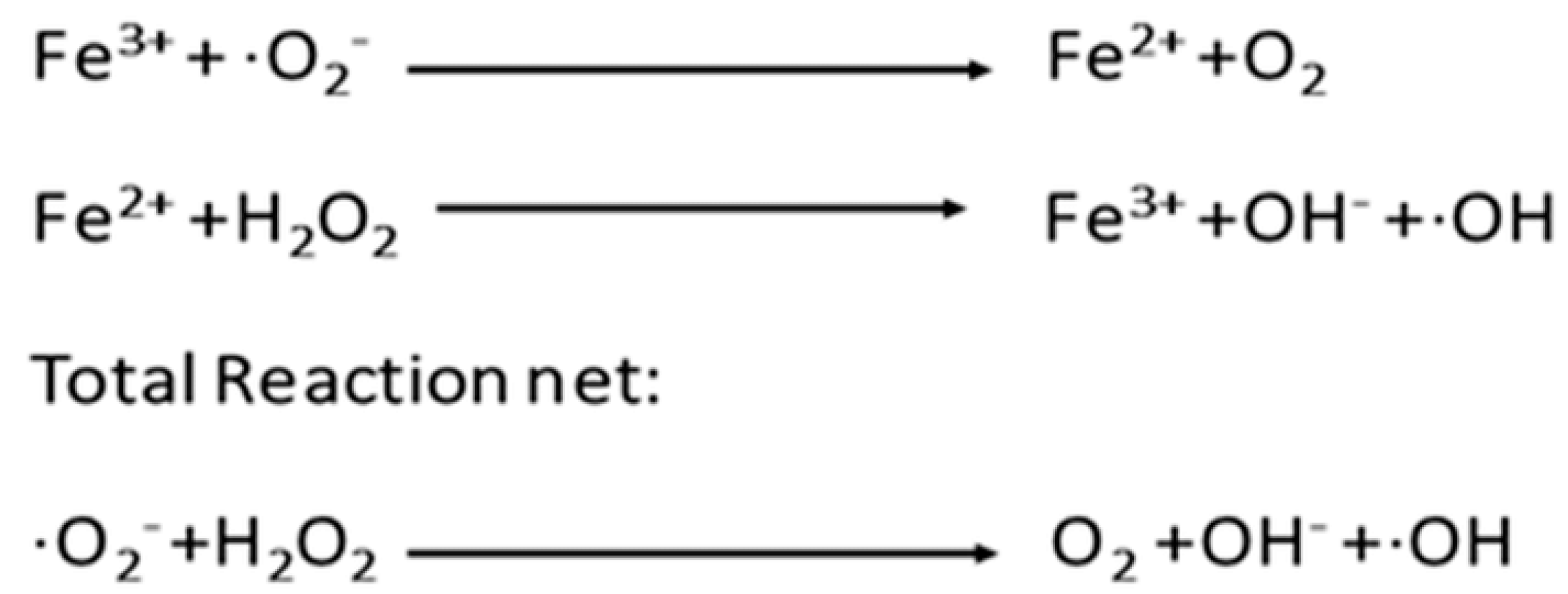
1.3. Chemical Containment
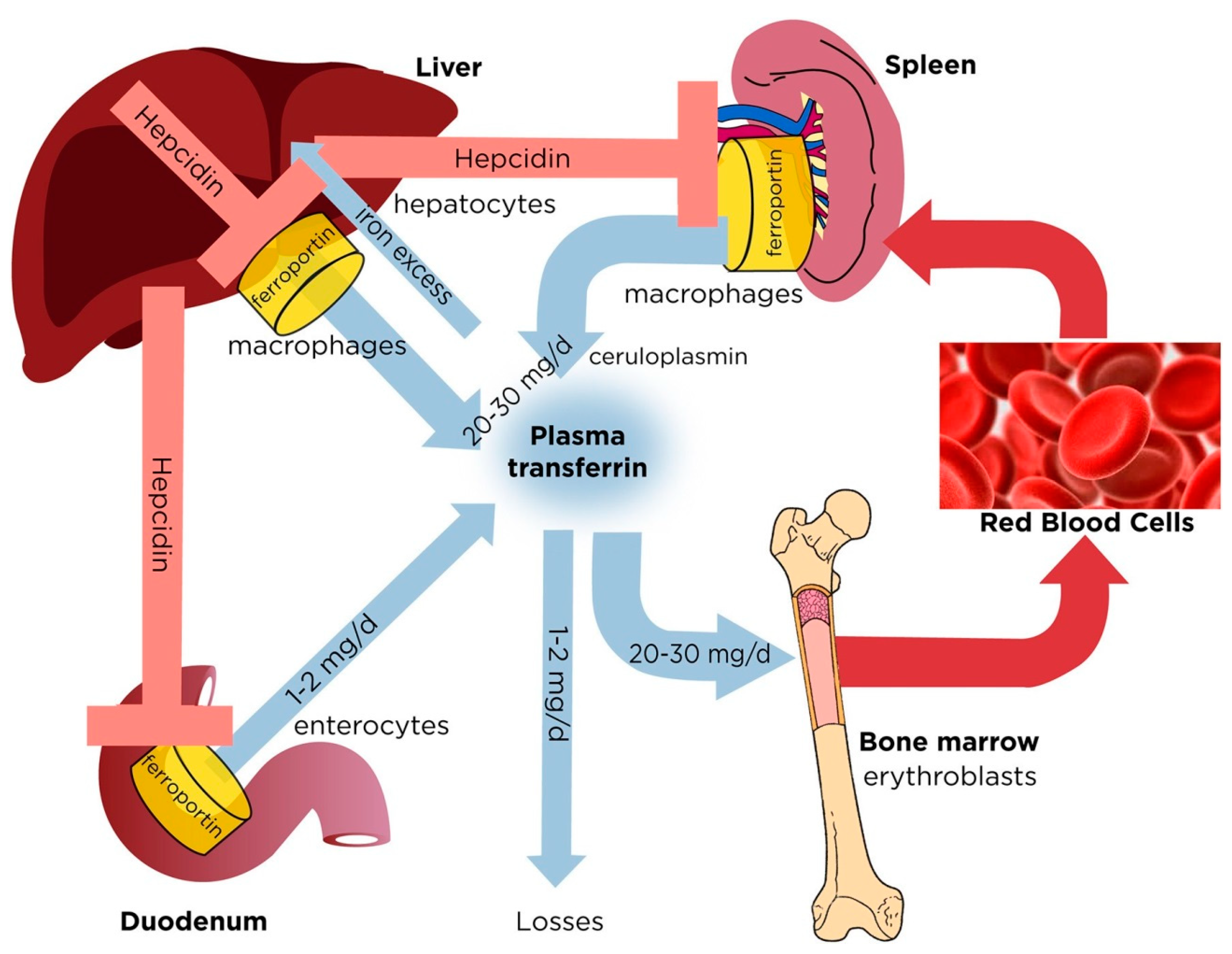
2. Regulation of Normal Iron Homeostasis
3. Iron Dysregulation Underlying Disease
3.1. The Nature of Iron Dysregulation
3.2. Infections
3.2.1. Pathogenic Microbes
3.2.2. Bacterial Sepsis
3.2.3. Parasitic Pathogens
3.2.4. Viral Infections
3.3. Cancer
3.4. Ferroptotic Cell Death
3.5. Inflammatory Diseases
3.5.1. Ocular
3.5.2. Lung Fibrosis
3.5.3. Kidney
3.6. Diabetes
3.7. Cardiovascular
3.8. Autoimmune
3.9. Neurological
3.10. Iron Overload
3.11. Cirrhosis
3.12. Anemia of Chronic Infection and Inflammation
4. Therapeutic Options
4.1. Iron Restriction
4.2. Iron Chelators
4.3. Hepcidin and Agonists
5. Future Needs and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Andreini, C.; Putignano, V.; Rosato, A.; Banci, L. The human iron-proteome. Metallomics 2018, 10, 1223–1231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, R.J.P. The mechanism of cytochrome oxidase and other reaction centres for electron/proton pumping. FEBS Lett. 1987, 226, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beinert, H.; Kennedy, M.C.; Stout, C.D. Aconitase as iron-sulfur protein, enzyme, and iron-regulatory protein. Chem. Rev. 1996, 96, 2335–2373. [Google Scholar] [CrossRef]
- Jordan, A.; Reichard, P. Ribonucleotide reductases. Annu. Rev. Biochem. 1998, 67, 71–98. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C. Essential functions of iron-requiring proteins in DNA replication, repair and cell cycle control. Protein Cell 2014, 5, 750–760. [Google Scholar] [CrossRef] [Green Version]
- McDonough, M.A.; Loenarz, C.; Chowdhury, R.; Clifton, I.J.; Schofield, C.J. Structural studies on human 2-oxoglutarate dependent oxygenases. Curr. Opin. Struct. Biol. 2010, 20, 659–672. [Google Scholar] [CrossRef]
- Parker, M.W.; Blake, C.C.F. Iron- and manganese-containing superoxide dismutases can be distinguished by analysis of their primary structures. FEBS Lett. 1988, 229, 377–382. [Google Scholar] [CrossRef] [Green Version]
- Alfonso-Prieto, M.; Biarnés, X.; Vidossich, P.; Rovira, C. The molecular mechanism of the catalase reaction. J. Am. Chem. Soc. 2009, 131, 11751–11761. [Google Scholar] [CrossRef] [PubMed]
- Hayyan, M.; Hashim, M.A.; Alnashef, I.M. Superoxide Ion: Generation and Chemical Implications. Chem. Rev. 2016, 116, 3029–3085. [Google Scholar] [CrossRef] [Green Version]
- Ray, P.D.; Huang, B.W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef] [Green Version]
- Minakami, R.; Sumimoto, H. Phagocytosis-coupled activation of the superoxide-producing phagocyte oxidase, a member of the NADPH oxidase (Nox) family. Int. J. Hematol. 2006, 84, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B.; Adhikary, A.; Dingfelder, M.; Dizdaroglu, M. Hydroxyl radical is a significant player in oxidative DNA damage: In vivo. Chem. Soc. Rev. 2021, 50, 8355–8360. [Google Scholar] [CrossRef] [PubMed]
- Fang, F.C. Antimicrobial reactive oxygen and nitrogen species: Concepts and controversies. Nat. Rev. Microbiol. 2004, 2, 820–832. [Google Scholar] [CrossRef] [PubMed]
- Yiannikourides, A.; Latunde-Dada, G. A Short Review of Iron Metabolism and Pathophysiology of Iron Disorders. Medicines 2019, 6, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kakhlon, O.; Cabantchik, Z.I. The labile iron pool: Characterization, measurement, and participation in cellular processes. Free Radic. Biol. Med. 2002, 33, 1037–1046. [Google Scholar] [CrossRef] [PubMed]
