
Antioxidant Properties of Oral Antithrombotic Therapies in Atherosclerotic Disease and Atrial Fibrillation

 , , ,
, , ,  ,
,  ,
, 
Abstract
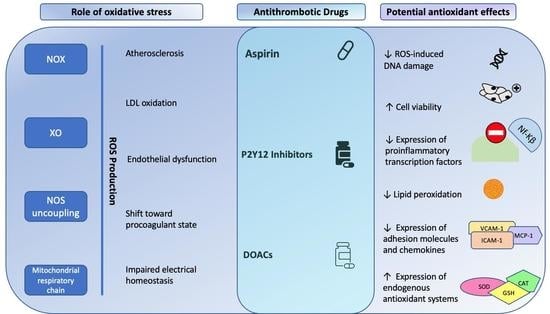
:
1. Introduction
2. Role of Oxidative Stress in Thrombosis
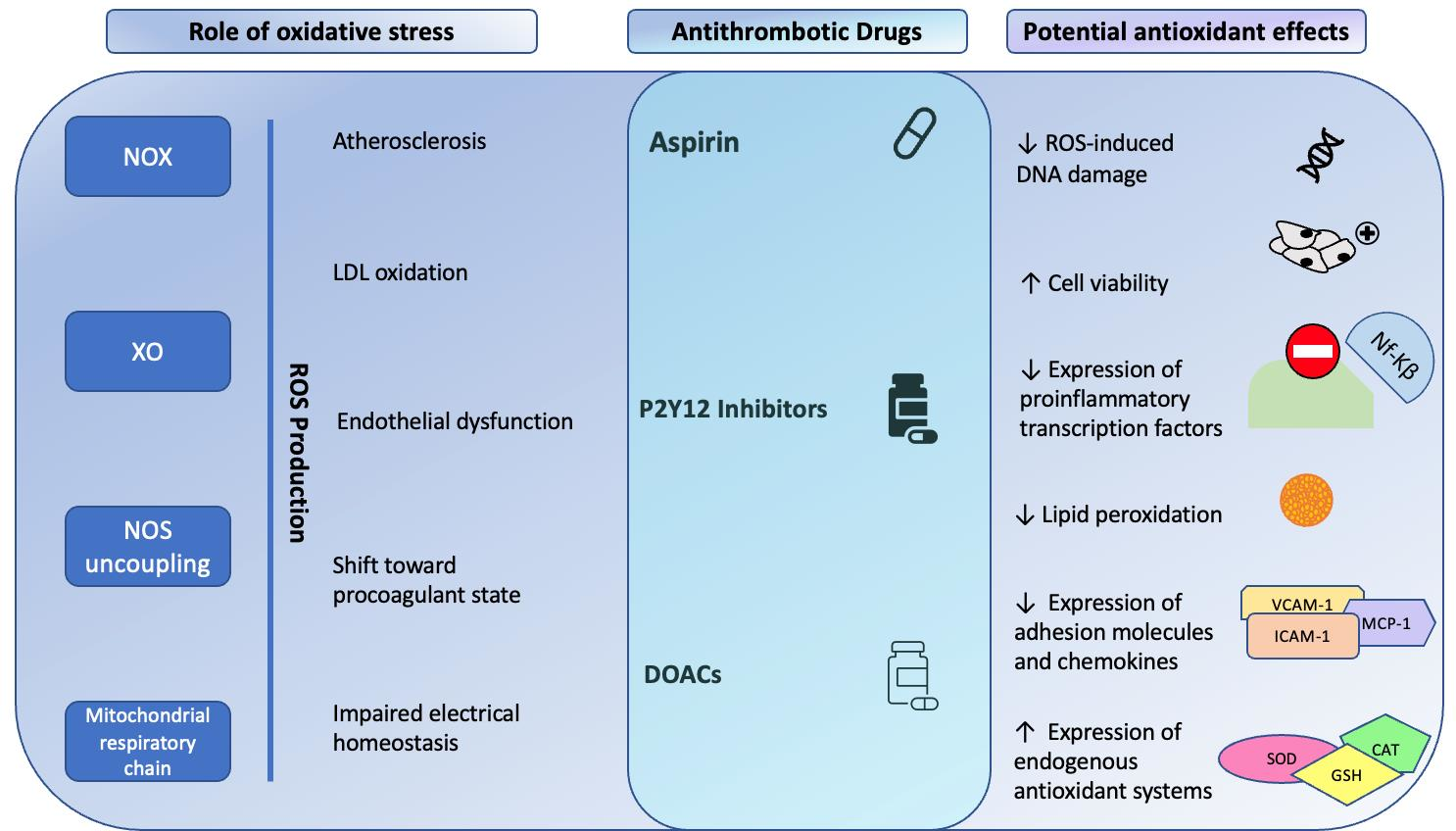
2.1. Role of Oxidative Stress in Atherogenesis
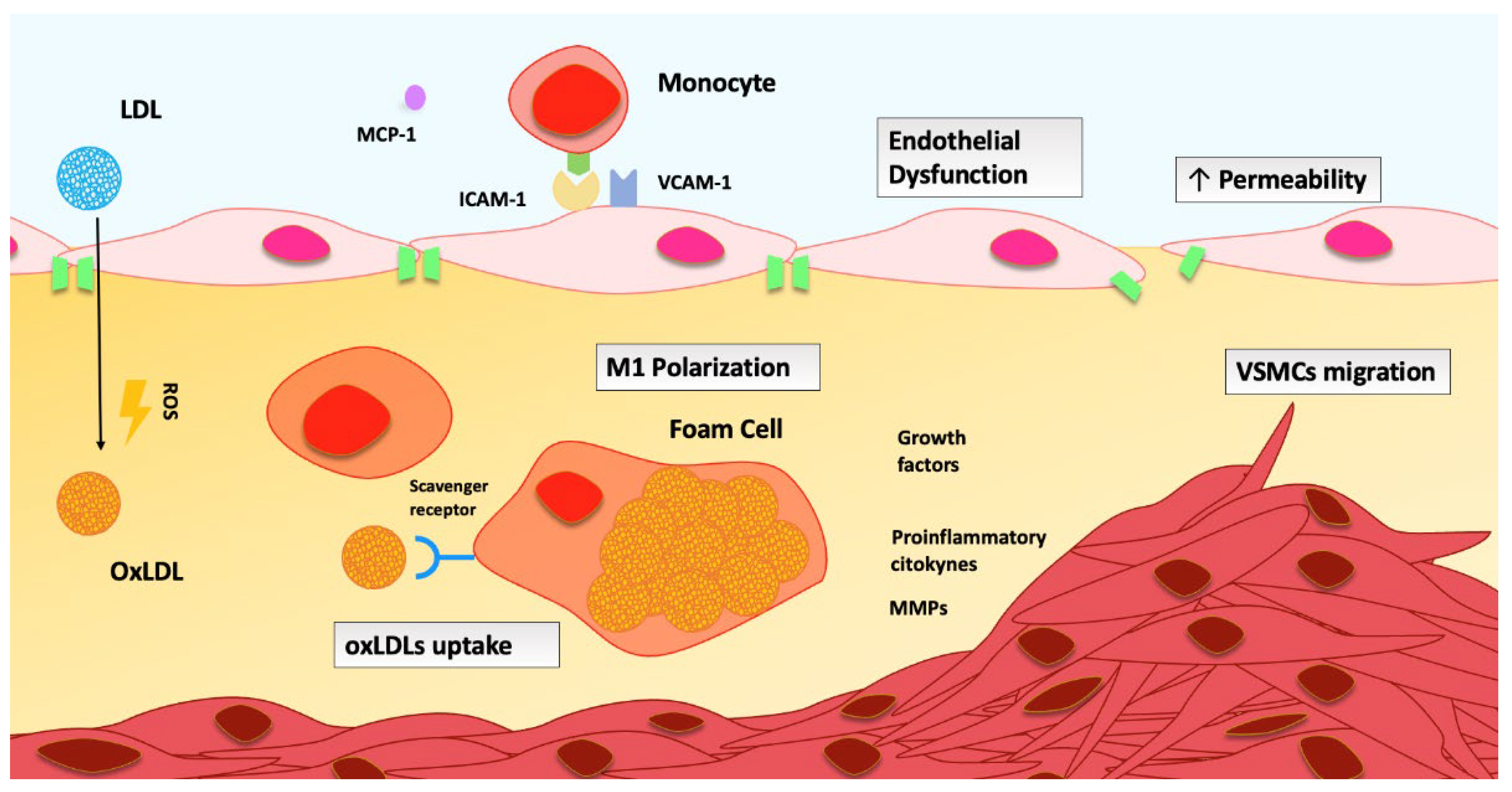
2.2. Role of Oxidative Stress in Venous Thrombosis
2.3. Role of Oxidative Stress in Atrial Fibrillation
3. Antioxidant Effects of Antiplatelet Drugs
3.1. Aspirin
3.2. P2Y12 Inhibitors
4. Antioxidant Effects of DOACs
4.1. Rivaroxaban
4.2. Apixaban
4.3. Edoxaban
4.4. Dabigatran
5. Clinical Implications
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dubois-Deruy, E.; Peugnet, V.; Turkieh, A.; Pinet, F. Oxidative Stress in Cardiovascular Diseases. Antioxidants 2020, 9, 864. [Google Scholar] [CrossRef] [PubMed]
- Ibanez, B.; James, S.; Agewall, S.; Antunes, M.J.; Bucciarelli-Ducci, C.; Bueno, H.; Caforio, A.L.P.; Crea, F.; Goudevenos, J.A.; Halvorsen, S.; et al. 2017 ESC Guidelines for the management of acute myocardial infarction in patients presenting with ST-segment elevation: The Task Force for the management of acute myocardial infarction in patients presenting with ST-segment elevation of the European Society of Cardiology (ESC). Eur. Heart J. 2018, 39, 119–177. [Google Scholar] [PubMed]
- Knuuti, J.; Wijns, W.; Saraste, A.; Capodanno, D.; Barbato, E.; Funck-Brentano, C.; Prescott, E.; Storey, R.F.; Deaton, C.; Cuisset, T.; et al. 2019 ESC Guidelines for the diagnosis and management of chronic coronary syndromes. Eur. Heart J. 2020, 41, 407–477. [Google Scholar] [CrossRef] [PubMed]
- Collet, J.P.; Thiele, H.; Barbato, E.; Barthélémy, O.; Bauersachs, J.; Bhatt, D.L.; Dendale, P.; Dorobantu, M.; Edvardsen, T.; Folliguet, T.; et al. 2020 ESC Guidelines for the management of acute coronary syndromes in patients presenting without persistent ST-segment elevation. Eur. Heart J. 2021, 42, 1289–1367. [Google Scholar] [CrossRef]
- Aboyans, V.; Ricco, J.B.; Bartelink, M.E.L.; Björck, M.; Brodmann, M.; Cohnert, T.; Collet, J.P.; Czerny, M.; De Carlo, M.; Debus, S.; et al. 2017 ESC Guidelines on the Diagnosis and Treatment of Peripheral Arterial Diseases, in collaboration with the European Society for Vascular Surgery (ESVS): Document covering atherosclerotic disease of extracranial carotid and vertebral, mesenteric, renal, upper and lower extremity arteriesEndorsed by: The European Stroke Organization (ESO)The Task Force for the Diagnosis and Treatment of Peripheral Arterial Diseases of the European Society of Cardiology (ESC) and of the European Society for Vascular Surgery (ESVS). Eur. Heart J. 2018, 39, 763–816. [Google Scholar]
- Hindricks, G.; Potpara, T.; Dagres, N.; Arbelo, E.; Bax, J.J.; Blomström-Lundqvist, C.; Boriani, G.; Castella, M.; Dan, G.A.; Dilaveris, P.E.; et al. 2020 ESC Guidelines for the diagnosis and management of atrial fibrillation developed in collaboration with the European Association for Cardio-Thoracic Surgery (EACTS): The Task Force for the diagnosis and management of atrial fibrillation of the European Society of Cardiology (ESC) Developed with the special contribution of the European Heart Rhythm Association (EHRA) of the ESC. Eur. Heart J. 2021, 42, 373–498. [Google Scholar]
- Ortel, T.L.; Neumann, I.; Ageno, W.; Beyth, R.; Clark, N.P.; Cuker, A.; Hutten, B.A.; Jaff, M.R.; Manja, V.; Schulman, S.; et al. American Society of Hematology 2020 guidelines for management of venous thromboembolism: Treatment of deep vein thrombosis and pulmonary embolism. Blood Adv. 2020, 4, 4693–4738. [Google Scholar] [CrossRef]
- Kleindorfer, D.O.; Towfighi, A.; Chaturvedi, S.; Cockroft, K.M.; Gutierrez, J.; Lombardi-Hill, D.; Kamel, H.; Kernan, W.N.; Kittner, S.J.; Leira, E.C.; et al. 2021 Guideline for the Prevention of Stroke in Patients with Stroke and Transient Ischemic Attack: A Guideline from the American Heart Association/American Stroke Association. Stroke 2021, 52, e364–e467. [Google Scholar] [CrossRef]
- Hicks, T.; Stewart, F.; Eisinga, A. NOACs versus warfarin for stroke prevention in patients with AF: A systematic review and meta-analysis. Open Heart 2016, 3, e000279. [Google Scholar] [CrossRef]
- Ruff, C.T.; Giugliano, R.P.; Braunwald, E.; Hoffman, E.B.; Deenadayalu, N.; Ezekowitz, M.D.; Camm, A.J.; Weitz, J.I.; Lewis, B.S.; Parkhomenko, A.; et al. Comparison of the efficacy and safety of new oral anticoagulants with warfarin in patients with atrial fibrillation: A meta-analysis of randomised trials. Lancet 2014, 383, 955–962. [Google Scholar] [CrossRef]
- Van der Hulle, T.; Kooiman, J.; den Exter, P.L.; Dekkers, O.M.; Klok, F.A.; Huisman, M.V. Effectiveness and safety of novel oral anticoagulants as compared with vitamin K antagonists in the treatment of acute symptomatic venous thromboembolism: A systematic review and meta-analysis. J. Thromb. Haemost. 2014, 12, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Russo, V.; Cattaneo, D.; Giannetti, L.; Bottino, R.; Laezza, N.; Atripaldi, U.; Clementi, E. Pharmacokinetics of Direct Oral Anticoagulants in Patients with Atrial Fibrillation and Extreme Obesity. Clin. Ther. 2021, 43, e255–e263. [Google Scholar] [CrossRef]
- Mai, V.; Marceau-Ferron, E.; Bertoletti, L.; Lacasse, Y.; Bonnet, S.; Lega, J.C.; Provencher, S. Direct oral anticoagulants in the treatment of acute venous thromboembolism in patients with obesity: A systematic review with meta-analysis. Pharmacol. Res. 2021, 163, 105317. [Google Scholar] [CrossRef] [PubMed]
- Russo, V.; Rago, A.; Papa, A.A.; Meo, F.D.; Attena, E.; Golino, P.; D’Onofrio, A.; Nigro, G. Use of Non-Vitamin K Antagonist Oral Anticoagulants in Atrial Fibrillation Patients with Malignancy: Clinical Practice Experience in a Single Institution and Literature Review. Semin. Thromb. Hemost. 2018, 44, 370–376. [Google Scholar]
- Russo, V.; Bottino, R.; Rago, A.; Micco, P.D.; D’ Onofrio, A.; Liccardo, B.; Golino, P.; Nigro, G. Atrial Fibrillation and Malignancy: The Clinical Performance of Non-Vitamin K Oral Anticoagulants-A Systematic Review. Semin. Thromb. Hemost. 2019, 45, 205–214. [Google Scholar] [PubMed]
- Russo, V.; Carbone, A.; Rago, A.; Golino, P.; Nigro, G. Direct Oral Anticoagulants in Octogenarians with Atrial Fibrillation: It Is Never Too Late. J. Cardiovasc. Pharmacol. 2019, 73, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Grymonprez, M.; Steurbaut, S.; De Backer, T.L.; Petrovic, M.; Lahousse, L. Effectiveness and Safety of Oral Anticoagulants in Older Patients with Atrial Fibrillation: A Systematic Review and Meta-Analysis. Front. Pharmacol. 2020, 11, 583311. [Google Scholar] [CrossRef]
- Carbone, A.; Santelli, F.; Bottino, R.; Attena, E.; Mazzone, C.; Parisi, V.; D’Andrea, A.; Golino, P.; Nigro, G.; Russo, V. Prevalence and clinical predictors of inappropriate direct oral anticoagulant dosage in octagenarians with atrial fibrillation. Eur. J. Clin. Pharmacol. 2022, 78, 879–886. [Google Scholar] [CrossRef]
- Carbone, A.; Bottino, R.; Attena, E.; Parisi, V.; Conte, M.; D’Andrea, A.; Imbalzano, E.; Golino, P.; Russo, V. Clinical impact of oral anticoagulation among octogenarians with atrial fibrillation and anaemia. J. Thromb. Thrombolysis 2023, 55, 222–227. [Google Scholar] [CrossRef]
- Russo, V.; Attena, E.; Di Maio, M.; Carbone, A.; Parisi, V.; Rago, A.; Grieco, F.V.; Buonauro, A.; Golino, P.; Nigro, G. Non-vitamin K vs vitamin K oral anticoagulants in patients aged >80 year with atrial fibrillation and low body weight. Eur. J. Clin. Investig. 2020, 50, e13335. [Google Scholar] [CrossRef]
- Russo, V.; Attena, E.; Di Maio, M.; Mazzone, C.; Carbone, A.; Parisi, V.; Rago, A.; D’Onofrio, A.; Golino, P.; Nigro, G. Clinical profile of direct oral anticoagulants versus vitamin K anticoagulants in octogenarians with atrial fibrillation: A multicentre propensity score matched real-world cohort study. J. Thromb. Thrombolysis 2020, 49, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Russo, V.; Attena, E.; Mazzone, C.; Esposito, F.; Parisi, V.; Bancone, C.; Rago, A.; Nigro, G.; Sangiuolo, R.; D’ Onofrio, A. Nonvitamin K Antagonist Oral Anticoagulants Use in Patients with Atrial Fibrillation and Bioprosthetic Heart Valves/Prior Surgical Valve Repair: A Multicenter Clinical Practice Experience. Semin. Thromb. Hemost. 2018, 44, 364–369. [Google Scholar]
