
Reactive Sulfur Species Omics Analysis in the Brain Tissue of the 5xFAD Mouse Model of Alzheimer’s Disease


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Animals
2.3. Evaluation of Cognitive Function by a Behavior Analysis Test (Y-Maze Test)
2.4. Imaging Analysis
2.5. Quantification of the Total Polysulfide Content Using Alkaline/Reductive Sulfur Elimination Protocol
2.6. Quantification of the Total Sulfur Content by Acid-Circulating Decomposition and Inductively Coupled Plasma Optical Emission Spectroscopy
2.7. Evaluation of RSS-Producing Enzyme Expression Levels by Western Blotting
2.8. Quantitative Sulfur Metabolomics Using LC-ESI–MS/MS with TME-IAM
2.9. TME-IAM Switching Assay
2.10. LC-ESI–MS/MS
2.11. Statistical Analysis
3. Results
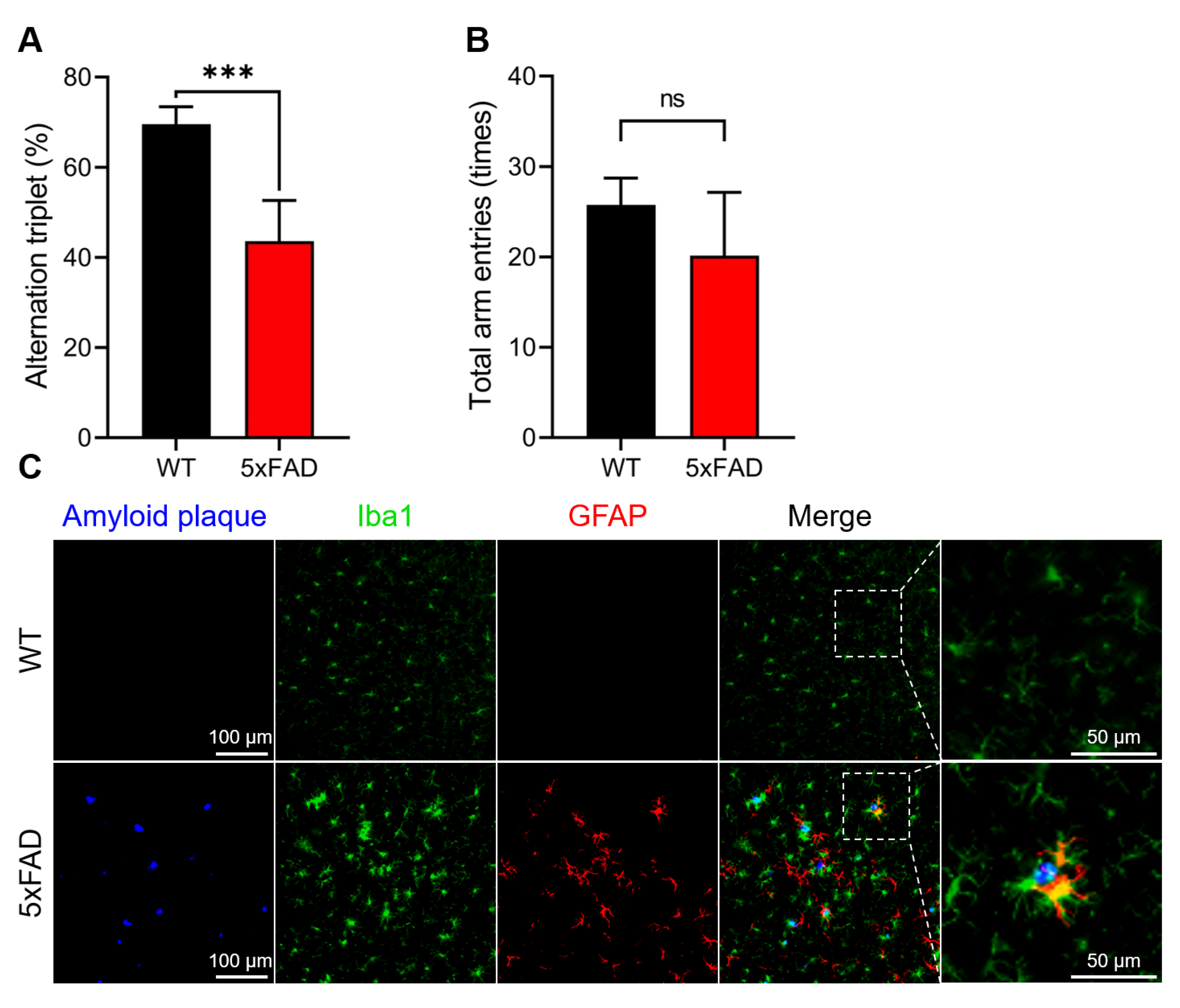
3.1. Confirmation of AD Onset in the 5xFAD Mice
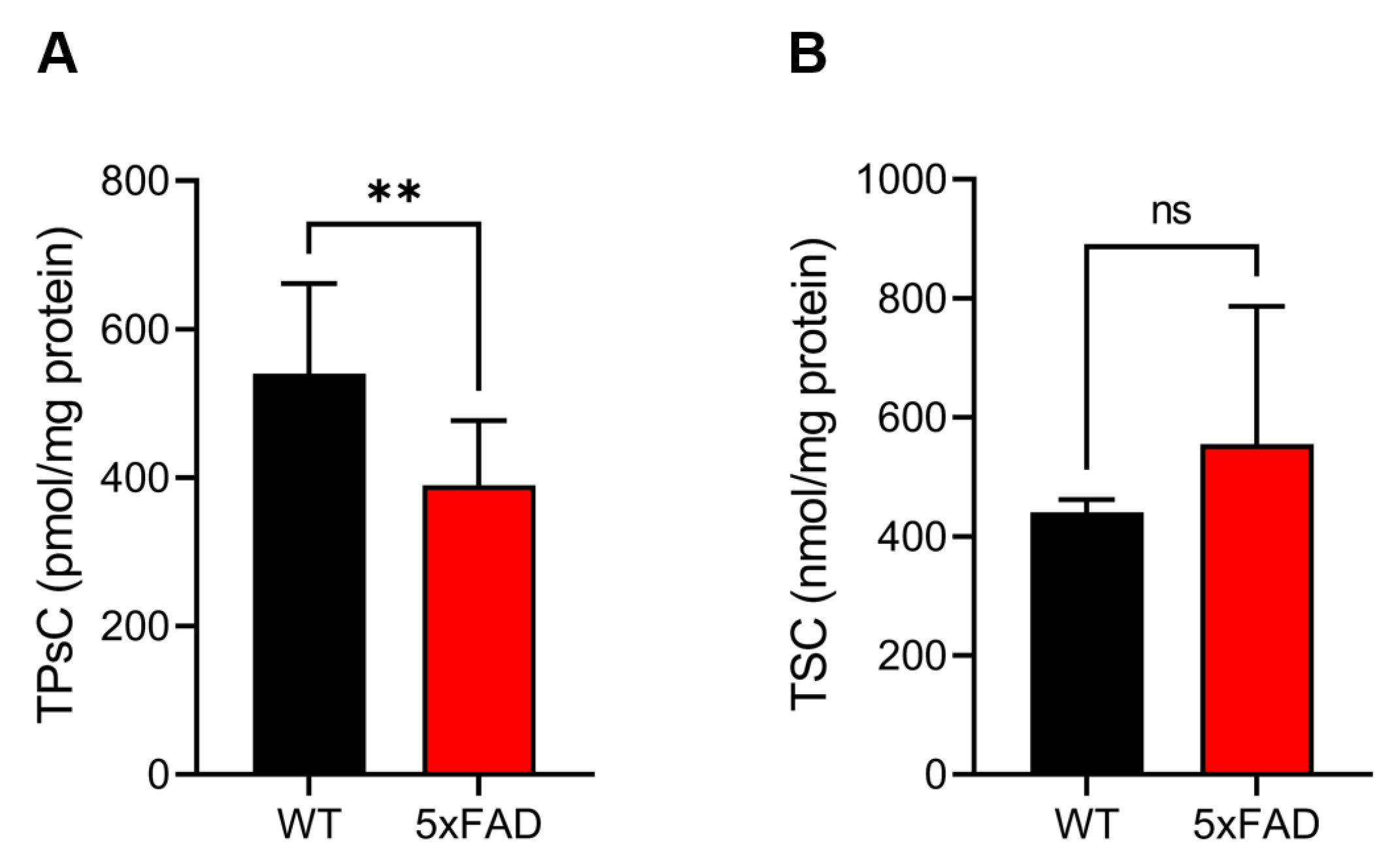
3.2. Quantification of the TPsC Using Alkaline/Reductive Sulfur Elimination Protocol and the TSC by Acid-Circulating Decomposition System
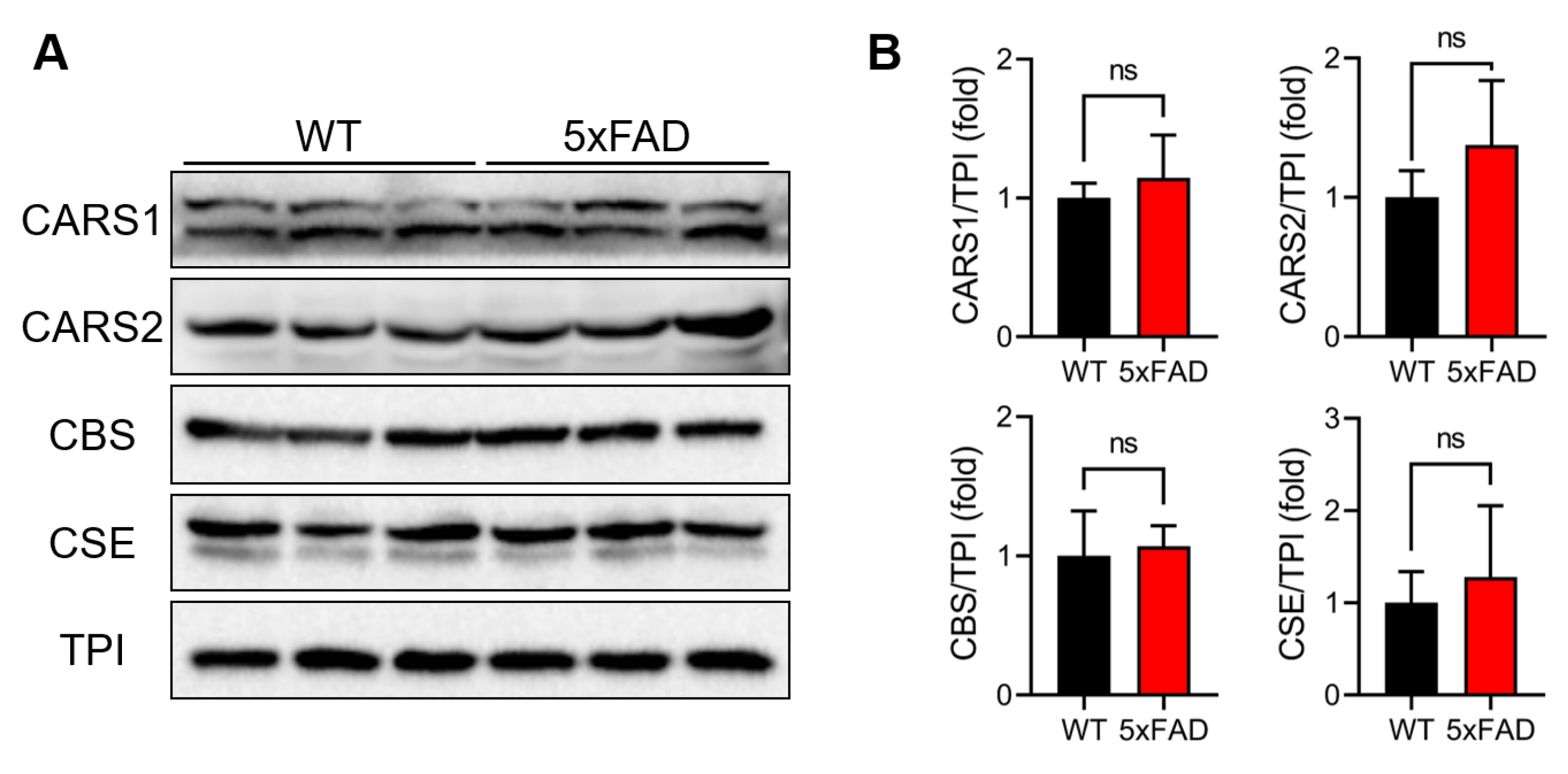
3.3. Evaluation of RSS-Producing Enzyme Expression Levels
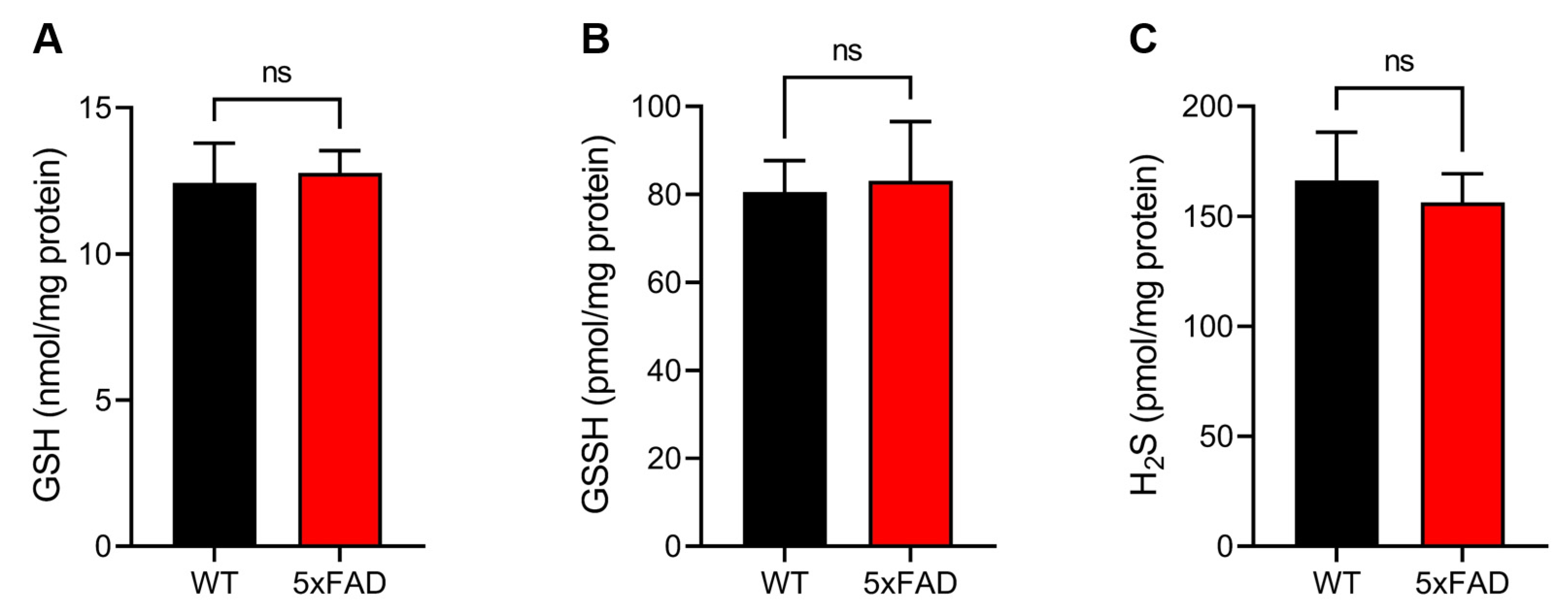
3.4. Quantitative Sulfur Metabolomics Using LC-ESI–MS/MS with TME-IAM
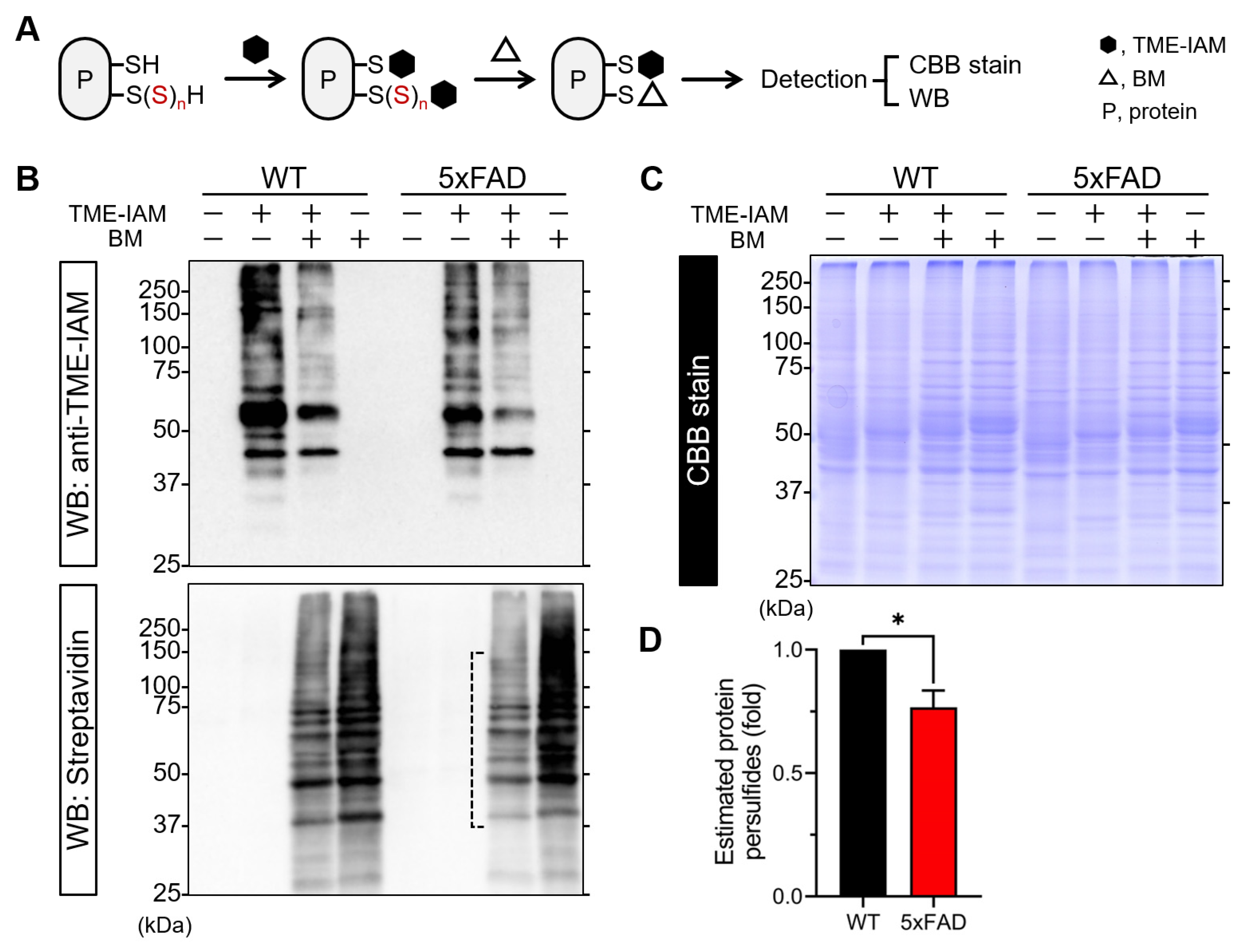
3.5. Evaluation of Protein Polysulfides by TME-IAM Switching Assay
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- 2021 Alzheimer’s Disease Facts and Figures. Alzheimers Dement. 2021, 17, 327–406. [CrossRef] [PubMed]
- Finder, V.H. Alzheimer’s Disease: A General Introduction and Pathomechanism. J. Alzheimers Dis. 2010, 22, S5–S19. [Google Scholar] [CrossRef] [PubMed]
- Pimplikar, S.W. Reassessing the Amyloid Cascade Hypothesis of Alzheimer’s Disease. Int. J. Biochem. Cell Biol. 2009, 41, 1261–1268. [Google Scholar] [CrossRef] [PubMed]
- Marcus, D.L.; Thomas, C.; Rodriguez, C.; Simberkoff, K.; Tsai, J.S.; Strafaci, J.A.; Freedman, M.L. Increased Peroxidation and Reduced Antioxidant Enzyme Activity in Alzheimer’s Disease. Exp. Neurol. 1998, 150, 40–44. [Google Scholar] [CrossRef]
- Simpson, D.S.A.; Oliver, P.L. ROS Generation in Microglia: Understanding Oxidative Stress and Inflammation in Neurodegenerative Disease. Antioxidants 2020, 9, 743. [Google Scholar] [CrossRef]