- Ezraty, B.; Barras, F. The “liaisons dangereuses” between iron and antibiotics. FEMS Microbiol. Rev. 2016, 40, 418–435. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Steyger, P. Synergistic ototoxicity due to noise exposure and aminoglycoside antibiotics. Noise Health 2009, 11, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Pantopoulos, K. Inherited Disorders of Iron Overload. Front. Nutr. 2018, 5, 103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nemeth, E.; Ganz, T. Hepcidin and Iron in Health and Disease. Annu. Rev. Med. 2022, 74, 261–277. [Google Scholar] [CrossRef]
- Vinchi, F. Non-Transferrin-Bound Iron in the Spotlight: Novel Mechanistic Insights into the Vasculotoxic and Atherosclerotic Effect of Iron. Antioxid. Redox Signal. 2021, 35, 387–414. [Google Scholar] [CrossRef]
- Holbein, B.E.; Ang, M.T.C.; Allan, D.S.; Chen, W.; Lehmann, C. Iron-withdrawing anti-infectives for new host-directed therapies based on iron dependence, the Achilles’ heel of antibiotic-resistant microbes. Environ. Chem. Lett. 2021, 19, 2789–2808. [Google Scholar] [CrossRef]
- Akinc, A.; Chan-Daniels, A.; Sehgal, A.; Foster, D.; Bettencourt, B.R.; Hettinger, J.; Racie, T.; Aubin, J.; Kuchimanchi, S.; Epstein-Barash, H.; et al. Targeting the Hepcidin Pathway with RNAi Therapeutics for the Treatment of Anemia. Blood 2011, 118, 688. [Google Scholar] [CrossRef]
- Cabantchik, Z.I.; Breuer, W.; Zanninelli, G.; Cianciulli, P. LPI-labile plasma iron in iron overload. Best Pract. Res. Clin. Haematol. 2005, 18, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Cabantchik, Z.I. Labile iron in cells and body fluids: Physiology, pathology, and pharmacology. Front. Pharmacol. 2014, 5, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bullen, J.; Griffiths, E.; Rogers, H.; Ward, G. Sepsis: The critical role of iron. Microbes Infect. 2000, 2, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Mach, J.; Sutak, R. Iron in parasitic protists-from uptake to storage and where we can interfere. Metallomics 2020, 12, 1335–1347. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, S. The role of iron in viral infections. Front. Biosci. (Landmark Ed.) 2020, 25, 893–911. [Google Scholar] [CrossRef] [PubMed]
- Torti, S.V.; Manz, D.; Paul, B.; Blanchette-Farra, N.; Torti, F.M. Iron and cancer. Iron Hum. Dis. 2018, 38, 97–125. [Google Scholar] [CrossRef]
- Harrison, A.V.; Lorenzo, F.R.; McClain, D.A. Iron and the Pathophysiology of Diabetes. Annu. Rev. Physiol. 2023, 85, 339–362. [Google Scholar] [CrossRef]
- Li, S.; Zhang, X. Iron in Cardiovascular Disease: Challenges and Potentials. Front. Cardiovasc. Med. 2021, 8, 707138. [Google Scholar] [CrossRef]
- David, S.; Jhelum, P.; Ryan, F.; Jeong, S.Y.; Kroner, A. Dysregulation of Iron Homeostasis in the Central Nervous System and the Role of Ferroptosis in Neurodegenerative Disorders. Antioxid. Redox Signal. 2022, 37, 150–170. [Google Scholar] [CrossRef] [PubMed]
- Ogger, P.P.; Byrne, A.J. Lung fibrosis enters the iron age. J. Pathol. 2020, 252, 1–3. [Google Scholar] [CrossRef]
- Baker, J.F.; Ghio, A.J. Iron homoeostasis in rheumatic disease. Rheumatology 2009, 48, 1339–1344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, C.P.; Thévenod, F. Iron transport and the kidney. Biochim. Biophys. Acta-Gen. Subj. 2009, 1790, 724–730. [Google Scholar] [CrossRef] [PubMed]
- Loh, A.; Hadziahmetovic, M.; Dunaief, J.L. Iron homeostasis and eye disease. Biochim. Biophys. Acta-Gen. Subj. 2009, 1790, 637–649. [Google Scholar] [CrossRef] [Green Version]