- Russo, V.; Carbone, A.; Attena, E.; Rago, A.; Mazzone, C.; Proietti, R.; Parisi, V.; Scotti, A.; Nigro, G.; Golino, P.; et al. Clinical Benefit of Direct Oral Anticoagulants Versus Vitamin K Antagonists in Patients with Atrial Fibrillation and Bioprosthetic Heart Valves. Clin. Ther. 2019, 41, 2549–2557. [Google Scholar] [CrossRef] [PubMed]
- Melillo, E.; Rago, A.; Proietti, R.; Attena, E.; Carrella, M.; Golino, P.; D’Onofrio, A.; Nigro, G.; Russo, V. Atrial Fibrillation and Mitral Regurgitation: Clinical Performance of Direct Oral Anticoagulants in a Real-World Setting. J. Cardiovasc. Pharmacol. Ther. 2020, 25, 564–569. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Wang, Q.; Zhu, J.; Xiao, Q.; Zhang, L. Reactive oxygen species: Key regulators in vascular health and diseases. Br. J. Pharmacol. 2018, 175, 1279–1292. [Google Scholar] [CrossRef]
- Konior, A.; Schramm, A.; Czesnikiewicz-Guzik, M.; Guzik, T.J. NADPH oxidases in vascular pathology. Antioxid. Redox Signal. 2014, 20, 2794–2814. [Google Scholar] [CrossRef]
- Förstermann, U. Oxidative stress in vascular disease: Causes, defense mechanisms and potential therapies. Nat. Clin. Pract. Cardiovasc. Med. 2008, 5, 338–349. [Google Scholar] [CrossRef]
- Förstermann, U.; Xia, N.; Li, H. Roles of Vascular Oxidative Stress and Nitric Oxide in the Pathogenesis of Atherosclerosis. Circ. Res. 2017, 120, 713–735. [Google Scholar] [CrossRef]
- Martin-Ventura, J.L.; Rodrigues-Diez, R.; Martinez-Lopez, D.; Salaices, M.; Blanco-Colio, L.M.; Briones, A.M. Oxidative Stress in Human Atherothrombosis: Sources, Markers and Therapeutic Targets. Int. J. Mol. Sci. 2017, 18, 2315. [Google Scholar] [CrossRef]
- Barry-Lane, P.A.; Patterson, C.; van der Merwe, M.; Hu, Z.; Holland, S.M.; Yeh, E.T.; Runge, M.S. p47phox is required for atherosclerotic lesion progression in ApoE(−/−) mice. J. Clin. Investig. 2001, 108, 1513–1522. [Google Scholar] [CrossRef]
- Gray, S.P.; Di Marco, E.; Okabe, J.; Szyndralewiez, C.; Heitz, F.; Montezano, A.C.; de Haan, J.B.; Koulis, C.; El-Osta, A.; Andrews, K.L.; et al. NADPH oxidase 1 plays a key role in diabetes mellitus-accelerated atherosclerosis. Circulation 2013, 127, 1888–1902. [Google Scholar] [CrossRef] [PubMed]
- Kalinina, N.; Agrotis, A.; Tararak, E.; Antropova, Y.; Kanellakis, P.; Ilyinskaya, O.; Quinn, M.T.; Smirnov, V.; Bobik, A. Cytochrome b558-dependent NAD(P)H oxidase-phox units in smooth muscle and macrophages of atherosclerotic lesions. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 2037–2043. [Google Scholar] [CrossRef]
- Sorescu, D.; Weiss, D.; Lassègue, B.; Clempus, R.E.; Szöcs, K.; Sorescu, G.P.; Valppu, L.; Quinn, M.T.; Lambeth, J.D.; Vega, J.D.; et al. Superoxide production and expression of nox family proteins in human atherosclerosis. Circulation 2002, 105, 1429–1435. [Google Scholar] [CrossRef] [PubMed]
- Kleinschnitz, C.; Grund, H.; Wingler, K.; Armitage, M.E.; Jones, E.; Mittal, M.; Barit, D.; Schwarz, T.; Geis, C.; Kraft, P.; et al. Post-stroke inhibition of induced NADPH oxidase type 4 prevents oxidative stress and neurodegeneration. PLoS Biol. 2010, 8, e1000479. [Google Scholar] [CrossRef]
- Guzik, T.J.; Chen, W.; Gongora, M.C.; Guzik, B.; Lob, H.E.; Mangalat, D.; Hoch, N.; Dikalov, S.; Rudzinski, P.; Kapelak, B.; et al. Calcium- dependent NOX5 nicotinamide adenine dinucleotide phosphate oxidase contributes to vascular oxidative stress in human coronary artery disease. J. Am. Coll. Cardiol. 2008, 52, 1803–1809. [Google Scholar] [CrossRef]
- Manea, A.; Manea, S.A.; Gan, A.M.; Constantin, A.; Fenyo, I.M.; Raicu, M.; Muresian, H.; Simionescu, M. Human monocytes and macrophages express NADPH oxidase 5; a potential source of reactive oxygen species in atherosclerosis. Biochem. Biophys. Res. Commun. 2015, 461, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Guzik, T.J.; Sadowski, J.; Guzik, B.; Jopek, A.; Kapelak, B.; Przybylowski, P.; Wierzbicki, K.; Korbut, R.; Harrison, D.G.; Channon, K.M. Coronary artery superoxide production and nox isoform expression in human coronary artery disease. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 333–339. [Google Scholar] [CrossRef]
- Vendrov, A.E.; Hakim, Z.S.; Madamanchi, N.R.; Rojas, M.; Madamanchi, C.; Runge, M.S. Atherosclerosis is attenuated by limiting superoxide generation in both macrophages and vessel wall cells. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2714–2721. [Google Scholar] [CrossRef]
- Landmesser, U.; Spiekermann, S.; Preuss, C.; Sorrentino, S.; Fischer, D.; Manes, C.; Mueller, M.; Drexler, H. Angiotensin II induces endothelial xanthine oxidase activation: Role for endothelial dysfunction in patients with coronary disease. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 943–948. [Google Scholar] [CrossRef]
- White, C.R.; Darley-Usmar, V.; Berrington, W.R.; McAdams, M.; Gore, J.Z.; Thompson, J.A.; Parks, D.A.; Tarpey, M.M.; Freeman, B.A. Circulating plasma xanthine oxidase contributes to vascular dysfunction in hypercholesterolemic rabbits. Proc. Natl. Acad. Sci. USA 1996, 93, 8745–8749. [Google Scholar] [CrossRef]
- Guthikonda, S.; Sinkey, C.; Barenz, T.; Haynes, W.G. Xanthine oxidase inhibition reverses endothelial dysfunction in heavy smokers. Circulation 2003, 107, 416–421. [Google Scholar] [CrossRef]
- Schröder, K.; Vecchione, C.; Jung, O.; Schreiber, J.G.; Shiri-Sverdlov, R.; van Gorp, P.J.; Busse, R.; Brandes, R.P. Xanthine oxidase inhibitor tungsten prevents the development of atherosclerosis in ApoE knockout mice fed a Western-type diet. Free Radic. Biol. Med. 2006, 41, 1353–1360. [Google Scholar] [CrossRef]
- Förstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Förstermann, U. Uncoupling of endothelial NO synthase in atherosclerosis and vascular disease. Curr. Opin. Pharmacol. 2013, 13, 161–167. [Google Scholar] [CrossRef]
- Wohlfart, P.; Xu, H.; Endlich, A.; Habermeier, A.; Closs, E.I.; Hübschle, T.; Mang, C.; Strobel, H.; Suzuki, T.; Kleinert, H.; et al. Antiatherosclerotic effects of small-molecular-weight compounds enhancing endothelial nitric-oxide synthase (eNOS) expression and preventing eNOS uncoupling. J. Pharmacol. Exp. Ther. 2008, 325, 370–379. [Google Scholar] [CrossRef]
- Alp, N.J.; McAteer, M.A.; Khoo, J.; Choudhury, R.P.; Channon, K.M. Increased endothelial tetrahydrobiopterin synthesis by targeted transgenic GTP- cyclohydrolase I overexpression reduces endothelial dysfunction and atherosclerosis in ApoE-knockout mice. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 445–450. [Google Scholar] [CrossRef] [PubMed]
- Antoniades, C.; Shirodaria, C.; Crabtree, M.; Rinze, R.; Alp, N.; Cunnington, C.; Diesch, J.; Tousoulis, D.; Stefanadis, C.; Leeson, P.; et al. Altered plasma versus vascular biopterins in human atherosclerosis reveal relationships between endothelial nitric oxide synthase coupling, endothelial function, and inflammation. Circulation 2007, 116, 2851–2859. [Google Scholar] [CrossRef]
- Kuhlencordt, P.J.; Chen, J.; Han, F.; Astern, J.; Huang, P.L. Genetic deficiency of inducible nitric oxide synthase reduces atherosclerosis and lowers plasma lipid peroxides in apolipoprotein E-knockout mice. Circulation 2001, 103, 3099–3104. [Google Scholar] [CrossRef]
- Peoples, J.N.; Saraf, A.; Ghazal, N.; Pham, T.T.; Kwong, J.Q. Mitochondrial dysfunction and oxidative stress in heart disease. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ballinger, S.W.; Patterson, C.; Knight-Lozano, C.A.; Burow, D.L.; Conklin, C.A.; Hu, Z.; Reuf, J.; Horaist, C.; Lebovitz, R.; Hunter, G.C.; et al. Mitochondrial integrity and function in atherogenesis. Circulation 2002, 106, 544–549. [Google Scholar] [CrossRef]
- Zeini, M.; López-Fontal, R.; Través, P.G.; Benito, G.; Hortelano, S. Differential sensitivity to apoptosis among the cells that contribute to the atherosclerotic disease. Biochem. Biophys. Res. Commun. 2007, 363, 444–450. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, M.; Runge, M.S.; Faraci, F.M.; Heistad, D.D. MnSOD deficiency increases endothelial dysfunction in ApoE-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2331–2336. [Google Scholar] [CrossRef]
- Masselli, E.; Pozzi, G.; Vaccarezza, M.; Mirandola, P.; Galli, D.; Vitale, M.; Carubbi, C.; Gobbi, G. ROS in Platelet Biology: Functional Aspects and Methodological Insights. Int. J. Mol. Sci. 2020, 21, 4866. [Google Scholar] [CrossRef]
- Loscalzo, J. Oxidative stress in endothelial cell dysfunction and thrombosis. Pathophysiol. Haemost. Thromb. 2002, 32, 359–360. [Google Scholar] [CrossRef]