- Kishimoto, Y.; Kasamatsu, S.; Yanai, S.; Endo, S.; Akaike, T.; Ihara, H. 8-Nitro-CGMP Attenuates Context-Dependent Fear Memory in Mice. Biochem. Biophys. Res. Commun. 2019, 511, 141–147. [Google Scholar] [CrossRef]
- Heneka, M.T.; Carson, M.J.; Khoury, J.E.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s Disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- Zhao, Q.-F.; Yu, J.-T.; Tan, L. S-Nitrosylation in Alzheimer’s Disease. Mol. Neurobiol. 2015, 51, 268–280. [Google Scholar] [CrossRef]
- Li, L.; Rose, P.; Moore, P.K. Hydrogen Sulfide and Cell Signaling. Annu. Rev. Pharmacol. Toxicol. 2011, 51, 169–187. [Google Scholar] [CrossRef]
- Eto, K.; Asada, T.; Arima, K.; Makifuchi, T.; Kimura, H. Brain Hydrogen Sulfide Is Severely Decreased in Alzheimer’s Disease. Biochem. Biophys. Res. Commun. 2002, 293, 1485–1488. [Google Scholar] [CrossRef]
- Giuliani, D.; Ottani, A.; Zaffe, D.; Galantucci, M.; Strinati, F.; Lodi, R.; Guarini, S. Hydrogen Sulfide Slows down Progression of Experimental Alzheimer’s Disease by Targeting Multiple Pathophysiological Mechanisms. Neurobiol. Learn. Mem. 2013, 104, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Xing, H.; Cai, J.; Zhang, H.; Xu, S. H2S Exposure-Induced Oxidative Stress Promotes LPS-Mediated Hepatocyte Autophagy through the PI3K/AKT/TOR Pathway. Ecotoxicol. Environ. Saf. 2021, 209, 111801. [Google Scholar] [CrossRef]
- Ida, T.; Sawa, T.; Ihara, H.; Tsuchiya, Y.; Watanabe, Y.; Kumagai, Y.; Suematsu, M.; Motohashi, H.; Fujii, S.; Matsunaga, T.; et al. Reactive Cysteine Persulfides and S-Polythiolation Regulate Oxidative Stress and Redox Signaling. Proc. Natl. Acad. Sci. USA 2014, 111, 7606–7611. [Google Scholar] [CrossRef]
- Hamid, H.A.; Tanaka, A.; Ida, T.; Nishimura, A.; Matsunaga, T.; Fujii, S.; Morita, M.; Sawa, T.; Fukuto, J.M.; Nagy, P.; et al. Polysulfide Stabilization by Tyrosine and Hydroxyphenyl-Containing Derivatives That Is Important for a Reactive Sulfur Metabolomics Analysis. Redox Biol. 2019, 21, 101096. [Google Scholar] [CrossRef]
- Bogdándi, V.; Ida, T.; Sutton, T.R.; Bianco, C.; Ditrói, T.; Koster, G.; Henthorn, H.A.; Minnion, M.; Toscano, J.P.; van der Vliet, A.; et al. Speciation of Reactive Sulfur Species and Their Reactions with Alkylating Agents: Do We Have Any Clue about What Is Present inside the Cell? Br. J. Pharmacol. 2019, 176, 646–670. [Google Scholar] [CrossRef] [PubMed]