- Chen, H. Iron metabolism in non-alcoholic fatty liver disease: A promising therapeutic target. Liver Res. 2022, 6, 203–213. [Google Scholar] [CrossRef]
- Schaible, U.E.; Kaufmann, S.H.E. Iron and microbial infection. Nat. Rev. Microbiol. 2004, 2, 946–953. [Google Scholar] [CrossRef]
- Troxell, B.; Hassan, H.M. Transcriptional regulation by Ferric Uptake Regulator (Fur) in pathogenic bacteria. Front. Cell. Infect. Microbiol. 2013, 4, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murdoch, C.C.; Skaar, E.P. Nutritional immunity: The battle for nutrient metals at the host-pathogen interface. Nat. Rev. Microbiol. 2022, 20, 657–670. [Google Scholar] [CrossRef]
- Hanstock, H.G.; Edwards, J.P.; Walsh, N.P. Tear lactoferrin and lysozyme as clinically relevant biomarkers of mucosal immune competence. Front. Immunol. 2019, 10, 1178. [Google Scholar] [CrossRef] [PubMed]
- Holbein, B.E. Enhancement of Neisseria meningitidis infection in mice by addition of iron bound to transferrin. Infect. Immun. 1981, 34, 120–125. [Google Scholar] [CrossRef] [Green Version]
- Xiao, R.; Kisaalita, W.S. Iron acquisition from transferrin and lactoferrin by Pseudomonas aeruginosa pyoverdin. Microbiology 1997, 143, 2509–2515. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, S.; Okujo, N.; Kataoka, H.; Narimatsu, S. Siderophore-mediated utilization of transferrin- and lactoferrin-bound iron by Acinetobacter baumannii. J. Health Sci. 1999, 45, 297–302. [Google Scholar] [CrossRef] [Green Version]
- Giardina, B.J.; Shahzad, S.; Huang, W.; Wilks, A. Heme uptake and utilization by hypervirulent Acinetobacter baumannii LAC-4 is dependent on a canonical heme oxygenase (abHemO). Arch. Biochem. Biophys. 2019, 672, 108066. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.E.; Rock, J.D.; Ridley, K.A.; Williams, P.H.; Ketley, J.M. Utilization of lactoferrin-bound and transferrin-bound iron by Campylobacter jejuni. J. Bacteriol. 2008, 190, 1900–1911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Modun, B.; Evans, R.W.; Joannou, C.L.; Williams, P. Receptor-mediated recognition and uptake of iron from human transferrin by Staphylococcus aureus and Staphylococcus epidermidis. Infect. Immun. 1998, 66, 3591–3596. [Google Scholar] [CrossRef] [Green Version]
- Skaar, E.P.; Schneewind, O. Iron-regulated surface determinants (Isd) of Staphylococcus aureus: Stealing iron from heme. Microbes Infect. 2004, 6, 390–397. [Google Scholar] [CrossRef] [PubMed]
- Clemens, D.L.; Horwitz, M.A. The Mycobacterium tuberculosis phagosome interacts with early endosomes and is accessible to exogenously administered transferrin. J. Exp. Med. 1996, 184, 1349–1355. [Google Scholar] [CrossRef] [Green Version]
- Knight, S.A.B.; Vilaire, G.; Lesuisse, E.; Dancis, A. Iron acquisition from transferrin by Candida albicans depends on the reductive pathway. Infect. Immun. 2005, 73, 5482–5492. [Google Scholar] [CrossRef] [Green Version]
- Hissen, A.H.T.; Chow, J.M.T.; Pinto, L.J.; Moore, M.M. Survival of Aspergillus fumigatus in Serum Involves Removal of Iron from Transferrin: The Role of Siderophores. Infect. Immun. 2004, 72, 1402–1408. [Google Scholar] [CrossRef] [Green Version]