- Essex, D.W. The role of thiols and disulfides in platelet function. Antioxid. Redox Signal. 2004, 6, 736–746. [Google Scholar] [CrossRef] [PubMed]
- Litvinov, R.I.; Weisel, J.W. Role of red blood cells in haemostasis and thrombosis. ISBT Sci. Ser. 2017, 12, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Whelihan, M.F.; Zachary, V.; Orfeo, T.; Mann, K.G. Prothrombin activation in blood coagulation: The erythrocyte contribution to thrombin generation. Blood 2012, 120, 3837–3845. [Google Scholar] [CrossRef] [PubMed]
- Tokarev, A.A.; Butylin, A.A.; Ataullakhanov, F.I. Platelet adhesion from shear blood flow is controlled by near-wall rebounding collisions with erythrocytes. Biophys. J. 2011, 100, 799–808. [Google Scholar] [CrossRef]
- Skalen, K.; Gustafsson, M.; Rydberg, E.K.; Hulten, L.M.; Wiklund, O.; Innerarity, T.L.; Boren, J. Subendothelial retention of atherogenic lipoproteins in early atherosclerosis. Nature 2002, 417, 750–754. [Google Scholar] [CrossRef] [PubMed]
- Joris, I.; Zand, T.; Nunnari, J.J.; Krolikowski, F.J.; Majno, G. Studies on the pathogenesis of atherosclerosis. I. Adhesion and emigration of mononuclear cells in the aorta of hypercholesterolemic rats. Am. J. Pathol. 1983, 113, 341–358. [Google Scholar] [PubMed]
- Steinberg, D. Atherogenesis in perspective: Hypercholesterolemia and inflammation as partners in crime. Nat. Med. 2002, 8, 1211–1217. [Google Scholar] [CrossRef] [PubMed]
- Mundi, S.; Massaro, M.; Scoditti, E.; Carluccio, M.A.; van Hinsbergh, V.W.M.; Iruela-Arispe, M.L.; De Caterina, R. Endothelial permeability, LDL deposition, and cardiovascular risk factors-a review. Cardiovasc. Res. 2018, 114, 35–52. [Google Scholar] [CrossRef] [PubMed]
- Osterud, B.; Bjorklid, E. Role of monocytes in atherogenesis. Physiol. Rev. 2003, 83, 1069–1112. [Google Scholar] [CrossRef] [PubMed]
- Galkina, E.; Ley, K. Immune and Inflammatory Mechanisms of Atherosclerosis. Annu. Rev. Immunol. 2009, 27, 165–197. [Google Scholar] [CrossRef]
- Libby, P.; Ridker, P.M.; Hansson, G.K. Progress and challenges in translating the biology of atherosclerosis. Nature 2011, 473, 317–325. [Google Scholar] [CrossRef]
- Leitinger, N.; Schulman, I.G. Phenotypic polarization of macrophages in ath- erosclerosis. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1120–1126. [Google Scholar] [CrossRef]
- Potter, M.D.; Barbero, S.; Cheresh, D.A. Tyrosine phosphorylation of VE-cadherin prevents binding of p120- and beta-catenin and maintains the cellular mesenchymal state. J. Biol. Chem. 2005, 280, 31906–31912. [Google Scholar] [CrossRef]
- Kevil, C.G.; Oshima, T.; Alexander, B.; Coe, L.L.; Alexander, J.S. H2O2-mediated permeability: Role of MAPK and occludin. Am. J. Physiol. Cell Physiol. 2000, 279, C21–C30. [Google Scholar] [CrossRef]
- Staal, F.J.; Anderson, M.T.; Staal, G.E.; Herzenberg, L.A.; Gitler, C.; Herzenberg, L.A. Redox regulation of signal transduction: Tyrosine phosphorylation and calcium influx. Proc. Natl. Acad. Sci. USA 1994, 91, 3619–3622. [Google Scholar] [CrossRef]
- Kisseleva, T.; Song, L.; Vorontchikhina, M.; Feirt, N.; Kitajewski, J.; Schindler, C. NF-kappaB regulation of endothelial cell function during LPS-induced toxemia and cancer. J. Clin. Investig. 2006, 116, 2955–2963. [Google Scholar] [CrossRef]
- Mitra, R.; O’Neil, G.L.; Harding, I.C.; Cheng, M.J.; Mensah, S.A.; Ebong, E.E. Glycocalyx in Atherosclerosis-Relevant Endothelium Function and as a Therapeutic Target. Curr. Atheroscler. Rep. 2017, 19, 63. [Google Scholar] [CrossRef] [PubMed]
- Vink, H.; Constantinescu, A.A.; Spaan, J.A. Oxidized lipoproteins degrade the endothelial surface layer: Implications for platelet-endothelial cell adhesion. Circulation 2000, 101, 1500–1502. [Google Scholar] [CrossRef]
- Keller, R.; Pratt, B.M.; Furthmayr, H.; Madri, J.A. Aortic endothelial cell proteoheparan sulfate. II. Modulation by extracellular matrix. Am. J. Pathol. 1987, 128, 299–306. [Google Scholar] [PubMed]
- Olgac, U.; Poulikakos, D.; Saur, S.C.; Alkadhi, H.; Kurtcuoglu, V. Patient-specific three- dimensional simulation of LDL accumulation in a human left coronary artery in its healthy and atherosclerotic states. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H1969–H1982. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zennadi, R. Oxidative Stress and Thrombosis during Aging: The Roles of Oxidative Stress in RBCs in Venous Thrombosis. Int. J. Mol. Sci. 2020, 21, 4259. [Google Scholar] [CrossRef]
- Golino, P.; Ragni, M.; Cirillo, P.; Avvedimento, V.E.; Feliciello, A.; Esposito, N.; Scognamiglio, A.; Trimarco, B.; Iaccarino, G.; Condorelli, M.; et al. Effects of tissue factor induced by oxygen free radicals on coronary flow during reperfusion. Nat. Med. 1996, 2, 35–40. [Google Scholar] [CrossRef]
- Herkert, O.; Diebold, I.; Brandes, R.P.; Hess, J.; Busse, R.; Görlach, A. NADPH oxidase mediates tissue factor-dependent surface procoagulant activity by thrombin in human vascular smooth muscle cells. Circulation 2002, 105, 2030–2036. [Google Scholar] [CrossRef]
- Jacobi, J.; Kristal, B.; Chezar, J.; Shaul, S.M.; Sela, S. Exogenous superoxide mediates pro-oxidative, proinflammatory, and procoagulatory changes in primary endothelial cell cultures. Free Radic. Biol. Med. 2005, 39, 1238–1248. [Google Scholar] [CrossRef]
- Banfi, C.; Brioschi, M.; Barbieri, S.S.; Eligini, S.; Barcella, S.; Tremoli, E.; Colli, S.; Mussoni, L. Mitochondrial reactive oxygen species: A common pathway for PAR1- and PAR2-mediated tissue factor induction in human endothelial cells. J. Thromb. Haemost. 2009, 7, 206–216. [Google Scholar] [CrossRef]
- Djordjevic, T.; Pogrebniak, A.; BelAiba, R.S.; Bonello, S.; Wotzlaw, C.; Acker, H.; Hess, J.; Görlach, A. The expression of the NADPH oxidase subunit p22phox is regulated by a redox-sensitive pathway in endothelial cells. Free Radic. Biol. Med. 2005, 38, 616–630. [Google Scholar] [CrossRef]
- Ohkura, N.; Hiraishi, S.; Itabe, H.; Hamuro, T.; Kamikubo, Y.; Takano, T.; Matsuda, J.; Horie, S. Oxidized phospholipids in oxidized low-density lipoprotein reduce the activity of tissue factor pathway inhibitor through association with its carboxy-terminal region. Antioxid. Redox Signal. 2004, 6, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Gutmann, C.; Siow, R.; Gwozdz, A.M.; Saha, P.; Smith, A. Reactive Oxygen Species in Venous Thrombosis. Int. J. Mol. Sci. 2020, 21, 1918. [Google Scholar] [CrossRef]
- Glaser, C.B.; Morser, J.; Clarke, J.H.; Blasko, E.; McLean, K.; Kuhn, I.; Chang, R.J.; Lin, J.H.; Vilander, L.; Andrews, W.H.; et al. Oxidation of a specific methionine in thrombomodulin by activated neutrophil products blocks cofactor activity. A potential rapid mechanism for modulation of coagulation. J. Clin. Investig. 1992, 90, 2565–2573. [Google Scholar] [CrossRef] [PubMed]
- Van Patten, S.M.; Hanson, E.; Bernasconi, R.; Zhang, K.; Manavalan, P.; Cole, E.S.; McPherson, J.M.; Edmunds, T. Oxidation of methionine residues in antithrombin. Effects on biological activity and heparin binding. J. Biol. Chem. 1999, 274, 10268–10276. [Google Scholar] [CrossRef]
- Upchurch, G.R., Jr.; Ramdev, N.; Walsh, M.T.; Loscalzo, J. Prothrombotic Consequences of the Oxidation of Fibrinogen and their Inhibition by Aspirin. J. Thromb. Thrombolysis 1998, 5, 9–14. [Google Scholar] [CrossRef]
- Sovari, A.A.; Dudley, S.C., Jr. Reactive oxygen species-targeted therapeutic interventions for atrial fibrillation. Front. Physiol. 2012, 3, 311. [Google Scholar] [CrossRef] [PubMed]
- Dai, D.F.; Rabinovitch, P.S. Cardiac aging in mice and humans: The role of mitochondrial oxidative stress. Trends Cardiovasc. Med. 2009, 19, 213–220. [Google Scholar] [CrossRef]
- Huang, C.X.; Liu, Y.; Xia, W.F.; Tang, Y.H.; Huang, H. Oxidative stress: A possible pathogenesis of atrial fibrillation. Med. Hypotheses. 2009, 72, 466–467. [Google Scholar] [CrossRef]
- Korantzopoulos, P.; Kolettis, T.M.; Galaris, D.; Goudevenos, J.A. The role of oxidative stress in the pathogenesis and perpetuation of atrial fibrillation. Int. J. Cardiol. 2007, 115, 135–143. [Google Scholar] [CrossRef]
- Van Wagoner, D.R. Electrophysiological Remodeling in Human Atrial Fibrillation. Pacing Clin. Electrophysiol. 2003, 26, 1572–1575. [Google Scholar] [CrossRef]