- Kasamatsu, S.; Ida, T.; Koga, T.; Asada, K.; Motohashi, H.; Ihara, H.; Akaike, T. High-Precision Sulfur Metabolomics Innovated by a New Specific Probe for Trapping Reactive Sulfur Species. Antioxid. Redox Signal. 2021, 34, 1407–1419. [Google Scholar] [CrossRef] [PubMed]
- Sawa, T.; Takata, T.; Matsunaga, T.; Ihara, H.; Motohashi, H.; Akaike, T. Chemical Biology of Reactive Sulfur Species: Hydrolysis-Driven Equilibrium of Polysulfides as a Determinant of Physiological Functions. Antioxid. Redox Signal. 2022, 36, 327–336. [Google Scholar] [CrossRef]
- Akaike, T.; Ida, T.; Wei, F.-Y.; Nishida, M.; Kumagai, Y.; Alam, M.M.; Ihara, H.; Sawa, T.; Matsunaga, T.; Kasamatsu, S.; et al. Cysteinyl-TRNA Synthetase Governs Cysteine Polysulfidation and Mitochondrial Bioenergetics. Nat. Commun. 2017, 8, 1177. [Google Scholar] [CrossRef]
- Kasamatsu, S.; Kinno, A.; Hishiyama, J.; Akaike, T.; Ihara, H. Development of Methods for Quantitative Determination of the Total and Reactive Polysulfides: Reactive Polysulfide Profiling in Vegetables. Food Chem. 2023, 413, 135610. [Google Scholar] [CrossRef]
- Quan, F.-S.; Lee, G.-J. Analytical Methods for Detection of Gasotransmitter Hydrogen Sulfide Released from Live Cells. Biomed. Res. Int. 2021, 2021, 5473965. [Google Scholar] [CrossRef]
- Olson, K.R.; DeLeon, E.R.; Liu, F. Controversies and Conundrums in Hydrogen Sulfide Biology. Nitric Oxide 2014, 41, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Marutani, E.; Morita, M.; Hirai, S.; Kai, S.; Grange, R.M.H.; Miyazaki, Y.; Nagashima, F.; Traeger, L.; Magliocca, A.; Ida, T.; et al. Sulfide Catabolism Ameliorates Hypoxic Brain Injury. Nat. Commun. 2021, 12, 3108. [Google Scholar] [CrossRef] [PubMed]
- Switzer, C.H.; Guttzeit, S.; Eykyn, T.R.; Eaton, P. Cysteine Trisulfide Oxidizes Protein Thiols and Induces Electrophilic Stress in Human Cells. Redox Biol. 2021, 47, 102155. [Google Scholar] [CrossRef] [PubMed]