- Olwal, C.O.; Nganyewo, N.N.; Tapela, K.; Djomkam Zune, A.L.; Owoicho, O.; Bediako, Y.; Duodu, S. Parallels in Sepsis and COVID-19 Conditions: Implications for Managing Severe COVID-19. Front. Immunol. 2021, 12, 744425. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Wu, J.; Zhang, X.; Wu, X.; Zhao, Y.; Ren, J. Iron homeostasis and disorders revisited in the sepsis. Free Radic. Biol. Med. 2021, 165, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, C.; Islam, S.; Jarosch, S.; Zhou, J.; Hoskin, D.; Greenshields, A.; Al-Banna, N.; Sharawy, N.; Sczcesniak, A.; Kelly, M.; et al. The utility of iron chelators in the management of inflammatory disorders. Mediat. Inflamm. 2015, 2015, 516740. [Google Scholar] [CrossRef] [Green Version]
- Lan, P.; Pan, K.H.; Wang, S.J.; Shi, Q.C.; Yu, Y.X.; Fu, Y.; Chen, Y.; Jiang, Y.; Hua, X.T.; Zhou, J.C.; et al. High Serum Iron level is Associated with Increased Mortality in Patients with Sepsis. Sci. Rep. 2018, 8, 11072. [Google Scholar] [CrossRef] [Green Version]
- Tacke, F.; Nuraldeen, R.; Koch, A.; Strathmann, K.; Hutschenreuter, G.; Trautwein, C.; Strnad, P. Iron parameters determine the prognosis of critically ill patients. Crit. Care Med. 2016, 44, 1049–1058. [Google Scholar] [CrossRef]
- Wilson, M.E.; Vorhies, R.W.; Andersen, K.A.; Britigan, B.E. Acquisition of iron from transferrin and lactoferrin by the protozoan Leishmania chagasi. Infect. Immun. 1994, 62, 3262–3269. [Google Scholar] [CrossRef] [Green Version]
- Clark, M.A.; Goheen, M.M.; Cerami, C. Influence of host iron status on Plasmodium falciparum infection. Front. Pharmacol. 2014, 5, 84. [Google Scholar] [CrossRef] [PubMed]
- Lalonde, R.G.; Holbein, B.E. Role of iron in trypanosoma cruzi infection of mice. J. Clin. Investig. 1984, 73, 470–476. [Google Scholar] [CrossRef]
- Wiciński, M.; Liczner, G.; Cadelski, K.; Kołnierzak, T.; Nowaczewska, M.; Malinowski, B. Anemia of chronic diseases: Wider diagnostics—Better treatment? Nutrients 2020, 12, 1784. [Google Scholar] [CrossRef]
- Wei, Y.; Ye, W.; Zhao, W. Serum iron levels decreased in patients with HBV-related hepatocellular carcinoma, as a risk factor for the prognosis of HBV-related HCC. Front. Physiol. 2018, 9, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kakizaki, S.; Takagi, H.; Horiguchi, N.; Toyoda, M.; Takayama, H.; Nagamine, T.; Mori, M.; Kato, N. Iron enhances hepatitis C virus replication in cultured human hepatocytes. Liver 2000, 20, 125–128. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.C.; Bayeva, M.; Taiwo, B.; Palella, F.J.; Hope, T.J.; Ardehali, H. Short communication: High cellular iron levels are associated with increased HIV infection and replication. AIDS Res. Hum. Retrovir. 2015, 31, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Mao, W.L.; Hu, Y.; Lou, Y.F.; Chen, Y.M.; Zhang, J.W. Abnormal serum iron markers in chronic hepatitis B virus infection may be because of liver injury. Eur. J. Gastroenterol. Hepatol. 2015, 27, 130–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torti, S.V.; Torti, F.M. Iron and cancer: More ore to be mined. Nat. Rev. Cancer 2013, 13, 342–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, T.; Sempos, C.T.; Freudenheim, J.L.; Muti, P.; Smit, E. Serum iron, copper and zinc concentrations and risk of cancer mortality in US adults. Ann. Epidemiol. 2004, 14, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Greenshields, A.L.; Power Coombs, M.R.; Fernando, W.; Holbein, B.E.; Hoskin, D.W. DIBI, a novel 3-hydroxypyridin-4-one chelator iron-binding polymer, inhibits breast cancer cell growth and functions as a chemosensitizer by promoting S-phase DNA damage. BioMetals 2019, 32, 909–921. [Google Scholar] [CrossRef]