- Mihm, M.J.; Yu, F.; Carnes, C.A.; Reiser, P.J.; McCarthy, P.M.; Van Wagoner, D.R.; Bauer, J.A. Impaired myofibrillar energetics and oxidative injury during human atrial fibrillation. Circulation 2001, 104, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Vest, J.A.; Wehrens, X.H.; Reiken, S.R.; Lehnart, S.E.; Dobrev, D.; Chandra, P.; Danilo, P.; Ravens, U.; Rosen, M.R.; Marks, A.R. Defective cardiac ryanodine receptor regulation during atrial fibrillation. Circulation 2005, 111, 2025–2032. [Google Scholar] [CrossRef] [PubMed]
- Neef, S.; Dybkova, N.; Sossalla, S.; Ort, K.R.; Fluschnik, N.; Neumann, K.; Seipelt, R.; Schöndube, F.A.; Hasenfuss, G.; Maier, L.S. CaMKII-dependent diastolic SR Ca2+ leak and elevated diastolic Ca2+ levels in right atrial myocardium of patients with atrial fibrillation. Circ. Res. 2010, 106, 1134–1144. [Google Scholar] [CrossRef]
- Zhang, Y.; Fraser, J.A.; Jeevaratnam, K.; Hao, X.; Hothi, S.S.; Grace, A.A.; Lei, M.; Huang, C.L. Acute atrial arrhythmogenicity and altered Ca(2+) homeostasis in murine RyR2-P2328S hearts. Cardiovasc. Res. 2011, 89, 794–804. [Google Scholar] [CrossRef]
- Shan, J.; Xie, W.; Betzenhauser, M.; Reiken, S.; Chen, B.X.; Wronska, A.; Marks, A.R. Calcium leak through ryanodine receptors leads to atrial fibrillation in 3 mouse models of catecholaminergic polymorphic ventricular tachycardia. Circ. Res. 2012, 111, 708–717. [Google Scholar] [CrossRef]
- Purohit, A.; Rokita, A.G.; Guan, X.; Chen, B.; Koval, O.M.; Voigt, N.; Neef, S.; Sowa, T.; Gao, Z.; Luczak, E.D.; et al. Oxidized CaMKII triggers atrial fibrillation. Circulation 2013, 128, 1748–1757. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Santulli, G.; Reiken, S.R.; Yuan, Q.; Osborne, B.W.; Chen, B.X.; Marks, A.R. Mitochondrial oxidative stress promotes atrial fibrillation. Sci. Rep. 2015, 5, 11427. [Google Scholar] [CrossRef]
- Podhaisky, H.P.; Abate, A.; Polte, T.; Oberle, S.; Schröder, H. Aspirin protects endothelial cells from oxidative stress—Possible synergism with vitamin E. FEBS Lett. 1997, 417, 349–351. [Google Scholar] [CrossRef]
- Oberle, S.; Polte, T.; Abate, A.; Podhaisky, H.P.; Schröder, H. Aspirin increases ferritin synthesis in endothelial cells: A novel antioxidant pathway. Circ. Res. 1998, 82, 1016–1020. [Google Scholar] [CrossRef]
- Grosser, N.; Abate, A.; Oberle, S.; Vreman, H.J.; Dennery, P.A.; Becker, J.C.; Pohle, T.; Seidman, D.S.; Schröder, H. Heme oxygenase-1 induction may explain the antioxidant profile of aspirin. Biochem. Biophys. Res. Commun. 2003, 308, 956–960. [Google Scholar] [CrossRef]
- Balla, G.; Jacob, H.S.; Balla, J.; Rosenberg, M.; Nath, K.; Apple, F.; Eaton, J.W.; Vercellotti, G.M. Ferritin: A cytoprotective antioxidant strategem of endothelium. J. Biol. Chem. 1992, 267, 18148–18153. [Google Scholar] [CrossRef] [PubMed]
- Juckett, M.B.; Balla, J.; Balla, G.; Jessurun, J.; Jacob, H.S.; Vercellotti, G.M. Ferritin protects endothelial cells from oxidized low density lipoprotein in vitro. Am. J. Pathol. 1995, 147, 782–789. [Google Scholar]
- Stocker, R.; Yamamoto, Y.; McDonagh, A.F.; Glazer, A.N.; Ames, B.N. Bilirubin is an antioxidant of possible physiological importance. Science 1987, 235, 1043–1046. [Google Scholar] [CrossRef] [PubMed]
- Jian, Z.; Tang, L.; Yi, X.; Liu, B.; Zhang, Q.; Zhu, G.; Wang, G.; Gao, T.; Li, C. Aspirin induces Nrf2-mediated transcriptional activation of haem oxygenase-1 in protection of human melanocytes from H2O2-induced oxidative stress. J. Cell. Mol. Med. 2016, 20, 1307–1318. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Chen, S.; Zhou, Z.; Dai, H.; Wang, H.; Li, Y.; Zhou, K.; Shen, Z.; Guo, Y.; Liu, C.; et al. Aspirin suppresses neuronal apoptosis, reduces tissue inflammation, and restrains astrocyte activation by activating the Nrf2/HO-1 signaling pathway. Neuroreport 2018, 29, 524–531. [Google Scholar]
- Otterbein, L.E.; Bach, F.H.; Alam, J.; Soares, M.; Tao Lu, H.; Wysk, M.; Davis, R.J.; Flavell, R.A.; Choi, A.M. Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway. Nat. Med. 2000, 6, 422–428. [Google Scholar] [CrossRef]
- Immenschuh, S.; Ramadori, G. Gene regulation of heme oxygenase-1 as a therapeutic target. Biochem. Pharmacol. 2000, 60, 1121–1128. [Google Scholar] [CrossRef]
- Lee, J.S.; Surh, Y.J. Nrf2 as a novel molecular target for chemoprevention. Cancer Lett. 2005, 224, 171–184. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-κB signaling in inflammation. Signal. Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef]
- Chen, C.M.; Tung, Y.T.; Wei, C.H.; Lee, P.Y.; Chen, W. Anti-Inflammatory and Reactive Oxygen Species Suppression through Aspirin Pretreatment to Treat Hyperoxia-Induced Acute Lung Injury in NF-κB-Luciferase Inducible Transgenic Mice. Antioxidants 2020, 9, 429. [Google Scholar] [CrossRef]
- Jorda, A.; Aldasoro, M.; Aldasoro, C.; Guerra-Ojeda, S.; Iradi, A.; Vila, J.M.; Campos-Campos, J.; Valles, S.L. Action of low doses of Aspirin in Inflammation and Oxidative Stress induced by aβ1-42 on Astrocytes in primary culture. Int. J. Med. Sci. 2020, 17, 834–843. [Google Scholar] [CrossRef]
- Yang, J.J.; Li, P.; Wang, F.; Liang, W.J.; Ma, H.; Chen, Y.; Ma, Z.M.; Li, Q.Z.; Peng, Q.S.; Zhang, Y.; et al. Activation of activator protein 2 alpha by aspirin alleviates atherosclerotic plaque growth and instability in vivo. Oncotarget 2016, 7, 52729–52739. [Google Scholar] [CrossRef]
- Shi, X.; Ding, M.; Dong, Z.; Chen, F.; Ye, J.; Wang, S.; Leonard, S.S.; Castranova, V.; Vallyathan, V. Antioxidant properties of aspirin: Characterization of the ability of aspirin to inhibit silica-induced lipid peroxidation, DNA damage, NF-kappaB activation, and TNF-alpha production. Mol. Cell. Biochem. 1999, 199, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Prasad, K.; Lee, P. Suppression of oxidative stress as a mechanism of reduction of hypercholesterolemic atherosclerosis by aspirin. J. Cardiovasc. Pharmacol. Ther. 2003, 8, 61–69. [Google Scholar] [CrossRef] [PubMed]
- John, A.; Amiri, L.; Shafarin, J.; Howarth, F.C.; Raza, H. Effect of Aspirin on Mitochondrial Dysfunction and Stress in the Pancreas and Heart of Goto-Kakizaki Diabetic Rats. Life 2021, 11, 902. [Google Scholar] [CrossRef]
- John, A.; Amiri, L.; Shafarin, J.; Tariq, S.; Adeghate, E.; Howarth, F.C.; Raza, H. Alterations in Energy Metabolism, Mitochondrial Function and Redox Homeostasis in GK Diabetic Rat Tissues Treated with Aspirin. Life 2022, 12, 104. [Google Scholar] [CrossRef] [PubMed]
- Caballero, F.; Gerez, E.; Batlle, A.; Vazquez, E. Preventive aspirin treatment of streptozotocin induced diabetes: Blockage of oxidative status and revertion of heme enzymes inhibition. Chem. Biol. Interact. 2000, 126, 215–225. [Google Scholar] [CrossRef]
- De Cristóbal, J.; Madrigal, J.L.; Lizasoain, I.; Lorenzo, P.; Leza, J.C.; Moro, M.A. Aspirin inhibits stress-induced increase in plasma glutamate, brain oxidative damage and ATP fall in rats. Neuroreport 2002, 13, 217–221. [Google Scholar] [CrossRef]
- Chávez, E.; Castro-Sánchez, L.; Shibayama, M.; Tsutsumi, V.; Pérez Salazar, E.; Moreno, M.G.; Muriel, P. Effects of acetyl salycilic acid and ibuprofen in chronic liver damage induced by CCl4. J. Appl. Toxicol. 2012, 32, 51–59. [Google Scholar] [CrossRef]
- Wróbel, M.; Góralska, J.; Jurkowska, H.; Sura, P. Similar effect of sodium nitroprusside and acetylsalicylic acid on antioxidant system improvement in mouse liver but not in the brain. Biochimie 2017, 135, 181–185. [Google Scholar] [CrossRef]
- Wu, R.; Lamontagne, D.; de Champlain, J. Antioxidative properties of acetylsalicylic Acid on vascular tissues from normotensive and spontaneously hypertensive rats. Circulation 2002, 105, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Yin, D.; Sadekova, N.; Deschepper, C.F.; de Champlain, J.; Girouard, H. Protective effects of aspirin from cardiac hypertrophy and oxidative stress in cardiomyopathic hamsters. Oxid. Med. Cell. Longev. 2012, 2012, 761710. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Zhai, H.; Huang, L.; Li, H.; Xu, Y.; Qiao, X.; Sun, S.; Wu, Y. Aspirin protects dopaminergic neurons against lipopolysaccharide-induced neurotoxicity in primary midbrain cultures. J. Mol. Neurosci. 2012, 46, 153–161. [Google Scholar] [CrossRef] [PubMed]
- De La Cruz, J.P.; Guerrero, A.; González-Correa, J.A.; Arrebola, M.M.; Sánchez de la Cuesta, F. Antioxidant effect of acetylsalicylic and salicylic acid in rat brain slices subjected to hypoxia. J. Neurosci. Res. 2004, 75, 280–290. [Google Scholar] [CrossRef]
- Kiliçoğlu Aydin, B.; Eren, Z. The Effects of Aspirin and Vitamin E on Blood Antioxidant Enzymes of Rats during Experimental Liver Ischemia-Reperfusion. J. Appl. Biol. Sci. 2007, 1, 51–56. [Google Scholar]
