
Nrf2 as a Therapeutic Target in the Resistance to Targeted Therapies in Melanoma


 ,
,  and
and 
Abstract
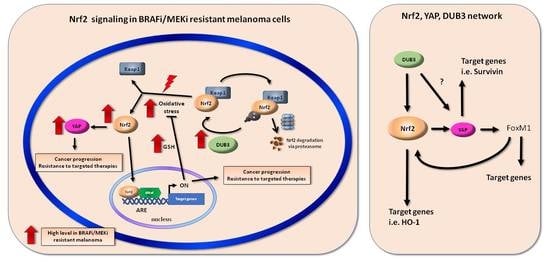
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cells, Culture Conditions
2.3. Generation of D4M Subclones Resistant to Dabrafenib, Trametinib, or Dual Resistance to Dabrafenib/Trametinib
2.4. MTT Assay
2.5. Anchorage-Independent Growth Assay
2.5.1. Sphere Formation Assay
2.5.2. Soft Agar Colony Formation Assay
2.6. Cell Apoptosis
2.7. Cell Invasion Assay
2.8. Angiogenesis Assay
2.9. Measurement of the Cell Redox Status
2.10. GSH Content
2.11. Protein Extraction and Western Blot Analysis
2.12. Quantitative Reverse Transcription Polymerase Chain Reaction (qRT-PCR)
2.13. Cell Transfection with siRNA against Nrf2 or DUB3
2.14. Statistics
3. Results
3.1. Generation and Characterization of D4M Cell Lines Resistant to Dabrafenib, Trametinib, or Dual Resistance to Dabrafenib/Trametinib
3.1.1. Cell Viability
3.1.2. Anchorage-Independent Cell Growth
3.1.3. Apoptosis
3.1.4. Cell Migration and Angiogenesis
3.2. Analysis of the Redox Status and Nrf2 Expression in D4M Cell Lines Resistant to Dabrafenib, Trametinib, or Dual Resistance to Dabrafenib/Trametinib
3.3. Nrf2 Affects YAP Expression in D4M Cell Lines Resistant to Dabrafenib, Trametinib, or Dual Resistance to Dabrafenib/Trametinib
3.4. Mechanism of Nrf2 Activity Regulation in D4M Cell Lines Resistant to Dabrafenib, Trametinib, or Dual RESISTANCE to Dabrafenib/Trametinib
3.5. Inhibition of Nrf2 or DUB3 Expression Sensitizes Resistant Melanoma D4M to Targeted Therapies
3.6. Nrf2 and YAP Expression in A375 Cell Line Resistant to Dabrafenib
3.7. Mechanisms of Nrf2 Gene Expression Control in an A375 Cell Line Resistant to Dabrafenib
3.8. Inhibition of Nrf2 or DUB3 Expression Sensitizes Resistant A375 Melanoma Cells to Targeted Therapy
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Matthews, N.H.; Li, W.Q.; Qureshi, A.A.; Weinstock, M.A.; Cho, E. Epidemiology of Melanoma. In Cutaneous Melanoma: Etiology and Therapy [Internet]; Ward, W.H., Farma, J.M., Eds.; Codon Publications: Brisbane, Australia, 2017; Volume 1. [Google Scholar]
- Saginala, K.; Barsouk, A.; Aluru, J.S.; Rawla, P.; Barsouk, A. Epidemiology of Melanoma. Med. Sci. 2021, 9, 63. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef]
- Niezgoda, A.; Niezgoda, P.; Czajkowski, R. Novel Approaches to Treatment of Advanced Melanoma: A Review on Targeted Therapy and Immunotherapy. Biomed. Res. Int. 2015, 2015, 851387. [Google Scholar] [CrossRef] [Green Version]
- Quaglino, P.; Fava, P.; Tonella, L.; Rubatto, M.; Ribero, S.; Fierro, M.T. Treatment of Advanced Metastatic Melanoma. Dermatol. Pract. Concept. 2011, 11, e2021164S. [Google Scholar] [CrossRef]
- Cheng, L.; Lopez-Beltran, A.; Massari, F.; MacLennan, G.T.; Montironi, R. Molecular testing for BRAF mutations to inform melanoma treatment decisions: A move toward precision medicine. Mod. Pathol. 2018, 31, 24–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert, C.; Karaszewska, B.; Schachter, J.; Rutkowski, P.; Mackiewicz, A.; Stroiakovski, D.; Lichinitser, M.; Dummer, R.; Grange, F.; Mortier, L.; et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N. Engl. J. Med. 2015, 372, 30–39. [Google Scholar] [CrossRef] [Green Version]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.J.; et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: A multicentre, double-blind, phase 3 randomised controlled trial. Lancet 2015, 386, 444–451. [Google Scholar] [CrossRef]