- Shen, Y.; Li, X.; Dong, D.; Zhang, B.; Xue, Y.; Shang, P. Transferrin receptor 1 in cancer: A new sight for cancer therapy. Am. J. Cancer Res. 2018, 8, 916–931. [Google Scholar]
- Candelaria, P.V.; Leoh, L.S.; Penichet, M.L.; Daniels-Wells, T.R. Antibodies Targeting the Transferrin Receptor 1 (TfR1) as Direct Anti-cancer Agents. Front. Immunol. 2021, 12, 607692. [Google Scholar] [CrossRef]
- Zhang, S.; Chang, W.; Wu, H.; Wang, Y.H.; Gong, Y.W.; Zhao, Y.L.; Liu, S.H.; Wang, H.Z.; Svatek, R.S.; Rodriguez, R.; et al. Pan-cancer analysis of iron metabolic landscape across the Cancer Genome Atlas. J. Cell. Physiol. 2020, 235, 1013–1024. [Google Scholar] [CrossRef]
- Lei, P.; Bai, T.; Sun, Y. Mechanisms of ferroptosis and relations with regulated cell death: A review. Front. Physiol. 2019, 10, 139. [Google Scholar] [CrossRef] [Green Version]
- Toyokuni, S.; Yanatori, I.; Kong, Y.; Zheng, H.; Motooka, Y.; Jiang, L. Ferroptosis at the crossroads of infection, aging and cancer. Cancer Sci. 2020, 111, 2665–2671. [Google Scholar] [CrossRef]
- Angeli, J.P.F.; Shah, R.; Pratt, D.A.; Conrad, M. Ferroptosis Inhibition: Mechanisms and Opportunities. Trends Pharmacol. Sci. 2017, 38, 489–498. [Google Scholar] [CrossRef]
- Yang, W.S.; Stockwell, B.R. Synthetic Lethal Screening Identifies Compounds Activating Iron-Dependent, Nonapoptotic Cell Death in Oncogenic-RAS-Harboring Cancer Cells. Chem. Biol. 2008, 15, 234–245. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Sheng, S.; Wang, W.; Dai, J.; Zhong, Y.; Ren, J.; Jiang, K.; Li, S.; Bian, X.; Liu, L. Molecular Mechanisms of Iron Mediated Programmed Cell Death and Its Roles in Eye Diseases. Front. Nutr. 2022, 9, 811469. [Google Scholar] [CrossRef] [PubMed]
- Chowers, I.; Wong, R.; Dentchev, T.; Farkas, R.H.; Iacovelli, J.; Gunatilaka, T.L.; Medeiros, N.E.; Presley, J.B.; Campochiaro, P.A.; Curcio, C.A.; et al. The iron carrier transferrin is upregulated in retinas from patients with age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2006, 47, 2135–2140. [Google Scholar] [CrossRef]
- Ali, M.K.; Kim, R.Y.; Brown, A.C.; Donovan, C.; Vanka, K.S.; Mayall, J.R.; Liu, G.; Pillar, A.L.; Jones-Freeman, B.; Xenaki, D.; et al. Critical role for iron accumulation in the pathogenesis of fibrotic lung disease. J. Pathol. 2020, 251, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Swaminathan, S.; Shah, S.V. Novel approaches targeted toward oxidative stress for the treatment of chronic kidney disease. Curr. Opin. Nephrol. Hypertens. 2008, 17, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Prinsen, B.H.C.M.T.; De Sain-van der Velden, M.G.M.; Kaysen, G.A.; Straver, H.W.H.C.; Van Rijn, H.J.M.; Stellaard, F.; Berger, R.; Rabelink, T.J. Transferrin synthesis is increased in nephrotic patients insufficiently to replace urinary losses. J. Am. Soc. Nephrol. 2001, 12, 1017–1025. [Google Scholar] [CrossRef] [PubMed]
- Theut, L.R.; Dsouza, D.L.; Grove, R.C.; Boesen, E.I. Evidence of Renal Iron Accumulation in a Male Mouse Model of Lupus. Front. Med. 2020, 7, 516. [Google Scholar] [CrossRef]
- Kochi, M.; Kohagura, K.; Shiohira, Y.; Iseki, K.; Ohya, Y. Chronic kidney disease, inflammation, and cardiovascular disease risk in rheumatoid arthritis. J. Cardiol. 2018, 71, 277–283. [Google Scholar] [CrossRef] [Green Version]
- Venkatesan, P.; Varghese, J.; Arthi, T.S.; James, J.V.; Anura, A.; Prasad, J.; Jacob, M. Evidence of dysregulated iron homeostasis in newly diagnosed diabetics, but not in pre-diabetics. J. Diabetes Complicat. 2021, 35, 107977. [Google Scholar] [CrossRef] [PubMed]