- Ayyadevara, S.; Bharill, P.; Dandapat, A.; Hu, C.; Khaidakov, M.; Mitra, S.; Shmookler Reis, R.J.; Mehta, J.L. Aspirin inhibits oxidant stress, reduces age-associated functional declines, and extends lifespan of Caenorhabditis elegans. Antioxid. Redox Signal. 2013, 18, 481–490. [Google Scholar] [CrossRef]
- Dimitrovska, M.; Dervisevik, M.; Cipanovska, N.; Gerazova, K.; Dinevska-Kjovkarovska, S.; Miova, B. Physiological and pharmacological inductors of HSP70 enhance the antioxidative defense mechanisms of the liver and pancreas in diabetic rats. Can. J. Physiol. Pharmacol. 2018, 96, 158–164. [Google Scholar] [CrossRef]
- Veres, G.; Benke, K.; Stengl, R.; Bai, Y.; Stark, K.A.; Sayour, A.A.; Radovits, T.; Loganathan, S.; Korkmaz-Icöz, S.; Karck, M.; et al. Aspirin Reduces Ischemia-Reperfusion Injury Induced Endothelial Cell Damage of Arterial Grafts in a Rodent Model. Antioxidants 2022, 11, 177. [Google Scholar] [CrossRef]
- Frydrychowski, P.; Michałek, M.; Bil-Lula, I.; Chełmecka, E.; Kafel, A.; Noszczyk-Nowak, A.; Stygar, D. Cardioprotective Effect of Acetylsalicylic Acid in the Myocardial Ischemia-Reperfusion Model on Oxidative Stress Markers Levels in Heart Muscle and Serum. Antioxidants 2022, 11, 1432. [Google Scholar] [CrossRef]
- Hsu, C.S.; Li, Y. Aspirin potently inhibits oxidative DNA strand breaks: Implications for cancer chemoprevention. Biochem. Biophys. Res. Commun. 2002, 293, 705–709. [Google Scholar]
- Ristimäe, T.; Zilmer, M.; Zilmer, K.; Kairane, C.; Kullisaar, T.; Teesalu, R. Effect of low-dose aspirin on the markers of oxidative stress. Cardiovasc. Drugs Ther. 1999, 13, 485–490. [Google Scholar] [CrossRef]
- Kurban, S.; Mehmetoglu, I. Effects of acetylsalicylic acid on serum paraoxonase activity, Ox-LDL, coenzyme Q10 and other oxidative stress markers in healthy volunteers. Clin. Biochem. 2010, 43, 287–290. [Google Scholar] [CrossRef]
- Berg, K.; Langaas, M.; Ericsson, M.; Pleym, H.; Basu, S.; Nordrum, I.S.; Vitale, N.; Haaverstad, R. Acetylsalicylic acid treatment until surgery reduces oxidative stress and inflammation in patients undergoing coronary artery bypass grafting. Eur. J. Cardiothorac. Surg. 2013, 43, 1154–1163. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Shan, X.F.; Wang, X.L.; Dong, W.W.; Li, Z.; Liu, X.H.; Zhang, W.; Xing, K.; Chang, F.J. Endothelial damage effects of circulating microparticles from patients with stable angina are reduced by aspirin through ERK/p38 MAPKs pathways. Cardiovasc. Ther. 2017, 35, e12273. [Google Scholar] [CrossRef] [PubMed]
- Franchi, F.; Angiolillo, D.J. Novel antiplatelet agents in acute coronary syndrome. Nat. Rev. Cardiol. 2015, 12, 30–47. [Google Scholar] [CrossRef]
- Ferri, N.; Corsini, A.; Bellosta, S. Pharmacology of the new P2Y12 receptor inhibitors: Insights on pharmacokinetic and pharmacodynamic properties. Drugs 2013, 73, 1681–1709. [Google Scholar] [CrossRef]
- Dayoub, E.J.; Seigerman, M.; Tuteja, S.; Kobayashi, T.; Kolansky, D.M.; Giri, J.; Groeneveld, P.W. Trends in Platelet Adenosine Diphosphate P2Y12 Receptor Inhibitor Use and Adherence Among Antiplatelet-Naive Patients After Percutaneous Coronary Intervention, 2008–2016. JAMA Intern. Med. 2018, 178, 943–950. [Google Scholar] [CrossRef] [PubMed]
- Faridi, K.F.; Garratt, K.N.; Kennedy, K.F.; Maddox, T.M.; Secemsky, E.A.; Butala, N.M.; Yeh, R.W. Physician and Hospital Utilization of P2Y12 Inhibitors in ST-Segment-Elevation Myocardial Infarction in the United States: A Study from the National Cardiovascular Data Registry’s Research to Practice Initiative. Circ. Cardiovasc. Qual. Outcomes 2020, 13, e006275. [Google Scholar] [CrossRef]
- Kanko, M.; Maral, H.; Akbas, M.H.; Ozden, M.; Bulbul, S.; Omay, O.; Yavuz, S.; Berki, K.T. Protective effects of clopidogrel on oxidant damage in a rat model of acute ischemia. Tohoku J. Exp. Med. 2005, 205, 133–139. [Google Scholar] [CrossRef]
- Hu, H.; Batteux, F.; Chéreau, C.; Kavian, N.; Marut, W.; Gobeaux, C.; Borderie, D.; Dinh-Xuan, A.T.; Weill, B.; Nicco, C. Clopidogrel protects from cell apoptosis and oxidative damage in a mouse model of renal ischaemia-reperfusion injury. J. Pathol. 2011, 225, 265–275. [Google Scholar] [CrossRef]
- Hadi, N.R.; Mohammad, B.I.; Ajeena, I.M.; Sahib, H.H. Antiatherosclerotic potential of clopidogrel: Antioxidant and anti-inflammatory approaches. Biomed. Res. Int. 2013, 2013, 790263. [Google Scholar] [CrossRef]
- Yang, H.; Zhao, P.; Tian, S. Clopidogrel Protects Endothelium by Hindering TNFα-Induced VCAM-1 Expression through CaMKKβ/AMPK/Nrf2 Pathway. J. Diabetes Res. 2016, 2016, 9128050. [Google Scholar] [CrossRef] [PubMed]
- An, X.; Jiang, G.; Cheng, C.; Lv, Z.; Liu, Y.; Wang, F. Inhibition of Platelets by Clopidogrel Suppressed Ang II-Induced Vascular Inflammation, Oxidative Stress, and Remodeling. J. Am. Heart Assoc. 2018, 7, e009600. [Google Scholar] [CrossRef] [PubMed]
- Korish, A.A. Clopidogrel Prophylaxis Abates Myocardial Ischemic Injury and Inhibits the Hyperlipidemia-Inflammation Loop in Hypercholestrolemic Mice. Arch. Med. Res. 2020, 51, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.; Shao, S.; Zhang, Y.; Liang, X.; Zhang, K.; Li, G. Beneficial Effects of Ticagrelor on Oxidized Low-Density Lipoprotein (ox-LDL)-Induced Apoptosis in Human Umbilical Vein Endothelial Cells. Med. Sci. Monit. 2019, 25, 9811–9819. [Google Scholar] [CrossRef]
- El-Mokadem, B.M.; El-Abhar, H.S.; Abdallah, D.M.; Awad, A.S.; Soubh, A.A. Epac-1/Rap-1 signaling pathway orchestrates the reno-therapeutic effect of ticagrelor against renal ischemia/reperfusion model. Biomed. Pharmacother. 2021, 139, 111488. [Google Scholar] [CrossRef]
- Bitirim, C.V.; Ozer, Z.B.; Aydos, D.; Genc, K.; Demirsoy, S.; Akcali, K.C.; Turan, B. Cardioprotective effect of extracellular vesicles derived from ticagrelor-pretreated cardiomyocyte on hyperglycemic cardiomyocytes through alleviation of oxidative and endoplasmic reticulum stress. Sci. Rep. 2022, 12, 5651. [Google Scholar] [CrossRef]
- Grzesk, G.; Kozinski, M.; Navarese, E.P.; Krzyzanowski, M.; Grzesk, E.; Kubica, A.; Siller-Matula, J.M.; Castriota, F.; Kubica, J. Ticagrelor, but not clopidogrel and prasugrel, prevents ADP-induced vascular smooth muscle cell contraction: A placebo-controlled study in rats. Thromb. Res. 2012, 130, 65–69. [Google Scholar] [CrossRef]
- Heitzer, T.; Rudolph, V.; Schwedhelm, E.; Karstens, M.; Sydow, K.; Ortak, M.; Tschentscher, P.; Meinertz, T.; Böger, R.; Baldus, S. Clopidogrel improves systemic endothelial nitric oxide bioavailability in patients with coronary artery disease: Evidence for antioxidant and antiinflammatory effects. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1648–1652. [Google Scholar] [CrossRef]
- Taher, M.A.; Nassir, E.S. Beneficial effects of clopidogrel on glycemic indices and oxidative stress in patients with type 2 diabetes. Saudi Pharm. J. 2011, 19, 107–113. [Google Scholar] [CrossRef]
- Rudolph, T.K.; Fuchs, A.; Klinke, A.; Schlichting, A.; Friedrichs, K.; Hellmich, M.; Mollenhauer, M.; Schwedhelm, E.; Baldus, S.; Rudolph, V. Prasugrel as opposed to clopidogrel improves endothelial nitric oxide bioavailability and reduces platelet-leukocyte interaction in patients with unstable angina pectoris: A randomized controlled trial. Int. J. Cardiol. 2017, 248, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Campo, G.; Vieceli Dalla Sega, F.; Pavasini, R.; Aquila, G.; Gallo, F.; Fortini, F.; Tonet, E.; Cimaglia, P.; Del Franco, A.; Pestelli, G.; et al. Biological effects of ticagrelor over clopidogrel in patients with stable coronary artery disease and chronic obstructive pulmonary disease. Thromb. Haemost. 2017, 117, 1208–1216. [Google Scholar] [CrossRef] [PubMed]
- Aquila, G.; Vieceli Dalla Sega, F.; Marracino, L.; Pavasini, R.; Cardelli, L.S.; Piredda, A.; Scoccia, A.; Martino, V.; Fortini, F.; Bononi, I.; et al. Ticagrelor Increases SIRT1 and HES1 mRNA Levels in Peripheral Blood Cells from Patients with Stable Coronary Artery Disease and Chronic Obstructive Pulmonary Disease. Int. J. Mol. Sci. 2020, 21, 1576. [Google Scholar] [CrossRef]
- Erdbruegger, U.; Haubitz, M.; Woywodt, A. Circulating endothelial cells: A novel marker of endothelial damage. Clin. Chim. Acta 2006, 373, 17–26. [Google Scholar] [CrossRef]
- McClung, J.A.; Kruger, A.L.; Ferraris, A.; Vanella, L.; Tsenovoy, P.; Weiss, M.B.; Abraham, N.G. Usefulness of clopidogrel to protect against diabetes-induced vascular damage. Am. J. Cardiol. 2010, 105, 1014–1018. [Google Scholar] [CrossRef]
