
Single-Cell Kinetic Modeling of β-Lapachone Metabolism in Head and Neck Squamous Cell Carcinoma


Abstract
:1. Introduction
2. Materials and Methods
2.1. Ordinary Differential Equation Model Construction
2.2. Sensitivity Analysis
2.3. Single-Cell RNA Sequencing Data Analysis
3. Results
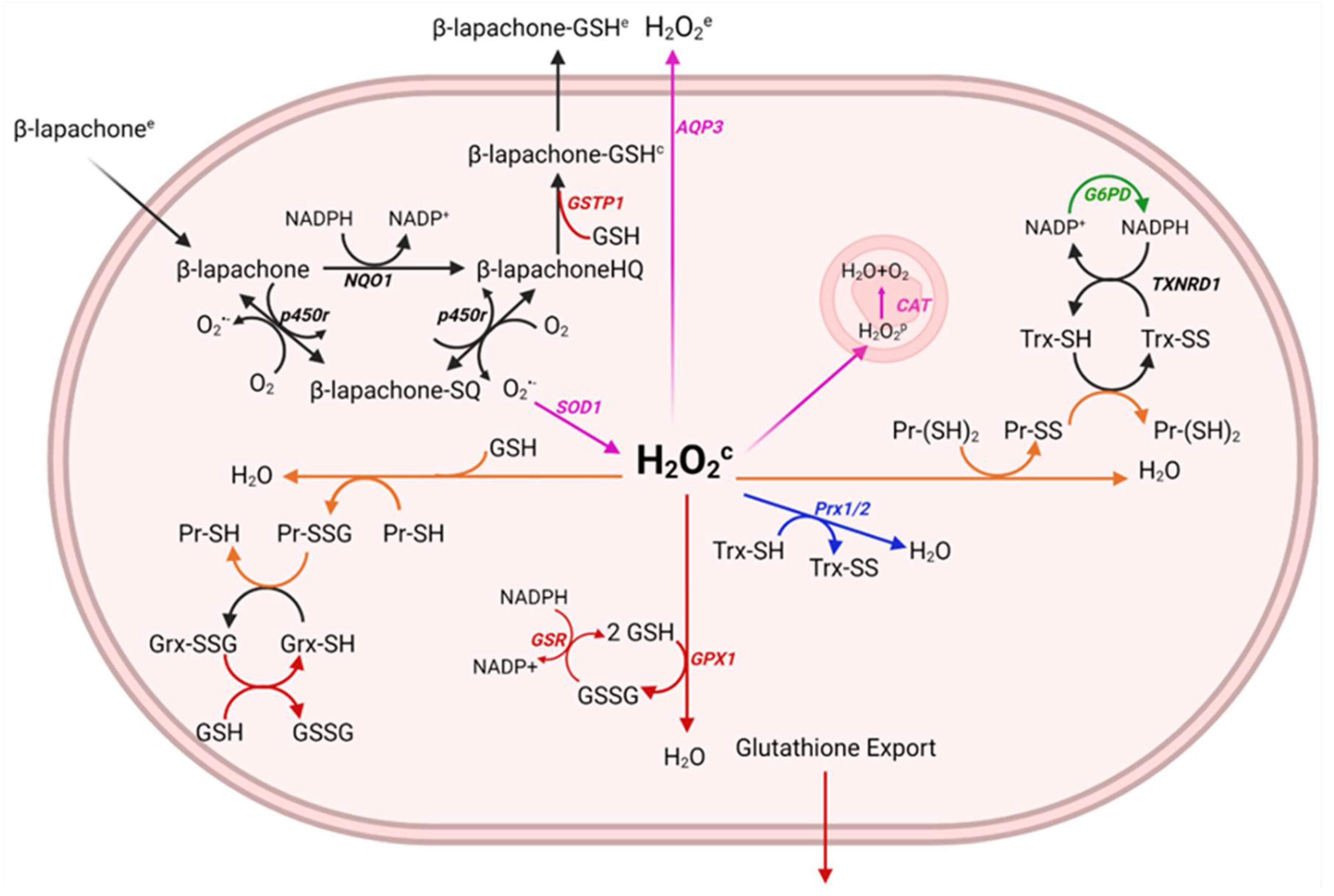
3.1. A Systems-Level Model of Superoxide and H2O2 Generation by Quinone Cycling
- (1)
- sets of critical H2O2-stabilizing antioxidant systems;
- (2)
- metabolism of the xenobiotic drug β-lapachone;
- (3)
- the permeation of key species across membranes of the cell, including organelle-specific transport.