- Dummer, R.; Ascierto, P.A.; Gogas, H.J.; Arance, A.; Mandala, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsova, I.; Gutzmer, R.; et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF-mutant melanoma (COLUMBUS): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2018, 19, 603–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kakadia, S.; Yarlagadda, N.; Awad, R.; Kundranda, M.; Niu, J.; Naraev, B.; Mina, L.; Dragovich, T.; Gimbel, M.; Mahmoud, F. Mechanisms of resistance to BRAF and MEK inhibitors and clinical update of US Food and Drug Administration-approved targeted therapy in advanced melanoma. OncoTargets Ther. 2018, 11, 7095–7107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moloney, J.N.; Cotter, T.G. ROS signalling in the biology of cancer. Semin. Cell Dev. Biol. 2018, 80, 50–64. [Google Scholar] [CrossRef]
- Pizzimenti, S.; Ribero, S.; Cucci, M.A.; Grattarola, M.; Monge, C.; Dianzani, C.; Barrera, G.; Muzio, G. Oxidative Stress-Related Mechanisms in Melanoma and in the Acquired Resistance to Targeted Therapies. Antioxidants 2021, 10, 1942. [Google Scholar] [CrossRef] [PubMed]
- de la Vega, M.R.; Chapman, E.; Zhang, D.D. NRF2 and the Hallmarks of Cancer. Cancer Cell 2018, 34, 21–43. [Google Scholar] [CrossRef]
- Carpenter, E.L.; Becker, A.L.; Indra, A.K. NRF2 and Key Transcriptional Targets in Melanoma Redox Manipulation. Cancers 2022, 14, 1531. [Google Scholar] [CrossRef]
- Malakoutikhah, Z.; Mohajeri, Z.; Dana, N.; Haghjooy Javanmard, S. The dual role of Nrf2 in melanoma: A systematic review. Mol. Cell Biol. 2023, 24, 5. [Google Scholar] [CrossRef]
- Milkovic, L.; Zarkovic, N.; Saso, L. Controversy about pharmacological modulation of Nrf2 for cancer therapy. Redox Biol. 2017, 12, 727–732. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Zhao, Z.; Meng, X.; Chen, H.; Fu, G. Migration and invasion in B16-F10 mouse melanoma cells are regulated by Nrf2 inhibition during treatment with ionizing radiation. Oncol. Lett. 2018, 16, 1959–1966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambrosio, L.; Argenziano, M.; Cucci, M.A.; Grattarola, M.; de Graaf, I.A.M.; Dianzani, C.; Barrera, G.; Sánchez Nieves, J.; Gomez, R.; Cavalli, R.; et al. Carbosilane Dendrimers Loaded with siRNA Targeting Nrf2 as a Tool to Overcome Cisplatin Chemoresistance in Bladder Cancer Cells. Antioxidants 2020, 9, 993. [Google Scholar] [CrossRef]
- Hintsala, H.R.; Jokinen, E.; Haapasaari, K.M.; Moza, M.; Ristimäki, A.; Soini, Y.; Koivunen, J.; Karihtala, P. Nrf2/Keap1 Pathway and Expression of Oxidative Stress Lesions 8-hydroxy-2′-deoxyguanosine and Nitrotyrosine in Melanoma. Anticancer Res. 2016, 36, 1497–1506. [Google Scholar] [PubMed]
- Miura, S.; Shibazaki, M.; Kasai, S.; Yasuhira, S.; Watanabe, A.; Inoue, T.; Kageshita, Y.; Tsunoda, K.; Takahashi, K.; Akasaka, T.; et al. A somatic mutation of the KEAP1 gene in malignant melanoma is involved in aberrant NRF2 activation and an increase in intrinsic drug resistance. J. Investig. Dermatol. 2014, 134, 553–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rocha, C.R.; Kajitani, G.S.; Quinet, A.; Fortunato, R.S.; Menck, C.F.M. NRF2 and glutathione are key resistance mediators to temozolomide in glioma and melanoma cells. Oncotarget 2016, 7, 48081–48092. [Google Scholar] [CrossRef] [Green Version]