- Bundhoo, S.; Sagan, E.; James, P.E.; Anderson, R.A. Clopidogrel results in favourable changes in nitric oxide metabolism in patients undergoing percutaneous coronary intervention. Thromb. Haemost. 2014, 111, 373–374. [Google Scholar] [CrossRef] [PubMed]
- Caliskan, A.; Yavuz, C.; Karahan, O.; Yazici, S.; Guclu, O.; Demirtas, S.; Mavitas, B. Factor-Xa inhibitors protect against systemic oxidant damage induced by peripheral-ischemia reperfusion. J. Thromb. Thrombolysis 2014, 37, 464–468. [Google Scholar] [CrossRef]
- Ishibashi, Y.; Matsui, T.; Ueda, S.; Fukami, K.; Yamagishi, S. Advanced glycation end products potentiate citrated plasma-evoked oxidative and inflammatory reactions in endothelial cells by up-regulating protease-activated receptor-1 expression. Cardiovasc. Diabetol. 2014, 13, 60. [Google Scholar] [CrossRef]
- Ishibashi, Y.; Matsui, T.; Fukami, K.; Ueda, S.; Okuda, S.; Yamagishi, S. Rivaroxaban inhibits oxidative and inflammatory reactions in advanced glycation end product-exposed tubular cells by blocking thrombin/protease-activated receptor-2 system. Thromb. Res. 2015, 135, 770–773. [Google Scholar] [CrossRef]
- Gul Utku, O.; Akbay Karatay, E.; Erdal, H.; Arhan, M.; Onal, I.K.; Ibis, M.; Ekinci, O.; Yilmaz Demirtas, C.; Unal, S. Rivaroxaban Induces Mucosal Healing in a Rat Model of Trinitrobenzene Sulfonic Acid-Induced Colitis. Med. Princ. Pract. 2015, 24, 470–476. [Google Scholar] [CrossRef]
- Ellinghaus, P.; Perzborn, E.; Hauenschild, P.; Gerdes, C.; Heitmeier, S.; Visser, M.; Summer, H.; Laux, V. Expression of pro-inflammatory genes in human endothelial cells: Comparison of rivaroxaban and dabigatran. Thromb. Res. 2016, 142, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Vilaseca, M.; García-Calderó, H.; Lafoz, E.; García-Irigoyen, O.; Avila, M.A.; Reverter, J.C.; Bosch, J.; Hernández-Gea, V.; Gracia-Sancho, J.; García-Pagán, J.C. The anticoagulant rivaroxaban lowers portal hypertension in cirrhotic rats mainly by deactivating hepatic stellate cells. Hepatology 2017, 65, 2031–2044. [Google Scholar] [CrossRef]
- Maeda, M.; Tsuboi, T.; Hayashi, T. An Inhibitor of Activated Blood Coagulation Factor X Shows Anti-Endothelial Senescence and Anti-Atherosclerotic Effects. J. Vasc. Res. 2019, 56, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Shafiey, S.I.; Abo-Saif, A.A.; Abo-Youssef, A.M.; Mohamed, W.R. Protective effects of rivaroxaban against cisplatin-induced testicular damage in rats: Impact on oxidative stress, coagulation, and p-NF-κB/VCAM-1 signaling. Food Chem. Toxicol. 2022, 169, 113419. [Google Scholar] [CrossRef] [PubMed]
- Abedalqader, N.N.; Rababa’h, A.M.; Ababneh, M. The protective effect of rivaroxaban with or without aspirin on inflammation, oxidative stress, and platelet reactivity in isoproterenol-induced cardiac injury in rats. Naunyn Schmiedebergs Arch. Pharmacol. 2023, 396, 337–351. [Google Scholar] [CrossRef]
- Imano, H.; Kato, R.; Tanikawa, S.; Yoshimura, F.; Nomura, A.; Ijiri, Y.; Yamaguchi, T.; Izumi, Y.; Yoshiyama, M.; Hayashi, T. Factor Xa inhibition by rivaroxaban attenuates cardiac remodeling due to intermittent hypoxia. J. Pharmacol. Sci. 2018, 137, 274–282. [Google Scholar] [CrossRef]
- Imam, F.; Al-Harbi, N.O.; Khan, M.R.; Qamar, W.; Alharbi, M.; Alshamrani, A.A.; Alhamami, H.N.; Alsaleh, N.B.; Alharbi, K.S. Protective Effect of RIVA Against Sunitinib-Induced Cardiotoxicity by Inhibiting Oxidative Stress-Mediated Inflammation: Probable Role of TGF-β and Smad Signaling. Cardiovasc. Toxicol. 2020, 20, 281–290. [Google Scholar] [CrossRef]
- Al-Harbi, N.O.; Imam, F.; Alharbi, M.M.; Khan, M.R.; Qamar, W.; Afzal, M.; Algahtani, M.; Alobaid, S.; Alfardan, A.S.; Alshammari, A.; et al. Role of rivaroxaban in sunitinib-induced renal injuries via inhibition of oxidative stress-induced apoptosis and inflammation through the tissue nacrosis factor-α induced nuclear factor-κappa B signaling pathway in rats. J. Thromb. Thrombolysis 2020, 50, 361–370. [Google Scholar] [CrossRef]
- Abdelzaher, W.Y.; Mohammed, H.H.; Welson, N.N.; Batiha, G.E.; Baty, R.S.; Abdel-Aziz, A.M. Rivaroxaban Modulates TLR4/Myd88/NF-Kβ Signaling Pathway in a Dose-Dependent Manner with Suppression of Oxidative Stress and Inflammation in an Experimental Model of Depression. Front. Pharmacol. 2021, 12, 715354. [Google Scholar] [CrossRef] [PubMed]
- Moñux, G.; Zamorano-León, J.J.; Marqués, P.; Sopeña, B.; García-García, J.M.; Laich de Koller, G.; Calvo-Rico, B.; García-Fernandez, M.A.; Serrano, J.; López-Farré, A. FXa inhibition by rivaroxaban modifies mechanisms associated with the pathogenesis of human abdominal aortic aneurysms. Br. J. Clin. Pharmacol. 2017, 83, 2661–2670. [Google Scholar] [CrossRef] [PubMed]
- Eikelboom, J.W.; Connolly, S.J.; Bosch, J.; Dagenais, G.R.; Hart, R.G.; Shestakovska, O.; Diaz, R.; Alings, M.; Lonn, E.M.; Anand, S.S.; et al. Rivaroxaban with or without Aspirin in Stable Cardiovascular Disease. N. Engl. J. Med. 2017, 377, 1319–1330. [Google Scholar] [CrossRef] [PubMed]
- Russo, V.; Fabiani, D.; Leonardi, S.; Attena, E.; D’Alterio, G.; Cotticelli, C.; Rago, A.; Sarpa, S.; Maione, B.; D’Onofrio, A.; et al. Dual Pathway Inhibition with Rivaroxaban and Aspirin Reduces Inflammatory Biomarkers in Atherosclerosis. J. Cardiovasc. Pharmacol. 2023, 81, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Torramade-Moix, S.; Palomo, M.; Vera, M.; Jerez, D.; Moreno-Castaño, A.B.; Zafar, M.U.; Rovira, J.; Diekmann, F.; Garcia-Pagan, J.C.; Escolar, G.; et al. Apixaban Downregulates Endothelial Inflammatory and Prothrombotic Phenotype in an In Vitro Model of Endothelial Dysfunction in Uremia. Cardiovasc. Drugs Ther. 2021, 35, 521–532. [Google Scholar] [CrossRef]
- Narita, Y.; Hamamura, K.; Kashiyama, M.; Utsumi, S.; Kakizoe, Y.; Kondo, Y.; Ishitsuka, Y.; Jono, H.; Irie, T.; Mukoyama, M.; et al. Edoxaban Exerts Antioxidant Effects Through FXa Inhibition and Direct Radical-Scavenging Activity. Int. J. Mol. Sci. 2019, 20, 4140. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Ohashi, K.; Ogawa, H.; Otaka, N.; Kawanishi, H.; Takikawa, T.; Ozaki, Y.; Takahara, K.; Tatsumi, M.; Takefuji, M.; et al. Factor Xa inhibitor, edoxaban ameliorates renal injury after subtotal nephrectomy by reducing epithelial-mesenchymal transition and inflammatory response. Physiol. Rep. 2022, 10, e15218. [Google Scholar] [CrossRef]
- Kadoglou, N.P.; Moustardas, P.; Katsimpoulas, M.; Kapelouzou, A.; Kostomitsopoulos, N.; Schafer, K.; Kostakis, A.; Liapis, C.D. The beneficial effects of a direct thrombin inhibitor, dabigatran etexilate, on the development and stability of atherosclerotic lesions in apolipoprotein E-deficient mice: Dabigatran etexilate and atherosclerosis. Cardiovasc. Drugs Ther. 2012, 26, 367–374. [Google Scholar] [CrossRef]
- Pingel, S.; Tiyerili, V.; Mueller, J.; Werner, N.; Nickenig, G.; Mueller, C. Thrombin inhibition by dabigatran attenuates atherosclerosis in ApoE deficient mice. Arch. Med. Sci. 2014, 10, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Tripathy, D.; Sanchez, A.; Yin, X.; Luo, J.; Martinez, J.; Grammas, P. Thrombin, a mediator of cerebrovascular inflammation in AD and hypoxia. Front. Aging Neurosci. 2013, 5, 19. [Google Scholar] [CrossRef]
- Iannucci, J.; Johnson, S.L.; Majchrzak, M.; Barlock, B.J.; Akhlaghi, F.; Seeram, N.P.; Sen, A.; Grammas, P. Short-term treatment with dabigatran alters protein expression patterns in a late-stage tau-based Alzheimer’s disease mouse model. Biochem. Biophys. Rep. 2020, 24, 100862. [Google Scholar] [CrossRef]
- Johnson, S.L.; Iannucci, J.; Seeram, N.P.; Grammas, P. Inhibiting thrombin improves motor function and decreases oxidative stress in the LRRK2 transgenic Drosophila melanogaster model of Parkinson’s disease. Biochem. Biophys. Res. Commun. 2020, 527, 532–538. [Google Scholar] [CrossRef] [PubMed]
- Yazici, S.; Karahan, O.; Oral, M.K.; Bayramoğlu, Z.; Unal, M.; Caynak, B.; Sagbas, E. Comparison of Renoprotective Effect of Dabigatran with Low-Molecular-Weight Heparin. Clin. Appl. Thromb. Hemost. 2016, 22, 361–365. [Google Scholar] [CrossRef]
- Durmaz, S.; Kurtoğlu, T.; Rahman, Ö.F.; Tataroğlu, C.; Yılmaz, M.; Barbarus, E.; Erkan, M.H. Direct oral anticoagulant agents attenuate temporary aortic occlusion-induced renal oxidative and inflammatory responses in rats. Turk. Gogus Kalp Damar Cerrahisi Derg. 2022, 30, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Woźniak, E.; Broncel, M.; Bukowska, B.; Gorzelak-Pabiś, P. The Protective Effect of Dabigatran and Rivaroxaban on DNA Oxidative Changes in a Model of Vascular Endothelial Damage with Oxidized Cholesterol. Int. J. Mol. Sci. 2020, 21, 1953. [Google Scholar] [CrossRef]