3.2. Head and Neck Squamous Cell Carcinoma Cells Exhibit Heterogeneity of Redox Gene Expression
3.3. Initializing Single-Cell ODE Models with scRNA-Seq
3.4. Sensitivity Analysis Shows H2O2 Production Is Insensitive to Individual Enzymatic Parameters
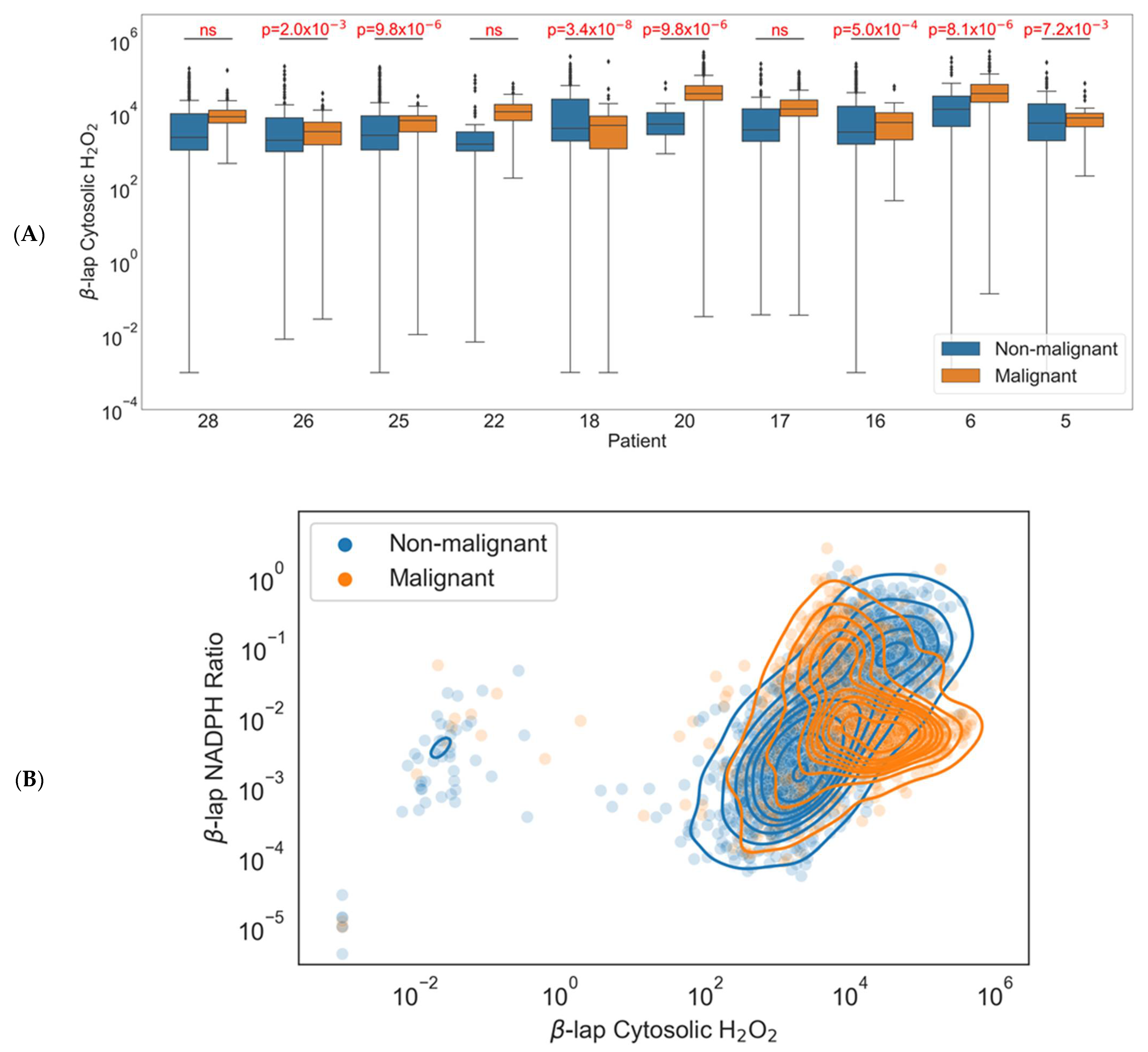
3.5. Comparison of H2O2 Accumulation in Healthy and Cancer Cells Identifies Patients with Greatest Potential for Targeted Therapy
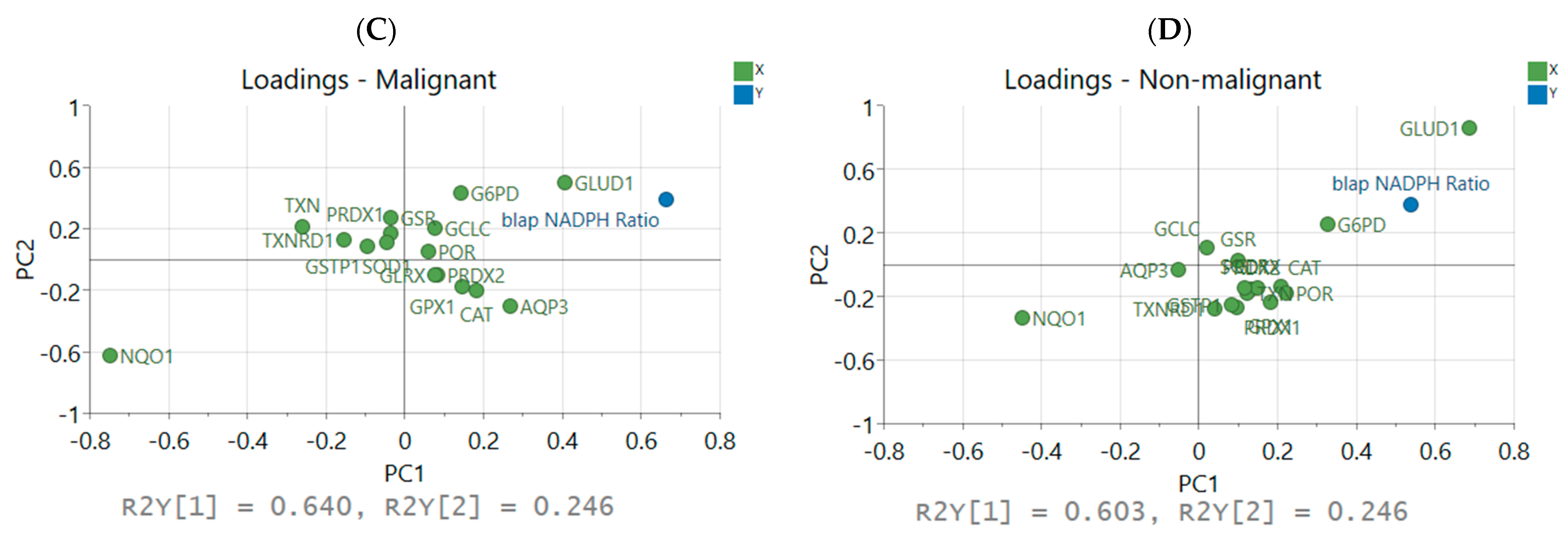
3.6. Initializing Single-Cell ODE Models with Patient HNSCC Scrna-Seq Identifies Proteins Correlated with NADPH Ratio
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Torabi, S.J.; Yarbrough, W.G.; Mehra, S.; Osborn, H.A.; Judson, B. Association of Human Papillomavirus Status at Head and Neck Carcinoma Subsites with Overall Survival. JAMA Otolaryngol. Head Neck Surg. 2018, 144, 519–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, S.; Yan, Z.; Wu, J.; Liu, X.; Guan, G.; Zou, C.; Guo, Q.; Zhu, C.; Liu, T.; Chen, C.; et al. Characterization of ROS Metabolic Equilibrium Reclassifies Pan-Cancer Samples and Guides Pathway Targeting Therapy. Front. Oncol. 2020, 10, 581197. [Google Scholar] [CrossRef]
- Son, Y.; Cheong, Y.-K.; Kim, N.-H.; Chung, H.-T.; Kang, D.G.; Pae, H.-O. Mitogen-Activated Protein Kinases and Reactive Oxygen Species: How Can ROS Activate MAPK Pathways? J. Signal Transduct. 2011, 2011, 792639. [Google Scholar] [CrossRef] [Green Version]
- Best, S.A.; de Souza, D.P.; Kersbergen, A.; Policheni, A.N.; Dayalan, S.; Tull, D.; Rathi, V.; Gray, D.H.; Ritchie, M.E.; McConville, M.J.; et al. Synergy between the KEAP1/NRF2 and PI3K Pathways Drives Non-Small-Cell Lung Cancer with an Altered Immune Microenvironment. Cell Metab. 2018, 27, 935–943.e4. [Google Scholar] [CrossRef] [Green Version]
- Koundouros, N.; Poulogiannis, G. Phosphoinositide 3-Kinase/Akt Signaling and Redox Metabolism in Cancer. Front. Oncol. 2018, 8, 160. [Google Scholar] [CrossRef] [PubMed]
- Johansson, K.; Cebula, M.; Rengby, O.; Dreij, K.; Carlström, K.E.; Sigmundsson, K.; Piehl, F.; Arnér, E.S.J. Cross Talk in HEK293 Cells between Nrf2, HIF, and NF-ΚB Activities upon Challenges with Redox Therapeutics Characterized with Single-Cell Resolution. Antioxid. Redox Signal. 2017, 26, 229–246. [Google Scholar] [CrossRef] [Green Version]