- Ihara, H.; Kasamatsu, S.; Kitamura, A.; Nishimura, A.; Tsutsuki, H.; Ida, T.; Ishizaki, K.; Toyama, T.; Yoshida, E.; Abdul Hamid, H.; et al. Exposure to Electrophiles Impairs Reactive Persulfide-Dependent Redox Signaling in Neuronal Cells. Chem. Res. Toxicol. 2017, 30, 1673–1684. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Ono, K.; Tsutsuki, H.; Ihara, H.; Islam, W.; Akaike, T.; Sawa, T. Enhanced Cellular Polysulfides Negatively Regulate TLR4 Signaling and Mitigate Lethal Endotoxin Shock. Cell Chem. Biol. 2019, 26, 686–698.e4. [Google Scholar] [CrossRef]
- Barayeu, U.; Schilling, D.; Eid, M.; Xavier da Silva, T.N.; Schlicker, L.; Mitreska, N.; Zapp, C.; Gräter, F.; Miller, A.K.; Kappl, R.; et al. Hydropersulfides Inhibit Lipid Peroxidation and Ferroptosis by Scavenging Radicals. Nat. Chem. Biol. 2022, 19, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; Van Eldik, L.; et al. Intraneuronal Beta-Amyloid Aggregates, Neurodegeneration, and Neuron Loss in Transgenic Mice with Five Familial Alzheimer’s Disease Mutations: Potential Factors in Amyloid Plaque Formation. J. Neurosci. 2006, 26, 10129–10140. [Google Scholar] [CrossRef] [PubMed]
- Kasamatsu, S.; Kakihana, Y.; Koga, T.; Yoshioka, H.; Ihara, H. Generation of Rat Monoclonal Antibody to Detect Hydrogen Sulfide and Polysulfides in Biological Samples. Antioxidants 2020, 9, 1160. [Google Scholar] [CrossRef]
- Holcomb, L.; Gordon, M.N.; McGowan, E.; Yu, X.; Benkovic, S.; Jantzen, P.; Wright, K.; Saad, I.; Mueller, R.; Morgan, D.; et al. Accelerated Alzheimer-Type Phenotype in Transgenic Mice Carrying Both Mutant Amyloid Precursor Protein and Presenilin 1 Transgenes. Nat. Med. 1998, 4, 97–100. [Google Scholar] [CrossRef]
- Ohno, M.; Sametsky, E.A.; Younkin, L.H.; Oakley, H.; Younkin, S.G.; Citron, M.; Vassar, R.; Disterhoft, J.F. BACE1 Deficiency Rescues Memory Deficits and Cholinergic Dysfunction in a Mouse Model of Alzheimer’s Disease. Neuron 2004, 41, 27–33. [Google Scholar] [CrossRef]
- Shinkai, Y.; Masuda, A.; Akiyama, M.; Xian, M.; Kumagai, Y. Cadmium-Mediated Activation of the HSP90/HSF1 Pathway Regulated by Reactive Persulfides/Polysulfides. Toxicol. Sci. 2017, 156, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Kunikata, H.; Ida, T.; Sato, K.; Aizawa, N.; Sawa, T.; Tawarayama, H.; Murayama, N.; Fujii, S.; Akaike, T.; Nakazawa, T. Metabolomic Profiling of Reactive Persulfides and Polysulfides in the Aqueous and Vitreous Humors. Sci. Rep. 2017, 7, 41984. [Google Scholar] [CrossRef]