- Song, K.; Wang, Y.; Sheng, J.; Ma, C.; Li, H. Effects of dabigatran regulates no-reflow phenomenon in acute myocardial infarction mice through anti-inflammatory and anti-oxidative activities and connective tissue growth factor expression. Mol. Med. Rep. 2018, 17, 580–585. [Google Scholar] [CrossRef]
- Mahmoud, N.I.; Messiha, B.A.S.; Salehc, I.G.; Abo-Saif, A.A.; Abdel-Bakky, M.S. Interruption of platelets and thrombin function as a new approach against liver fibrosis induced experimentally in rats. Life Sci. 2019, 231, 116522. [Google Scholar] [CrossRef] [PubMed]
- Youssef, M.E.; Abdel-Reheim, M.A.; Morsy, M.A.; El-Daly, M.; Atwa, G.M.K.; Yahya, G.; Cavalu, S.; Saber, S.; Ahmed Gaafar, A.G. Ameliorative Effect of Dabigatran on CFA-Induced Rheumatoid Arthritis via Modulating Kallikrein-Kinin System in Rats. Int. J. Mol. Sci. 2022, 23, 1029. [Google Scholar] [CrossRef]
- Ewees, M.G.E.; Abdel-Bakky, M.S.; Bayoumi, A.M.A.; Abo-Saif, A.A.; Altowayan, W.M.; Alharbi, K.S.; Messiha, B.A.S. Dabigatran mitigates cisplatin-mediated nephrotoxicity through down regulation of thrombin pathway. J. Adv. Res. 2021, 31, 127–136. [Google Scholar] [CrossRef]
- Saifi, M.A.; Annaldas, S.; Godugu, C. A direct thrombin inhibitor, dabigatran etexilate protects from renal fibrosis by inhibiting protease activated receptor-1. Eur. J. Pharmacol. 2021, 893, 173838. [Google Scholar] [CrossRef]
- Russo, V.; Attena, E.; Baroni, M.; Trotta, R.; Manu, M.C.; Kirchhof, P.; De Caterina, R. Clinical Performance of Oral Anticoagulants in Elderly with Atrial Fibrillation and Low Body Weight: Insight into Italian Cohort of PREFER-AF and PREFER-AF Prolongation Registries. J. Clin. Med. 2022, 11, 3751. [Google Scholar] [CrossRef]
- Carbone, A.; Bottino, R.; D’Andrea, A.; Russo, V. Direct Oral Anticoagulants for Stroke Prevention in Special Populations: Beyond the Clinical Trials. Biomedicines 2023, 11, 131. [Google Scholar] [CrossRef]
- Melillo, E.; Carbone, A.; Rago, A.; Papa, A.A.; D’ Onofrio, A.; Nigro, G.; Golino, P.; Russo, V. Update on Direct Oral Anticoagulants in Atrial Fibrillation Patients Undergoing Cardiac Interventional Procedures: From Clinical Trials to Real-World Evidence. J. Cardiovasc. Pharmacol. 2020, 75, 185–199. [Google Scholar] [CrossRef] [PubMed]
- Shoji, S.; Kuno, T.; Fujisaki, T.; Takagi, H.; Briasoulis, A.; Deharo, P.; Cuisset, T.; Latib, A.; Kohsaka, S. De-Escalation of Dual Antiplatelet Therapy in Patients with Acute Coronary Syndromes. J. Am. Coll. Cardiol. 2021, 78, 763–777. [Google Scholar] [CrossRef] [PubMed]
- Capodanno, D.; Baber, U.; Bhatt, D.L.; Collet, J.P.; Dangas, G.; Franchi, F.; Gibson, C.M.; Gwon, H.C.; Kastrati, A.; Kimura, T.; et al. P2Y12 inhibitor monotherapy in patients undergoing percutaneous coronary intervention. Nat. Rev. Cardiol. 2022, 19, 829–844. [Google Scholar] [CrossRef]
- Grześk, G.; Rogowicz, D.; Wołowiec, Ł.; Ratajczak, A.; Gilewski, W.; Chudzińska, M.; Sinkiewicz, A.; Banach, J. The Clinical Significance of Drug-Food Interactions of Direct Oral Anticoagulants. Int. J. Mol. Sci. 2021, 22, 8531. [Google Scholar] [CrossRef] [PubMed]
- Steffel, J.; Collins, R.; Antz, M.; Cornu, P.; Desteghe, L.; Haeusler, K.G.; Oldgren, J.; Reinecke, H.; Roldan-Schilling, V.; Rowell, N.; et al. 2021 European Heart Rhythm Association Practical Guide on the Use of Non-Vitamin K Antagonist Oral Anticoagulants in Patients with Atrial Fibrillation. Europace 2021, 23, 1612–1676. [Google Scholar]
- Grześk, G.; Woźniak-Wiśniewska, A.; Błażejewski, J.; Górny, B.; Wołowiec, Ł.; Rogowicz, D.; Nowaczyk, A. The Interactions of Nintedanib and Oral Anticoagulants-Molecular Mechanisms and Clinical Implications. Int. J. Mol. Sci. 2020, 22, 282. [Google Scholar] [CrossRef]
- Reilly, P.A.; Lehr, T.; Haertter, S.; Connolly, S.J.; Yusuf, S.; Eikelboom, J.W.; Ezekowitz, M.D.; Nehmiz, G.; Wang, S.; Wallentin, L.; et al. The effect of dabigatran plasma concentrations and patient characteristics on the frequency of ischemic stroke and major bleeding in atrial fibrillation patients: The RE-LY Trial (Randomized Evaluation of Long-Term Anticoagulation Therapy). J. Am. Coll. Cardiol. 2014, 63, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Ruff, C.T.; Giugliano, R.P.; Braunwald, E.; Morrow, D.A.; Murphy, S.A.; Kuder, J.F.; Deenadayalu, N.; Jarolim, P.; Betcher, J.; Shi, M.; et al. Association between edoxaban dose, concentration, anti-Factor Xa activity, and outcomes: An analysis of data from the randomised, double-blind ENGAGE AF-TIMI 48 trial. Lancet 2015, 385, 2288–2295. [Google Scholar] [CrossRef]
- Grześk, G. Therapeutic monitoring of direct oral anticoagulants—An 8-year observational study. Acta Haematol. Pol. 2021, 52, 446–452. [Google Scholar] [CrossRef]
- Zeitouni, M.; Giczewska, A.; Lopes, R.D.; Wojdyla, D.M.; Christersson, C.; Siegbahn, A.; De Caterina, R.; Steg, P.G.; Granger, C.B.; Wallentin, L.; et al. Clinical and Pharmacological Effects of Apixaban Dose Adjustment in the ARISTOTLE Trial. J. Am. Coll. Cardiol. 2020, 75, 1145–1155. [Google Scholar] [CrossRef]
- Russo, V.; Rago, A.; Laezza, N.; Di Micco, P.; Giannetti, L.; Atripaldi, L.; D’Onofrio, A.; Golino, P.; Nigro, G. Edoxaban in elderly patient with morbid obesity and atrial fibrillation: The role of plasma levels evaluation for selecting the appropriate dose. Monaldi Arch. Chest Dis. 2020, 90, 1224. [Google Scholar] [CrossRef] [PubMed]
- Renon, F.; Rago, A.; Liccardo, B.; D’Andrea, A.; Riegler, L.; Golino, P.; Nigro, G.; Russo, V. Direct Oral Anticoagulants Plasma Levels Measurement: Clinical Usefulness from Trials and Real-World Data. Semin. Thromb. Hemost. 2021, 47, 150–160. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Authors | Protocol | Patients | Target | Aspirin Dose | Results |
|---|---|---|---|---|---|
| Podhaisky [98] | Preclinical in vitro | - | Bovine pulmonary artery endothelial cells | - | ↓ H202-induced toxicity ↑ cell viability |
| Oberle [99] | Preclinical in vitro | - | Bovine pulmonary artery endothelial cells | - | ↑ Ferritin protein synthesis |
| Shi [113] | Preclinical in vitro | - | Mouse macrophage cells | - | ↓ silica-induced lipid peroxidation ↓ DNA strand breakage ↓ activation of NF-kB |
| Hsu [130] | Preclinical in vitro | - | ϕ-174 plasmid DNA | - | ↓ Oxidative stress-induced DNA damage |
| Grosser [100] | Preclinical in vitro | - | HUVECs | - | ↓ H202-mediated toxicity ↑ HO-1 protein expression |
| Wang [123] | Preclinical in vitro | - | ventral mesencephalic tissues of embryonic day 14–15 rats | - | ↓ NOX activity ↓ superoxide production by microglial cells ↓ intracellular ROS concentrations |
| Jian [104] | Preclinical in vitro | - | human primary melanocytes | - | ↑ cell viability intracellular ↓ ROS levels ↑ NRF2 nuclear translocation ↑ ARE-luciferase activity ↑ expression of HO-1 |
| Dimitrovska [127] | Preclinical in vitro | - | Adult male Wistar rats | - | ↑ CAT activity ↑ PARP and HSP70 |
| Wang [105] | Preclinical in vitro | - | Adult male Sprague-Dawley rats | - | ↑ Nrf2/HO-1 signaling pathway |
| Chen [110] | Preclinical in vitro | - | NF-kB–luciferase+/+transgenic mice | - | ↓ NF-kB expression ↓ ROS |
| Jorda [111] | Preclinical in vitro | - | Wilson rats | - | ↑ cell viability ↓ Cu/Zn-SOD and Mn-SOD ↓ NF-kB expression ↓ iNOS |
| John [115] | Preclinical in vitro | - | Goto-Kakizaki Diabetic Rats | - | ↓ ROS ↓ NOX activity ↓ lipid peroxidation ↓ CYP2E1 activity |
| John [116] | Preclinical in vitro | - | Goto-Kakizaki Diabetic Rats | - | ↓ Kidney ROS ↓ NOX activity ↓ lipid peroxidation ↑ SOD activity ↑ GSH |
| Veres [128] | Preclinical in vitro | - | Lewis rats | - | ↓ nitro-oxidative stress |
| De La Cruz [124] | Preclinical in vitro and ex vivo | - | Adult male Wistar rats | - | ↓ oxidative stress, ↓ iNOS activty ↓ TBARS formation, ↓ GSSG ↑ GP and GT activity |
| Wròbel [120] | Preclinical in vitro and ex vivo | - | male BALB/c mice | - | ↑ H2S-producing enzymes activity ↑ GSH/GSSG ratio |
| Wu [121] | Preclinical in vitro and in vivo | - | Male Sprague-Dawley, Wistar-Kyoto, and spontaneously hypertensive rats | - | ↓ O2 production ↓ NOX activity |
| Yang [112] | Preclinical in vitro and in vivo | - | ApoE−/−mice, vascular smooth-muscle cells | - | ↑ IkBα ↓ NOX activity ↓ F2-isoprostanes |