- Argenziano, M.; Bessone, F.; Dianzani, C.; Cucci, M.A.; Grattarola, M.; Pizzimenti, S.; Cavalli, R. Ultrasound-Responsive Nrf2-Targeting siRNA-Loaded Nanobubbles for Enhancing the Treatment of Melanoma. Pharmaceutics 2022, 14, 341. [Google Scholar] [CrossRef] [PubMed]
- McCord, J.M.; Gao, B.; Hybertson, B.M. The Complex Genetic and Epigenetic Regulation of the Nrf2 Pathways: A Review. Antioxidants 2023, 12, 366. [Google Scholar] [CrossRef] [PubMed]
- Ciamporcero, E.; Daga, M.; Pizzimenti, S.; Roetto, A.; Dianzani, C.; Compagnone, A.; Palmieri, A.; Ullio, C.; Cangemi, L.; Pili, R.; et al. Crosstalk between Nrf2 and YAP contributes to maintaining the antioxidant potential and chemoresistance in bladder cancer. Free Radic. Biol. Med. 2018, 115, 447–457. [Google Scholar] [CrossRef]
- Grattarola, M.; Cucci, M.A.; Roetto, A.; Dianzani, C.; Barrera, G.; Pizzimenti, S. Post-translational down-regulation of Nrf2 and YAP proteins, by targeting deubiquitinases, reduces growth and chemoresistance in pancreatic cancer cells. Free Radic. Biol. Med. 2021, 174, 202–210. [Google Scholar] [CrossRef]
- Harvey, K.F.; Zhang, X.; Thomas, D.M. The Hippo pathway and human cancer. Nat. Rev. Cancer 2013, 13, 246–257. [Google Scholar] [CrossRef]
- Zhao, B.; Ye, X.; Yu, J.; Li, L.; Li, W.; Li, S.; Yu, J.; Lin, J.D.; Wang, C.Y.; Chinnaiyan, A.M. KEAD mediates YAP-dependent gene induction and growth control. Genes Dev. 2008, 22, 1962–1971. [Google Scholar] [CrossRef] [Green Version]
- Mizuno, T.; Murakami, H.; Fujii, M.; Ishiguro, F.; Tanaka, I.; Kondo, Y.; Akatsuka, S.; Toyokuni, S.; Yokoi, K.; Osada, H.; et al. YAP induces malignant mesothelioma cell proliferation by upregulating transcription of cell cycle-promoting genes. Oncogene 2012, 31, 5117–5122. [Google Scholar] [CrossRef] [Green Version]
- Shao, D.; Zhai, P.; Del Re, D.P.; Sciarretta, S.; Yabuta, N.; Nojima, H.; Lim, D.S.; Pan, D.; Sadoshima, J.A. functional interaction between Hippo-YAP signalling and FoxO1 mediates the oxidative stress response. Nat. Commun. 2014, 5, 3315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Zhang, Z.Y.; Du, H.; Li, S.Z.; Tu, R.; Jia, Y.F.; Zheng, Z.; Song, X.M.; Du, R.L.; Zhang, X.D. DUB3 deubiquitinates and stabilizes NRF2 in chemotherapy resistance of colorectal cancer. Cell Death Differ. 2019, 26, 2300–2313. [Google Scholar] [CrossRef]
- Jung, B.J.; Yoo, H.S.; Shin, S.; Park, Y.J.; Jeon, S.M. Dysregulation of NRF2 in Cancer: From Molecular Mechanisms to Therapeutic Opportunities. Biomol. Ther. 2018, 26, 57–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hämälaïnen, M.; Teppo, H.R.; Skarp, S.; Haapasaari, K.; Porvari, K.; Vuopala, K.; Kietzmann, T.; Karihtala, P. NRF1 and NRF2 mRNA and Protein Expression Decrease Early during Melanoma Carcinogenesis: An Insight into Survival MicroRNAs. Oxid. Med. Cell. Longev. 2019, 2019, 2647068. [Google Scholar] [CrossRef] [Green Version]
- Barrera, G.; Cucci, M.A.; Grattarola, M.; Dianzani, C.; Muzio, G.; Pizzimenti, S. Control of Oxidative Stress in Cancer Chemoresistance: Spotlight on Nrf2 Role. Antioxidants 2021, 10, 510. [Google Scholar] [CrossRef] [PubMed]