- Movafagh, S.; Crook, S.; Vo, K. Regulation of Hypoxia-Inducible Factor-1a by Reactive Oxygen Species: New Developments in an Old Debate. J. Cell. Biochem. 2015, 116, 696–703. [Google Scholar] [CrossRef]
- Emanuele, S.; D’Anneo, A.; Calvaruso, G.; Cernigliaro, C.; Giuliano, M.; Lauricella, M. The Double-Edged Sword Profile of Redox Signaling: Oxidative Events As Molecular Switches in the Balance between Cell Physiology and Cancer. Chem. Res. Toxicol. 2018, 31, 201–210. [Google Scholar] [CrossRef]
- Badgley, M.A.; Kremer, D.M.; Maurer, H.C.; DelGiorno, K.E.; Lee, H.-J.; Purohit, V.; Sagalovskiy, I.R.; Ma, A.; Kapilian, J.; Firl, C.E.M.; et al. Cysteine Depletion Induces Pancreatic Tumor Ferroptosis in Mice. Science 2020, 368, 85–89. [Google Scholar] [CrossRef]
- Seibt, T.M.; Proneth, B.; Conrad, M. Role of GPX4 in Ferroptosis and Its Pharmacological Implication. Free Radic. Biol. Med. 2019, 133, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Adimora, N.J.; Jones, D.P.; Kemp, M.L. A Model of Redox Kinetics Implicates the Thiol Proteome in Cellular Hydrogen Peroxide Responses. Antioxid. Redox Signal. 2010, 13, 731–743. [Google Scholar] [CrossRef] [Green Version]
- Cloer, E.W.; Goldfarb, D.; Schrank, T.P.; Weissman, B.E.; Major, M.B. Nrf2 Activation in Cancer: From DNA to Protein. Cancer Res. 2019, 79, 889–898. [Google Scholar] [CrossRef] [Green Version]
- Panieri, E.; Telkoparan-Akillilar, P.; Suzen, S.; Saso, L. The NRF2/KEAP1 Axis in the Regulation of Tumor Metabolism: Mechanisms and Therapeutic Perspectives. Biomolecules 2020, 10, 791. [Google Scholar] [CrossRef] [PubMed]
- Harris, I.S.; DeNicola, G.M. The Complex Interplay between Antioxidants and ROS in Cancer. Trends Cell Biol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting Cancer Cells by ROS-Mediated Mechanisms: A Radical Therapeutic Approach? Nat. Rev. Drug. Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef]
- Roh, J.L.; Jang, H.; Kim, E.H.; Shin, D. Targeting of the Glutathione, Thioredoxin, and Nrf2 Antioxidant Systems in Head and Neck Cancer. Antioxid. Redox Signal. 2017, 27, 106–114. [Google Scholar] [CrossRef]