- Ono, K.; Akaike, T.; Sawa, T.; Kumagai, Y.; Wink, D.A.; Tantillo, D.J.; Hobbs, A.J.; Nagy, P.; Xian, M.; Lin, J.; et al. Redox Chemistry and Chemical Biology of H2S, Hydropersulfides, and Derived Species: Implications of Their Possible Biological Activity and Utility. Free Radic. Biol. Med. 2014, 77, 82–94. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.; Kasamatsu, S.; Matsunaga, T.; Akashi, S.; Ono, K.; Nishimura, A.; Morita, M.; Abdul Hamid, H.; Fujii, S.; Kitamura, H.; et al. Protein Polysulfidation-Dependent Persulfide Dioxygenase Activity of Ethylmalonic Encephalopathy Protein 1. Biochem. Biophys. Res. Commun. 2016, 480, 180–186. [Google Scholar] [CrossRef]
- Kajimura, M.; Fukuda, R.; Bateman, R.M.; Yamamoto, T.; Suematsu, M. Interactions of Multiple Gas-Transducing Systems: Hallmarks and Uncertainties of CO, NO, and H2S Gas Biology. Antioxid. Redox Signal. 2010, 13, 157–192. [Google Scholar] [CrossRef]
- Kimura, H. Hydrogen Sulfide: Its Production, Release and Functions. Amino Acids 2011, 41, 113–121. [Google Scholar] [CrossRef]
- Zhang, X.; Bian, J.-S. Hydrogen Sulfide: A Neuromodulator and Neuroprotectant in the Central Nervous System. ACS Chem. Neurosci. 2014, 5, 876–883. [Google Scholar] [CrossRef]
- Yang, G.; Zhao, K.; Ju, Y.; Mani, S.; Cao, Q.; Puukila, S.; Khaper, N.; Wu, L.; Wang, R. Hydrogen Sulfide Protects Against Cellular Senescence via S-Sulfhydration of Keap1 and Activation of Nrf2. Antioxid. Redox Signal. 2013, 18, 1906–1919. [Google Scholar] [CrossRef] [PubMed]
- Xuan, A.; Long, D.; Li, J.; Ji, W.; Zhang, M.; Hong, L.; Liu, J. Hydrogen Sulfide Attenuates Spatial Memory Impairment and Hippocampal Neuroinflammation in Beta-Amyloid Rat Model of Alzheimer’s Disease. J. Neuroinflammation 2012, 9, 687. [Google Scholar] [CrossRef]
- Sen, N.; Paul, B.D.; Gadalla, M.M.; Mustafa, A.K.; Sen, T.; Xu, R.; Kim, S.; Snyder, S.H. Hydrogen Sulfide-Linked Sulfhydration of NF-ΚB Mediates Its Antiapoptotic Actions. Mol. Cell 2012, 45, 13–24. [Google Scholar] [CrossRef]
- Dwyer, B.E.; Raina, A.K.; Perry, G.; Smith, M.A. Homocysteine and Alzheimer’s Disease: A Modifiable Risk? Free Radic. Biol. Med. 2004, 36, 1471–1475. [Google Scholar] [CrossRef] [PubMed]