- Giacco, F.; Brownlee, M. Oxidative stress and diabetic complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Cao, F.; Yin, H.L.; Huang, Z.J.; Lin, Z.T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Jiang, C.; Mei, G.; Zhao, Y.; Chen, L.; Liu, J.; Tang, Y.; Gao, C.; Yao, P. Quercetin alleviates ferroptosis of pancreatic β cells in type 2 diabetes. Nutrients 2020, 12, 2954. [Google Scholar] [CrossRef]
- Fang, X.; Ardehali, H.; Min, J.; Wang, F. The molecular and metabolic landscape of iron and ferroptosis in cardiovascular disease. Nat. Rev. Cardiol. 2023, 20, 7–23. [Google Scholar] [CrossRef]
- Fang, X.; Wang, H.; Han, D.; Xie, E.; Yang, X.; Wei, J.; Gu, S.; Gao, F.; Zhu, N.; Yin, X.; et al. Ferroptosis as a target for protection against cardiomyopathy. Proc. Natl. Acad. Sci. USA 2019, 116, 2672–2680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cronin, S.J.F.; Woolf, C.J.; Weiss, G.; Penninger, J.M. The Role of Iron Regulation in Immunometabolism and Immune-Related Disease. Front. Mol. Biosci. 2019, 6, 116. [Google Scholar] [CrossRef] [Green Version]
- Wincup, C.; Sawford, N.; Rahman, A. Pathological mechanisms of abnormal iron metabolism and mitochondrial dysfunction in systemic lupus erythematosus. Expert Rev. Clin. Immunol. 2021, 17, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Pfaender, S.; Grabrucker, A.M. Characterization of biometal profiles in neurological disorders. Metallomics 2014, 6, 960–977. [Google Scholar] [CrossRef]
- Hadzhieva, M.; Kirches, E.; Mawrin, C. Review: Iron metabolism and the role of iron in neurodegenerative disorders. Neuropathol. Appl. Neurobiol. 2014, 40, 240–257. [Google Scholar] [CrossRef]
- Das, N.; Raymick, J.; Sarkar, S. Role of metals in Alzheimer’s disease. Metab. Brain Dis. 2021, 36, 1627–1639. [Google Scholar] [CrossRef] [PubMed]
- Medeiros, M.S.; Schumacher-Schuh, A.; Cardoso, A.M.; Bochi, G.V.; Baldissarelli, J.; Kegler, A.; Santana, D.; Chaves, C.M.M.B.S.; Schetinger, M.R.C.; Moresco, R.N.; et al. Iron and oxidative stress in Parkinson’s disease: An observational study of injury biomarkers. PLoS ONE 2016, 11, e0146129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, J.X.; Sun, X.; Yan, X.L.; Guo, Z.N.; Yang, Y. Ferroptosis in Neurological Diseases. Front. Cell. Neurosci. 2020, 14, 218. [Google Scholar] [CrossRef]
- Ismaiel, A.; Srouji, N. Al Anemia of Chronic Disease: Epidemiology and Pathophysiological Mechanisms—Literature Review. Glob. J. Med. Ther. 2020, 2, 8–16. [Google Scholar] [CrossRef]
- Weiss, G.; Ganz, T.; Goodnough, L.T. Anemia of inflammation. Blood 2019, 133, 40–50. [Google Scholar] [CrossRef] [Green Version]
- Bao, W.; Rong, Y.; Rong, S.; Liu, L. Dietary iron intake, body iron stores, and the risk of type 2 diabetes: A systematic review and meta-analysis. BMC Med. 2012, 10, 119. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Real, J.M.; López-Bermejo, A.; Ricart, W. Cross-talk between iron metabolism and diabetes. Diabetes 2002, 51, 2348–2354. [Google Scholar] [CrossRef] [Green Version]
- Gumbau-Brisa, R.; Ang, M.T.C.; Holbein, B.E.; Bierenstiel, M. Enhanced Fe3+ binding through cooperativity of 3-hydroxypyridin-4-one groups within a linear co-polymer: Wrapping effect leading to superior antimicrobial activity. BioMetals 2020, 33, 339–351. [Google Scholar] [CrossRef]