- Glorieux, C.; Calderon, P.B. Catalase, a Remarkable Enzyme: Targeting the Oldest Antioxidant Enzyme to Find a New Cancer Treatment Approach. Biol. Chem. 2017, 398, 1095–1108. [Google Scholar] [CrossRef] [Green Version]
- Chakrabarti, G.; Moore, Z.R.; Luo, X.; Ilcheva, M.; Ali, A.; Padanad, M.; Zhou, Y.; Xie, Y.; Burma, S.; Scaglioni, P.P.; et al. Targeting Glutamine Metabolism Sensitizes Pancreatic Cancer to PARP-Driven Metabolic Catastrophe Induced by ß-Lapachone. Cancer Metab. 2015, 3, 12. [Google Scholar] [CrossRef] [Green Version]
- Jaiswal, A.K. Regulation of Genes Encoding NAD(P)H:Quinone Oxidoreductases. Free Radic. Biol. Med. 2000, 29, 254–262. [Google Scholar] [CrossRef]
- Nioi, P.; McMahon, M.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Identification of a Novel Nrf2-Regulated Antioxidant Response Element (ARE) in the Mouse NAD(P)H:Quinone Oxidoreductase 1 Gene: Reassessment of the ARE Consensus Sequence. Biochem. J. 2003, 374, 337. [Google Scholar] [CrossRef] [PubMed]
- Li, L.S.; Reddy, S.; Lin, Z.H.; Liu, S.; Park, H.; Chun, S.G.; Bornmann, W.G.; Thibodeaux, J.; Yan, J.; Chakrabarti, G.; et al. NQO1-Mediated Tumor-Selective Lethality and Radiosensitization for Head and Neck Cancer. Mol. Cancer Ther. 2016, 15, 1757–1767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerber, D.E.; Beg, M.S.; Fattah, F.; Frankel, A.E.; Fatunde, O.; Arriaga, Y.; Dowell, J.E.; Bisen, A.; Leff, R.D.; Meek, C.C.; et al. Phase 1 Study of ARQ 761, a β-Lapachone Analogue That Promotes NQO1-Mediated Programmed Cancer Cell Necrosis. Br. J. Cancer 2018, 119, 928–936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motea, E.A.; Huang, X.; Singh, N.; Kilgore, J.A.; Williams, N.S.; Xie, X.J.; Gerber, D.E.; Beg, M.S.; Bey, E.A.; Boothman, D.A. NQO1-Dependent, Tumor-Selective Radiosensitization of Non-Small Cell Lung Cancers. Clin. Cancer Res. 2019, 25, 2601–2609. [Google Scholar] [CrossRef] [Green Version]