- Giovinazzo, D.; Bursac, B.; Sbodio, J.I.; Nalluru, S.; Vignane, T.; Snowman, A.M.; Albacarys, L.M.; Sedlak, T.W.; Torregrossa, R.; Whiteman, M.; et al. Hydrogen Sulfide Is Neuroprotective in Alzheimer’s Disease by Sulfhydrating GSK3β and Inhibiting Tau Hyperphosphorylation. Proc. Natl. Acad. Sci. USA 2021, 118, e2017225118. [Google Scholar] [CrossRef] [PubMed]
- Clarke, R.; Smith, A.D.; Jobst, K.A.; Refsum, H.; Sutton, L.; Ueland, P.M. Folate, Vitamin B12, and Serum Total Homocysteine Levels in Confirmed Alzheimer Disease. Arch. Neurol. 1998, 55, 1449. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.-Y.; Wu, X.; Lu, T.; Cui, G.; Chen, G. Research Progress of Hydrogen Sulfide in Alzheimer′s Disease from Laboratory to Hospital: A Narrative Review. Med. Gas. Res. 2020, 10, 125–129. [Google Scholar] [CrossRef]
- Silva-Islas, C.A.; Chánez-Cárdenas, M.E.; Barrera-Oviedo, D.; Ortiz-Plata, A.; Pedraza-Chaverri, J.; Maldonado, P.D. Diallyl Trisulfide Protects Rat Brain Tissue against the Damage Induced by Ischemia-Reperfusion through the Nrf2 Pathway. Antioxidants 2019, 8, 410. [Google Scholar] [CrossRef]
- Yoshioka, Y.; Matsumura, S.; Morimoto, M.; Takemoto, Y.; Kishi, C.; Moriyama, T.; Zaima, N. Inhibitory Activities of Sulfur Compounds in Garlic Essential Oil against Alzheimer’s Disease-Related Enzymes and Their Distribution in the Mouse Brain. J. Agric. Food Chem. 2021, 69, 10163–10173. [Google Scholar] [CrossRef]
- Filipovic, M.R.; Zivanovic, J.; Alvarez, B.; Banerjee, R. Chemical Biology of H2S Signaling through Persulfidation. Chem. Rev. 2018, 118, 1253–1337. [Google Scholar] [CrossRef]
- Filipovic, M.R. Persulfidation (S-Sulfhydration) and H2S. Handb. Exp. Pharmacol. 2015, 230, 29–59. [Google Scholar] [CrossRef]
- Zivanovic, J.; Kouroussis, E.; Kohl, J.B.; Adhikari, B.; Bursac, B.; Schott-Roux, S.; Petrovic, D.; Miljkovic, J.L.; Thomas-Lopez, D.; Jung, Y.; et al. Selective Persulfide Detection Reveals Evolutionarily Conserved Antiaging Effects of S-Sulfhydration. Cell Metabol. 2019, 30, 1152–1170.e13. [Google Scholar] [CrossRef]
- Yang, C.; Devarie-Baez, N.O.; Hamsath, A.; Fu, X.; Xian, M. S-Persulfidation: Chemistry, Chemical Biology, and Significance in Health and Disease. Antioxid. Redox Signal. 2020, 33, 1092–1114. [Google Scholar] [CrossRef]