- Vlachodimitropoulou, E.; Chen, Y.L.; Garbowski, M.; Koonyosying, P.; Psaila, B.; Sola-Visner, M.; Cooper, N.; Hider, R.; Porter, J. Eltrombopag: A powerful chelator of cellular or extracellular iron(III) alone or combined with a second chelator. Blood 2017, 130, 1923–1933. [Google Scholar] [CrossRef] [Green Version]
- Foley, T.L.; Simeonov, A. Targeting iron assimilation to develop new antibacterials. Expert Opin. Drug Discov. 2012, 7, 831–847. [Google Scholar] [CrossRef] [PubMed]
- Parquet, M.d.C.; Savage, K.A.; Allan, D.S.; Davidson, R.J.; Holbein, B.E. Novel Iron-Chelator DIBI Inhibits Staphylococcus aureus Growth, Suppresses Experimental MRSA Infection in Mice and Enhances the Activities of Diverse Antibiotics in vitro. Front. Microbiol. 2018, 9, 1811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fokam, D.; Dickson, K.; Kamali, K.; Holbein, B.; Colp, P.; Stueck, A.; Zhou, J.; Lehmann, C. Iron chelation in murine models of systemic inflammation induced by gram-positive and gram-negative toxins. Antibiotics 2020, 9, 283. [Google Scholar] [CrossRef] [PubMed]
- Zeidan, A.M.; Griffiths, E.A. To chelate or not to chelate in MDS: That is the question! Blood Rev. 2018, 32, 368–377. [Google Scholar] [CrossRef] [PubMed]
- Badeli, H.; Baghersalimi, A.; Eslami, S.; Saadat, F.; Rad, A.H.; Basavand, R.; Papkiadeh, S.R.; Darbandi, B.; Kooti, W.; Peluso, I. Early kidney damage markers after Deferasirox treatment in patients with thalassemia major: A case-control study. Oxid. Med. Cell. Longev. 2019, 2019, 5461617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, T.; Zhang, X.; Liu, Y.; Wang, H.; Luo, J.; Luo, Y.; An, P. Effects of dietary polyphenol supplementation on iron status and erythropoiesis: A systematic review and meta-analysis of randomized controlled trials. Am. J. Clin. Nutr. 2021, 114, 780–793. [Google Scholar] [CrossRef] [PubMed]
- Casu, C.; Nemeth, E.; Rivella, S. Hepcidin agonists as therapeutic tools. Blood 2018, 131, 1790–1794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taher, A.; Kourakli-Symeonidis, A.; Tantiworawit, A.; Wong, P.; Szecsödy, P. S272: Safety and Preliminary Pharmacodynamic Effects of the Ferroportin Inhibitor Vamifeport (Vit-2763) in Patients with Non-Transfusion-Dependent Beta Thalassemia (Ntdt): Results from a Phase 2a Study. HemaSphere 2022, 6, 173–174. [Google Scholar] [CrossRef]
- Verstovsek, S.; Kuykendall, A.T.; Hoffman, R.; Ginzburg, Y.; Pemmaraju, N.; Valone, F.; Modi, N.B.; Khanna, S.; O’Connor, P.G.; Gupta, S.K.; et al. A Phase 3 Study of the Hepcidin Mimetic Rusfertide (PTG-300) in Patients with Polycythemia Vera. Blood 2021, 138, 1504. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Enzyme | Function | Reference |
|---|---|---|
| Cytochrome oxidase EC 7.1.1.9 | Energy production | Williams, 1987 [2] |
| Citrate aconitase EC 4.2.1.3 | TCA cycle | Beinert et al., 1996 [3] |
| Ribonucleotide reductase EC 2.7.7.56 | DNA synthesis | Jordan and Reichard, 1998 [4] |
| DNA polymerase EC 2.7.7.7 | DNA replication and repair | Zhang et al., 2014 [5] |
| Oxoglutarate oxygenase EC 1.13.12.19 | Lipid biosynthesis | McDonough et al., 2010 [6] |
| Superoxide dismutase EC 1.15.1.1 | Superoxide radical detoxification | Parker and Blake, 1988 [7] |
| Catalase EC 1.11.1.21 | Peroxide detoxification | Alfonso-Prieto et al., 2009 [8] |
| Disease Category | Disease | Example | Reference |
|---|---|---|---|
| Proliferative Cell Replication | Microbial Infection | Staphylococcal, Candidiasis | Holbein et al., 2021 [21] |