- Soysouvanh, F.; Giuliano, S.; Habel, N.; El-Hachem, N.; Pisibon, C.; Bertolotto, C.; Ballotti, R. An Update on the Role of Ubiquitination in Melanoma Development and Therapies. J. Clin. Med. 2021, 10, 1133. [Google Scholar] [CrossRef] [PubMed]
- Ohanna, M.; Biber, P.; Deckert, M. Emerging Role of Deubiquitinating Enzymes (DUBs) in Melanoma Pathogenesis. Cancers 2022, 14, 3371. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, M.H.; Steinberg, S.M.; Alexander, M.P.; Fisher, J.L.; Ernstoff, M.S.; Turk, M.J.; Mullins, D.W.; Brinckerhoff, C.E. Multiple murine BRaf(V600E) melanoma cell lines with sensitivity to PLX4032. Pigment Cell Melanoma Res. 2014, 27, 495–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cordaro, F.G.; De Presbiteris, A.L.; Camerlingo, R.; Mozzillo, N.; Pirozzi, G.; Cavalcanti, E.; Manca, A.; Palmieri, G.; Cossu, A.; Ciliberto, G.; et al. Phenotype characterization of human melanoma cells resistant to dabrafenib. Oncol. Rep. 2017, 38, 2741–2751. [Google Scholar] [CrossRef] [Green Version]
- Infante, J.R.; Fecher, L.A.; Alchook, G.S.; Nallapareddy, S.; Gordon, M.S.; Becerra, C.; DeMarini, D.J.; Cox, D.S.; Xu, Y.; Morris, S.R.; et al. Safety, pharmacokinetic, pharmacodynamic, and efficacy data for the oral MEK inhibitor trametinib: A phase 1 dose-escalation trial. Lancet Oncol. 2012, 13, 773–781. [Google Scholar] [CrossRef]
- Falchook, G.S.; Long, G.V.; Kurzrock, R.; Kim, K.B.; Arkenau, H.T.; Brown, M.P.; Hamid, O.; Infante, J.R.; Millward, M.; Pavlick, A.; et al. Dose selection, pharmacokinetics, and pharmacodynamics of BRAF inhibitor dabrafenib (GSK2118436). Clin. Cancer Res. 2014, 20, 4449–4458. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.; Wang, L.; Huang, S.; Heynen, G.J.J.E.; Prahallad, A.; Robert, C.; Haanen, J.; Blank, C.; Wesseling, J.; Willems, S.M.; et al. Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature 2014, 508, 118–122. [Google Scholar] [CrossRef]
- Dianzani, C.; Monge, C.; Miglio, G.; Serpe, L.; Martina, K.; Cangemi, L.; Ferraris, C.; Mioletti, S.; Osella, S.; Gigliotti, C.L.; et al. Nanoemulsions as Delivery Systems for Poly-Chemotherapy Aiming at Melanoma Treatment. Cancers 2020, 12, 1198. [Google Scholar] [CrossRef] [PubMed]
- Daga, M.; Ullio, C.; Argenziano, M.; Dianzani, C.; Cavalli, R.; Trotta, F.; Ferretti, C.; Zara, G.P.; Gigliotti, C.L.; Ciamporcero, E.S.; et al. GSH-targeted nanosponges increase doxorubicin-induced toxicity “in vitro” and “in vivo” in cancer cells with high antioxidant defenses. Free Radic. Biol. Med. 2016, 97, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Mori, S.; Chang, J.T.; Andrechek, E.R.; Matsumura, N.; Baba, T.; Yao, G.; Kim, J.W.; Gatza, M.; Murphy, S.; Nevins, J.R. Anchorage-independent cell growth signature identifies tumors with metastatic potential. Oncogene 2009, 28, 2796–2805. [Google Scholar] [CrossRef] [Green Version]
- Subbiah, V.; Baik, C.; Kirkwood, J.M. Clinical Development of BRAF plus MEK Inhibitor Combinations. Trends Cancer 2020, 6, 797–810. [Google Scholar] [CrossRef] [PubMed]
- Bellmann, L.; Cappellano, G.; Schachtl-Riess, J.F.; Prokopi, A.; Seretis, A.; Ortner, D.; Tripp, C.H.; Brinckerhoff, C.E.; Mullins, D.W.; Stoitzner, P. A TLR7 agonist strengthens T and NK cell function during BRAF-targeted therapy in a preclinical melanoma model. Int. J. Cancer 2020, 146, 1409–1420. [Google Scholar] [CrossRef]