| Frydrychowski [129] | Preclinical in vitro and in vivo | - | Female pigs | - | ↓ TOS, OSI, MDA ↑ TAC |
| Caballero [117] | Preclinical in vivo | - | Streptozotocin-induced diabetic male CF1 mice | - | ↓ accumulation of lipoperoxidative aldehydes |
| De Cristòbal [118] | Preclinical in vivo | - | Adult male Wistar rats | - | ↓ iNOS activity and expression ↓ TNF-a ↓ lipid peroxidation and concentration of GSSG |
| Prasad [114] | Preclinical in vivo | - | New Zealand white female rabbits | - | ↓ serum and aortic MDA ↓ white blood cell-chemiluminescence ↑ antioxidant reserve |
| Kiliçoğlu Aydin [125] | Preclinical in vivo | - | Spraque-Dawley rats | - | ↑ Erythrocyte CAT ↓ Erythrocyte SOD |
| Ayyadevara [126] | Preclinical in vivo | - | Caenorhabditis elegans | - | ↑ Transcript levels of antioxidant genes encoding SOD, CAT, GT ↑ resistance to exogenous peroxide ↓ ROS |
| Chaávez [119] | Preclinical in vivo and ex vivo | - | Wistar male rats | - | ↓ Lipid peroxidation ↑ GSH/GSSG ↑ GSH ↓ NF-kB |
| Wu [122] | Preclinical in vivo and ex vivo | - | Cardiomyopathic male hamsters | - | ↓ O2− production ↓ NOX activity |
| Ristimäe [131] | Non-blind, non-placebo-controlled study | 25 | Healthy middle-aged subjects | 100 mg | ↑ serum antioxidative capacity |
| Kurban [132] | Non-blind, non-placebo-controlled study | 30 | Healthy middle-aged subjects | 100–150 mg | (150 mg group): ↓ TOS ↓ OxLDLs |
| Cheng [134] | Non-blind, non-placebo-controlled study | 80 | 50 AS and 30 healthy middle-aged subjects | 100 mg | ↓ O2− ↓ NF-kB |
| Berg [133] | Prospective, randomized | 20 | Patients referred for first-time CABG | 160 mg | ↓ 8-iso-PGF2α |
| Authors | Protocol | Patients | Target | P2Y12 Drug and Dose | Results |
|---|---|---|---|---|---|
| Kanko [139] | Preclinical in vitro and in vivo | - | Male adult Sprague-Dawley rats | Clopidogrel | ↓ MDA ↑ GSH ↑ SOD activity |
| Hu [140] | Preclinical in vitro and in vivo | - | Healthy male BALB/c mice | Clopidogrel | ↓ apoptosis ↑ TAC |
| Hadi [141] | Preclinical in vitro | - | Domestic rabbits | Clopidogrel | ↓ MDA ↑ GSH |
| Yang [142] | Preclinical in vitro | - | HAECs | Clopidogrel | ↓ TNF-a ↓ ROS ↑ GSH ↑ HO-1 |
| An [143] | Preclinical in vitro | - | Wild-type male mice | Clopidogrel | ↓ NADPH |
| Korish [144] | Preclinical in vitro | - | Male mice | Clopidogrel | ↓ MDA ↑ catalase activity |
| Kang [145] | Preclinical in vitro | - | HUVECs | Ticagrelor | ↓ oxLDL-induced apoptosis |
| El-Mokadem [146] | Preclinical in vitro | - | Adults male Wistar rats | Ticagrelor | ↓ MDA ↓ TNF-a ↓ apoptosis |
| Bitirim [147] | Preclinical in vitro | - | High-glucose-incubated H9c2-cells | Ticagrelor | ↓ ROS ↓ apoptosis ↑ miR-499, miR-133a, miR-133b |
| McClung [155] | Non-blind, non-placebo-controlled study | 9 | Patients with DMII | Clopidogrel 75 mg | ↓ CECs ↑ EPCs |
| Bundhoo [156] | Non-blind, non-placebo prospective study | 58 | Patients undergoing PCI | Clopidogrel loading dose of 600 mg, then 75 mg od | ↑ TAC |
| Heitzer [149] | Prospective, randomized | 103 | Patients with stable CAD and chronic ASA therapy | Clopidogrel loading dose of 300 mg, then 75 mg od | ↓ urinary 8-iso-PG F2α |
| Taher [150] | Prospective, randomized | 42 | Patients with DMII | Clopidogrel 75 mg | ↓ MDA ↑ GSH |
| Campo [152] | Prospective, randomized | 46 | Patients with COPD and stable CAD requiring PCI | Clopidogrel loading dose if 300 mg, then 75 mg od, or ticagrelor loading dose of 180 mg, then 90 mg bid | ↓ ROS (ticagrelor group) |
| Aquila [153] | Prospective, randomized | 46 | Patients with COPD and stable CAD requiring PCI | Clopidogrel loading dose of 300 mg, then 75 mg od, or ticagrelor loading dose of 180 mg, then 90 mg bid | ↑ expression of SIRT1 and HES1 |
| Rudolph [151] | Randomized, active-controlled, double-blind trial | 45 | Patients undergoing PCI | Clopidogrel loading dose of 600 mg, then clopidogrel 75 mg od, or prasugrel 10 mg od | ↓ MPO |
| Authors | Protocol | Patients | Target | Dose | Results |
|---|---|---|---|---|---|
| Caliskan [157] | Preclinical in vitro | - | Male Sprague-Dawley Rats | - | ↓ MDA |
| Ishibashi [158] | Preclinical in vitro | - | HUVECs | - | ↓ ROS production ↓ MCP-1 ↓ ICAM-1 |
| Ishibashi [159] | Preclinical in vitro | - | Human proximal tubular cells | - | ↓ ROS production ↓ MCP-1 |
| Gul Utku [160] | Preclinical in vitro | - | Female Wistar rats | - | ↓ MDA ↓ MPO |
| Ellinghaus [161] | Preclinical in vitro | - | HUVECs | - | ↓ VCAM-1 ↓ ICAM-1, ↓ MCP-1 ↓ IL-8 ↓ CXCL1 ↓ CXCL2 ↓ TF |
| Vilaseca [162] | Preclinical in vitro | - | Wistar rats | - | ↓ ROS |
| Maeda [163] | Preclinical in vitro | - | HUVECs | - | ↓ ROS-induced senescence ↓ NOX subunits ↑ NOS |
| Shafiey [164] | Preclinical in vitro | - | Adult male Wistar rats | - | ↑ SOD ↑ GP ↓ MDA ↓ NO ↓ NF-kB |
| Abedalqader [165] | Preclinical in vitro | - | Adult male Wistar rats | - | ↓ TBARS |
| Imano [166] | Preclinical in vivo | - | Male C57BL/6J mice | - | ↓ ROS ↓ NF-kB |
| Imam [167] | Preclinical in vivo | - | Adult male Wistar rats | - | ↓ NOS ↑ GSH ↑ GR |
| Al-harbi [168] | Preclinical in vivo | - | Adult male Wistar rats | - | ↓ MDA ↑ GSH ↑ GR |
| Abdelzaher [169] | Preclinical in vivo | - | Adult male Wistar rats | - | ↓ MDA ↓ NF-Kb ↑ GSH ↑ SOD |
| Moñux [170] | Ex vivo | 6 | Abdominal aortic aneurysm sites with intraluminal mural thrombus | - | ↓ NOX subunits ↓ NOS2 ↓ ICAM-1 ↓ VCAM-1 |
| Authors | Protocol | Patients | Target | Dose | Results |
|---|---|---|---|---|---|
| Torramade-Moix [173] | Preclinical in vitro | - | HUVECs and HMEC-1 | - | ↓ ICAM-1 ↓ VCAM-1 ↓ ROS ↑ NOS |
| Authors | Protocol | Patients | Target | Dose | Results |
|---|---|---|---|---|---|
| Narita [174] | Preclinical in vitro | - | HK-2 cells | - | ↓ ROS |
| Fang [175] | Preclinical in vitro | - | Male wild-type mice and HK-2 cells | - | ↓ TNFa ↓ MCP-1 ↓ NOX subunits |
| Authors | Protocol | Patients | Target | Dose | Results |
|---|---|---|---|---|---|
| Kadoglou [176] | Preclinical in vitro | - | ApoE−/−mice | - | ↓ ROS |
| Tripathy [178] | Preclinical in vitro | - | Rat brain endothelial cell cultures | - | ↓ ROS ↓ MCP-1 |
| Pingel [177] | Preclinical in vitro | - | ApoE−/−mice | - | ↓ ROS |
| Yazici [181] | Preclinical in vitro | - | Male Sprague-Dawley rats | - | ↓ MDA |
| Song [184] | Preclinical in vitro | - | Male New Zealand White rabbits | - | ↓ NF-kB ↓ TNFa ↓ IL-1 ↑ CAT ↑ SOD |
| Wozńiak [183] | Preclinical in vitro | - | HUVECs | - | ↓ ROS-induced DNA strand breakage ↓ ROS |
| Mahmoud [185] | Preclinical in vitro | - | Adult male albino rats | - | ↓ MDA ↓ NOX ↓ TNFa ↓ IL-1 ↑ GSH |
| Iannucci [179] | Preclinical in vitro | - | Female transgenic Tg4510 AD mice | - | ↓ iNOS ↓ NOX |
| Johnson [180] | Preclinical in vitro | - | LRRK2 mutant Drosophila melanogaster | - | ↓ SOD ↓ NOX ↓ ROS |
| Ewees [187] | Preclinical in vitro | - | Male adult albino rats | - | ↓ MDA ↑ GSH ↓ NOX ↓ SOD |
| Saifi [188] | Preclinical in vitro | - | Male Swiss albino mice | - | ↓ ROS ↓ IL-1 ↓ TNFa ↑ GSH |
| Durmaz [182] | Preclinical in vitro | - | Male Wistar rats | - | ↑ TAC ↑ TOS ↓ TNFa ↓ IL-1 |
| Youssef [186] | Preclinical in vitro | - | Aimdualst female Wistar albino rats | - | ↓ MDA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Falco, L.; Tessitore, V.; Ciccarelli, G.; Malvezzi, M.; D’Andrea, A.; Imbalzano, E.; Golino, P.; Russo, V. Antioxidant Properties of Oral Antithrombotic Therapies in Atherosclerotic Disease and Atrial Fibrillation. Antioxidants 2023, 12, 1185. https://doi.org/10.3390/antiox12061185
Falco L, Tessitore V, Ciccarelli G, Malvezzi M, D’Andrea A, Imbalzano E, Golino P, Russo V. Antioxidant Properties of Oral Antithrombotic Therapies in Atherosclerotic Disease and Atrial Fibrillation. Antioxidants. 2023; 12(6):1185. https://doi.org/10.3390/antiox12061185
Chicago/Turabian StyleFalco, Luigi, Viviana Tessitore, Giovanni Ciccarelli, Marco Malvezzi, Antonello D’Andrea, Egidio Imbalzano, Paolo Golino, and Vincenzo Russo. 2023. "Antioxidant Properties of Oral Antithrombotic Therapies in Atherosclerotic Disease and Atrial Fibrillation" Antioxidants 12, no. 6: 1185. https://doi.org/10.3390/antiox12061185
APA StyleFalco, L., Tessitore, V., Ciccarelli, G., Malvezzi, M., D’Andrea, A., Imbalzano, E., Golino, P., & Russo, V. (2023). Antioxidant Properties of Oral Antithrombotic Therapies in Atherosclerotic Disease and Atrial Fibrillation. Antioxidants, 12(6), 1185. https://doi.org/10.3390/antiox12061185