| Bacterial Sepsis | Pseudomonas aeruginosa, Neisseria meningitidis | Bullen et al., 2000 [25] | |
| Parasitic Infection | Chagas disease, Malaria | Mach and Sutak, 2020 [26] | |
| Viral Infection | HIV, HBV, HCV, HCMV | Schmidt, 2020 [27] | |
| Cancer | Pancreatic, Liver, Lung | Torti et al., 2018 [28] | |
| Iron-Mediated Pathology | Diabetes | Type 2 Diabetes | Harrison et al., 2023 [29] |
| Cardiovascular | Cardiomyopathy | Li and Zhang, 2021 [30] | |
| Neurological | Alzheimer’s, ALS, MS, Parkinson’s | David et al., 2022 [31] | |
| Lung Fibrosis | Idiopathic Pulmonary Fibrosis | Ogger and Byme, 2020 [32] | |
| Autoimmune | Rheumatoid Arthritis, Lupus Erythematosus | Baker and Ghio, 2009 [33] | |
| Kidney | Fanconi Syndrome | Smith and Thévenod, 2009 [34] | |
| Retinal | Age-Related Macular degeneration | Loh et al., 2009 [35] | |
| Iron Overload | Hemochromatosis | Pantopoulos, 2018 [18] | |
| Cirrhosis | Non-Alcoholic Fatty Cirrhosis | Chen, 2022 [36] |
| Microbial Pathogen | Host Iron Source | Reference |
|---|---|---|
| Neisseria meningitidis | Transferrin | Holbein, 1981 [41] |
| Pseudomonas aeruginosa | Transferrin, Lactoferrin | Xiao and Kisaalita, 1997 [42] |
| Acinetobacter baumannii | Transferrin 1, Lactoferrin 1, Heme 2 | 1 Yamamoto et al., 1999 [43]; 2 Giardina et al., 2019 [44] |
| Campylobacter jejuni | Transferrin, Lactoferrin | Miller et al., 2008 [45] |
| Staphylococcus aureus | Transferrin 3, Heme 4 | 3 Modun et al., 1998 [46]; 4 Skaar and Schneewind, 2004 [47] |
| Mycobacterium tuberculosis | Transferrin | Clemens et al., 1996 [48] |
| Candida albicans | Transferrin | Knight et al., 2005 [49] |
| Aspergillus fumigatus | Transferrin | Hissen et al., 2004 [50] |
| Chelator | DFO Deferoxamine | DEF Deferasirox | DFP Deferiprone | DIBI Hydroxypyridinone |
|---|---|---|---|---|
| MW (Da) | 561 | 373 | 139 | 9000 avg. polymeric [98] |
| Fe(III): chelator binding complex | 1:1 | 1:2 | 1:3 | 3:1 [98] |
| Fe binding constant log K | 30.6 [99] | 36.5 [99] | 36.7 | 41.0 [98] |
| Hydrophobicity/hydrophilicity log P | −3 [99] | 4.3 [99] | −0.8 [99] | −1.87 a |
| FDA Approval | 1968 | 2005 | 2011 | In Development |
| Treatment Indication | Transfusional iron overload [100] | Transfusional iron overload [100] | Transfusional iron overload [100] | Anti-infective [101] Anti-inflammatory [102] |
| Serious limitations and toxicities | Promotes infections [21] | Toxicity, Renal failure [103,104] | Toxicity, Agranulocytosis [104] | None to date from oral and systemic animal testing b |
| Promotes microbial pathogen growth | Very Serious | Potentially Serious | Potentially Serious | No [21] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Holbein, B.E.; Lehmann, C. Dysregulated Iron Homeostasis as Common Disease Etiology and Promising Therapeutic Target. Antioxidants 2023, 12, 671. https://doi.org/10.3390/antiox12030671
Holbein BE, Lehmann C. Dysregulated Iron Homeostasis as Common Disease Etiology and Promising Therapeutic Target. Antioxidants. 2023; 12(3):671. https://doi.org/10.3390/antiox12030671
Chicago/Turabian StyleHolbein, Bruce E., and Christian Lehmann. 2023. "Dysregulated Iron Homeostasis as Common Disease Etiology and Promising Therapeutic Target" Antioxidants 12, no. 3: 671. https://doi.org/10.3390/antiox12030671
APA StyleHolbein, B. E., & Lehmann, C. (2023). Dysregulated Iron Homeostasis as Common Disease Etiology and Promising Therapeutic Target. Antioxidants, 12(3), 671. https://doi.org/10.3390/antiox12030671