- Bey, E.A.; Bentle, M.S.; Reinicke, K.E.; Dong, Y.; Yang, C.R.; Girard, L.; Minna, J.D.; Bornmann, W.G.; Gao, J.; Boothman, D.A. An NQO1- and PARP-1-Mediated Cell Death Pathway Induced in Non-Small-Cell Lung Cancer Cells by β-Lapachone. Proc. Natl. Acad. Sci. USA 2007, 104, 11832–11837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bey, E.A.; Reinicke, K.E.; Srougi, M.C.; Varnes, M.; Anderson, V.; Pink, J.J.; Li, L.S.; Patel, M.; Cao, L.; Moore, Z.; et al. Catalase Abrogates β-Lapachone-Induced PARP1 Hyperactivation-Directed Programmed Necrosis in NQO1-Positive Breast Cancers. Mol. Cancer Ther. 2013, 12, 2110–2120. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Dong, Y.; Bey, E.A.; Kilgore, J.A.; Bair, J.S.; Li, L.-S.; Patel, M.; Parkinson, E.I.; Wang, Y.; Williams, N.S.; et al. An NQO1 Substrate with Potent Antitumor Activity That Selectively Kills by PARP1-Induced Programmed Necrosis. Cancer Res. 2012, 72, 3038–3047. [Google Scholar] [CrossRef] [Green Version]
- Cao, L.; Li, L.S.; Spruell, C.; Xiao, L.; Chakrabarti, G.; Bey, E.A.; Reinicke, K.E.; Srougi, M.C.; Moore, Z.; Dong, Y.; et al. Tumor-Selective, Futile Redox Cycle-Induced Bystander Effects Elicited by NQO1 Bioactivatable Radiosensitizing Drugs in Triple-Negative Breast Cancers. Antioxid. Redox Signal. 2014, 21, 237–250. [Google Scholar] [CrossRef] [Green Version]
- Torrente, L.; Prieto-Farigua, N.; Falzone, A.; Elkins, C.M.; Boothman, D.A.; Haura, E.B.; DeNicola, G.M. Inhibition of TXNRD or SOD1 Overcomes NRF2-Mediated Resistance to β-Lapachone. Redox Biol. 2020, 30, 101440. [Google Scholar] [CrossRef]
- Puram, S.V.; Tirosh, I.; Parikh, A.S.; Patel, A.P.; Yizhak, K.; Gillespie, S.; Rodman, C.; Luo, C.L.; Mroz, E.A.; Emerick, K.S.; et al. Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell 2017, 171, 1611–1624.e24. [Google Scholar] [CrossRef] [Green Version]
- Tirosh, I.; Izar, B.; Prakadan, S.M.; Wadsworth, M.H.; Treacy, D.; Trombetta, J.J.; Rotem, A.; Rodman, C.; Lian, C.; Murphy, G.; et al. Dissecting the Multicellular Ecosystem of Metastatic Melanoma by Single-Cell RNA-Seq. Science 2016, 352, 189–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, Z.; Dai, Z.; Locasale, J.W. Metabolic Landscape of the Tumor Microenvironment at Single Cell Resolution. Nat. Commun. 2019, 10, 3763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, J.E.; Forshaw, T.E.; Boothman, D.A.; Furdui, C.M.; Kemp, M.L. Personalized Genome-Scale Metabolic Models Identify Targets of Redox Metabolism in Radiation-Resistant Tumors. Cell Syst. 2021, 12, 68–81.e11. [Google Scholar] [CrossRef]