- Dóka, É.; Pader, I.; Bíró, A.; Johansson, K.; Cheng, Q.; Ballagó, K.; Prigge, J.R.; Pastor-Flores, D.; Dick, T.P.; Schmidt, E.E.; et al. A Novel Persulfide Detection Method Reveals Protein Persulfide- and Polysulfide-Reducing Functions of Thioredoxin and Glutathione Systems. Sci. Adv. 2016, 2, e1500968. [Google Scholar] [CrossRef]
- Cuevasanta, E.; Möller, M.N.; Alvarez, B. Biological Chemistry of Hydrogen Sulfide and Persulfides. Arch. Biochem. Biophys. 2017, 617, 9–25. [Google Scholar] [CrossRef] [PubMed]
- Nagy, P. Mechanistic Chemical Perspective of Hydrogen Sulfide Signaling. Methods Enzymol. 2015, 554, 3–29. [Google Scholar] [CrossRef] [PubMed]
- Yadav, P.K.; Martinov, M.; Vitvitsky, V.; Seravalli, J.; Wedmann, R.; Filipovic, M.R.; Banerjee, R. Biosynthesis and Reactivity of Cysteine Persulfides in Signaling. J. Am. Chem. Soc. 2016, 138, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Greiner, R.; Pálinkás, Z.; Bäsell, K.; Becher, D.; Antelmann, H.; Nagy, P.; Dick, T.P. Polysulfides Link H2S to Protein Thiol Oxidation. Antioxid. Redox Signal. 2013, 19, 1749–1765. [Google Scholar] [CrossRef] [PubMed]
- Millikin, R.; Bianco, C.L.; White, C.; Saund, S.S.; Henriquez, S.; Sosa, V.; Akaike, T.; Kumagai, Y.; Soeda, S.; Toscano, J.P.; et al. The Chemical Biology of Protein Hydropersulfides: Studies of a Possible Protective Function of Biological Hydropersulfide Generation. Free Radic. Biol. Med. 2016, 97, 136–147. [Google Scholar] [CrossRef]
- Dóka, É.; Ida, T.; Dagnell, M.; Abiko, Y.; Luong, N.C.; Balog, N.; Takata, T.; Espinosa, B.; Nishimura, A.; Cheng, Q.; et al. Control of Protein Function through Oxidation and Reduction of Persulfidated States. Sci. Adv. 2020, 6, eaax8358. [Google Scholar] [CrossRef]
- Zhang, D.; Du, J.; Tang, C.; Huang, Y.; Jin, H. H2S-Induced Sulfhydration: Biological Function and Detection Methodology. Front. Pharmacol. 2017, 8, 608. [Google Scholar] [CrossRef]
- Sun, H.-J.; Wu, Z.-Y.; Nie, X.-W.; Bian, J.-S. Role of Hydrogen Sulfide and Polysulfides in Neurological Diseases: Focus on Protein S-Persulfidation. Curr. Neuropharmacol. 2021, 19, 868–884. [Google Scholar] [CrossRef]
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria Dysfunction in the Pathogenesis of Alzheimer’s Disease: Recent Advances. Mol. Neurodegener. 2020, 15, 30. [Google Scholar] [CrossRef]