- Oh, G.-S.; Kim, H.-J.; Choi, J.-H.; Shen, A.; Choe, S.-K.; Karna, A.; Lee, S.H.; Jo, H.-J.; Yang, S.-H.; Hwan Kwak, T.; et al. Pharmacological Activation of NQO1 Increases NAD þ Levels and Attenuates Cisplatin-Mediated Acute Kidney Injury in Mice. Kidney Int. 2013, 85, 547–560. [Google Scholar] [CrossRef] [Green Version]
- Lewis, J.E.; Costantini, F.; Mims, J.; Chen, X.; Furdui, C.M.; Boothman, D.A.; Kemp, M.L. Genome-Scale Modeling of NADPH-Driven β-Lapachone Sensitization in Head and Neck Squamous Cell Carcinoma. Antioxid. Redox Signal. 2018, 29, 937–952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hrovatin, K.; Fischer, D.S.; Theis, F.J. Toward Modeling Metabolic State from Single-Cell Transcriptomics. Mol. Metab. 2022, 57, 101396. [Google Scholar] [CrossRef] [PubMed]
- Schwanhüusser, B.; Busse, D.; Li, N.; Dittmar, G.; Schuchhardt, J.; Wolf, J.; Chen, W.; Selbach, M. Global Quantification of Mammalian Gene Expression Control. Nature 2011, 473, 337–342. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.; Jiang, L.; Fang, T.; Fang, F.; Liu, Y.; Zhao, Y.; You, Y.; Zhou, H.; Su, X.; Wang, J.; et al. β-Lapachone Selectively Kills Hepatocellular Carcinoma Cells by Targeting NQO1 to Induce Extensive DNA Damage and PARP1 Hyperactivation. Front. Oncol. 2021, 11, 4026. [Google Scholar] [CrossRef]
- Panieri, E.; Saso, L. Inhibition of the NRF2/KEAP1 axis: A promising therapeutic strategy to alter redox balance of cancer cells. Antioxid. Redox Signal. 2021, 34, 1428–1483. [Google Scholar] [CrossRef]
- Winski, S.; Koutalos, Y.; Bentley, D.; Ross, D. Subcellular Localization of NAD(P)H:Quinone Oxidoreductase 1 in Human Cancer Cells1|Cancer Research|American Association for Cancer Research. Available online: https://aacrjournals.org/cancerres/article/62/5/1420/509711/Subcellular-Localization-of-NAD-P-H-quinone (accessed on 22 February 2022).