- Baloyannis, S.J. Mitochondrial Alterations in Alzheimer’s Disease. J. Alzheimers Dis. 2006, 9, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; Karbowski, M. Mitochondrial Fission in Apoptosis. Nat. Rev. Mol. Cell Biol. 2005, 6, 657–663. [Google Scholar] [CrossRef]
- Knott, A.B.; Perkins, G.; Schwarzenbacher, R.; Bossy-Wetzel, E. Mitochondrial Fragmentation in Neurodegeneration. Nat. Rev. Neurosci. 2008, 9, 505–518. [Google Scholar] [CrossRef] [PubMed]
- Abiko, Y.; Yoshida, E.; Ishii, I.; Fukuto, J.M.; Akaike, T.; Kumagai, Y. Involvement of Reactive Persulfides in Biological Bismethylmercury Sulfide Formation. Chem. Res. Toxicol. 2015, 28, 1301–1306. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, A.; Shimoda, K.; Tanaka, T.; Toyama, T.; Nishiyama, K.; Shinkai, Y.; Numaga-Tomita, T.; Yamazaki, D.; Kanda, Y.; Akaike, T.; et al. Depolysulfidation of Drp1 Induced by Low-Dose Methylmercury Exposure Increases Cardiac Vulnerability to Hemodynamic Overload. Sci. Signal. 2019, 12, eaaw1920. [Google Scholar] [CrossRef]
- Vitvitsky, V.; Kumar, R.; Libiad, M.; Maebius, A.; Landry, A.P.; Banerjee, R. The Mitochondrial NADH Pool Is Involved in Hydrogen Sulfide Signaling and Stimulation of Aerobic Glycolysis. J. Biol. Chem. 2021, 296, 100736. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kinno, A.; Kasamatsu, S.; Akaike, T.; Ihara, H. Reactive Sulfur Species Omics Analysis in the Brain Tissue of the 5xFAD Mouse Model of Alzheimer’s Disease. Antioxidants 2023, 12, 1105. https://doi.org/10.3390/antiox12051105
Kinno A, Kasamatsu S, Akaike T, Ihara H. Reactive Sulfur Species Omics Analysis in the Brain Tissue of the 5xFAD Mouse Model of Alzheimer’s Disease. Antioxidants. 2023; 12(5):1105. https://doi.org/10.3390/antiox12051105
Chicago/Turabian StyleKinno, Ayaka, Shingo Kasamatsu, Takaaki Akaike, and Hideshi Ihara. 2023. "Reactive Sulfur Species Omics Analysis in the Brain Tissue of the 5xFAD Mouse Model of Alzheimer’s Disease" Antioxidants 12, no. 5: 1105. https://doi.org/10.3390/antiox12051105
APA StyleKinno, A., Kasamatsu, S., Akaike, T., & Ihara, H. (2023). Reactive Sulfur Species Omics Analysis in the Brain Tissue of the 5xFAD Mouse Model of Alzheimer’s Disease. Antioxidants, 12(5), 1105. https://doi.org/10.3390/antiox12051105