- Ma, J.; Lim, C.; Sacher, J.R.; van Houten, B.; Qian, W.; Wipf, P. Mitochondrial Targeted β-Lapachone Induces Mitochondrial Dysfunction and Catastrophic Vacuolization in Cancer Cells. Bioorg. Med. Chem. Lett. 2015, 25, 4828. [Google Scholar] [CrossRef] [Green Version]
- Thiagarajah, J.R.; Chang, J.; Goettel, J.A.; Verkman, A.S.; Lencer, W.I. Aquaporin-3 Mediates Hydrogen Peroxide-Dependent Responses to Environmental Stress in Colonic Epithelia. Proc. Natl. Acad. Sci. USA 2017, 114, 568–573. [Google Scholar] [CrossRef] [Green Version]
- Bienert, G.P.; Chaumont, F. Aquaporin-Facilitated Transmembrane Diffusion of Hydrogen Peroxide. Biochim. Biophys. Acta Gen. Subj 2014, 1840, 1596–1604. [Google Scholar] [CrossRef] [PubMed]
- Bienert, G.P.; Møller, A.L.B.; Kristiansen, K.A.; Schulz, A.; Møller, I.M.; Schjoerring, J.K.; Jahn, T.P. Specific Aquaporins Facilitate the Diffusion of Hydrogen Peroxide across Membranes. J. Biol. Chem. 2007, 282, 1183–1192. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, S.; Moniaga, C.S.; Nielsen, S.; Hara-Chikuma, M. Aquaporin-9 Facilitates Membrane Transport of Hydrogen Peroxide in Mammalian Cells. Biochem. Biophys. Res. Commun. 2016, 471, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Lähnemann, D.; Köster, J.; Szczurek, E.; McCarthy, D.J.; Hicks, S.C.; Robinson, M.D.; Vallejos, C.A.; Campbell, K.R.; Beerenwinkel, N.; Mahfouz, A.; et al. Eleven Grand Challenges in Single-Cell Data Science. Genome Biol. 2020, 21, 31. [Google Scholar] [CrossRef]
- Kim, T.H.; Zhou, X.; Chen, M. Demystifying “Drop-Outs” in Single-Cell UMI Data. Genome Biol. 2020, 21, 196. [Google Scholar] [CrossRef] [PubMed]
- Qiu, P. Embracing the Dropouts in Single-Cell RNA-Seq Analysis. Nat. Commun. 2020, 11, 1169. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reaction Name | Rate Term |
|---|---|
| β-lap permeation * | k34 * Acells * ([β-lapext]–[β-lapQ]) |
| β-lap reduction | k29 * [β-lapQ] * [NADPH] |
| β-lap semioxidation | k30 * [β-lapHQ] * [O2] |
| β-lap oxidation | k31 * [β-lapSQ] * [O2] |
| superoxide dismutase | k32 * [O2•−]2 |
| β-lap semireduction | k33 * [β-lapQ] * [NADPH] |
| β-lap semiquinone semireduction | k33 * [β-lapSQ] * [NADPH] |
| β-lap glutathionylation | k35 * [β-lapHQ] * [GSH] |
| Glutathionylated β-lap permeation * | k36 * Acells * [β-lapHQ-SG] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raddatz, A.D.; Furdui, C.M.; Bey, E.A.; Kemp, M.L. Single-Cell Kinetic Modeling of β-Lapachone Metabolism in Head and Neck Squamous Cell Carcinoma. Antioxidants 2023, 12, 741. https://doi.org/10.3390/antiox12030741
Raddatz AD, Furdui CM, Bey EA, Kemp ML. Single-Cell Kinetic Modeling of β-Lapachone Metabolism in Head and Neck Squamous Cell Carcinoma. Antioxidants. 2023; 12(3):741. https://doi.org/10.3390/antiox12030741
Chicago/Turabian StyleRaddatz, Andrew D., Cristina M. Furdui, Erik A. Bey, and Melissa L. Kemp. 2023. "Single-Cell Kinetic Modeling of β-Lapachone Metabolism in Head and Neck Squamous Cell Carcinoma" Antioxidants 12, no. 3: 741. https://doi.org/10.3390/antiox12030741
APA StyleRaddatz, A. D., Furdui, C. M., Bey, E. A., & Kemp, M. L. (2023). Single-Cell Kinetic Modeling of β-Lapachone Metabolism in Head and Neck Squamous Cell Carcinoma. Antioxidants, 12(3), 741. https://doi.org/10.3390/antiox12030741