- Meyskens, F.L., Jr.; McNulty, S.E.; Buckmeier, J.A.; Tohidian, N.B.; Spillane, T.J.; Kahlon, R.S.; Gonzalez, R.I. Aberrant redox regulation in human metastatic melanoma cells compared to normal melanocytes. Free Radic. Biol. Med. 2001, 31, 799–808. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, L.B.; Chandel, N.S. Mitochondrial reactive oxygen species and cancer. Cancer Metab. 2014, 2, 17. [Google Scholar] [CrossRef] [Green Version]
- Valavanidis, A.; Vlachogianni, T.; Fiotakis, K. Tobacco smoke: Involvement of reactive oxygen species and stable free radicals in mechanisms of oxidative damage, carcinogenesis and synergistic effects with other respirable particles. Int. J. Environ. Res. Public Health 2009, 6, 445–462. [Google Scholar] [CrossRef]
- Obrador, E.; Liu-Smith, F.; Dellinger, R.W.; Salvador, R.; Meyskens, F.L.; Estrela, J.M. Oxidative stress and antioxidants in the pathophysiology of malignant melanoma. Biol. Chem. 2019, 400, 589–612. [Google Scholar] [CrossRef] [Green Version]
- Hall, A.; Meyle, K.D.; Lange, M.K.; Klima, M.; Sanderhoff, M.; Dahl, C.; Abildgaard, C.; Thorup, K.; Moghimi, S.M.; Jensen, P.B.; et al. Dysfunctional oxidative phosphorylation makes malignant melanoma cells addicted to glycolysis driven by the (V600E)BRAF oncogene. Oncotarget 2013, 4, 584–599. [Google Scholar] [CrossRef] [Green Version]
- Ruocco, M.R.; Avagliano, A.; Granato, G.; Vigliar, E.; Masone, S.; Montagnani, S.; Arcucci, A. Metabolic flexibility in melanoma: A potential therapeutic target. Semin. Cancer Biol. 2019, 59, 187–207. [Google Scholar] [CrossRef] [PubMed]
- Audrito, V.; Managò, A.; Gaudino, F.; Deaglio, S. Targeting metabolic reprogramming in metastatic melanoma: The key role of nicotinamide phosphoribosyltransferase (NAMPT). Semin. Cell Dev. Biol. 2019, 98, 192–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corazao-Rozas, P.; Guerreschi, P.; Jendoubi, M.; André, F.; Jonneaux, A.; Scalbert, C.; Garçon, G.; Malet-Martino, M.; Balayssac, S.; Rocchi, S.; et al. Mitochondrial oxidative stress is the Achille’s heel of melanoma cells resistant to Braf-mutant inhibitor. Oncotarget 2013, 4, 1986–1998. [Google Scholar] [CrossRef] [Green Version]
- Khamari, R.; Trinh, A.; Gabert, P.E.; Corazao-Rozas, P.; Riveros-Cruz, S.; Balayssac, S.; Malet-Martino, M.; Dekiouk, S.; Joncquel Chevalier Curt, M.; Maboudou, P.; et al. Glucose metabolism and NRF2 coordinate the antioxidant response in melanoma resistant to MAPK inhibitors. Cell Death Dis. 2018, 9, 325. [Google Scholar] [CrossRef] [Green Version]
- Yuan, L.; Mishra, R.; Patel, H.; Abdulsalam, S.; Greis, K.D.; Kadekaro, A.L.; Merino, E.J.; Garrett, J.T. Utilization of Reactive Oxygen Species Targeted Therapy to Prolong the Efficacy of BRAF Inhibitors in Melanoma. J. Cancer 2018, 9, 4665–4676. [Google Scholar] [CrossRef]
- Wang, L.; Leite de Oliveira, R.; Huijberts, S.; Bosdriesz, E.; Pencheva, N.; Brunen, D.; Bosma, A.; Song, J.Y.; Zevenhoven, J.; Los-de Vries, G.T.; et al. An Acquired Vulnerability of Drug-Resistant Melanoma with Therapeutic Potential. Cell 2018, 173, 1413–1425. [Google Scholar] [CrossRef] [Green Version]
- Haq, R.; Shoag, J.; Andreu-Perez, P.; Yokoyama, S.; Edelman, H.; Rowe, G.C.; Frederick, D.T.; Hurley, A.D.; Nellore, A.; Kung, A.L.; et al. Oncogenic BRAF regulates oxidative metabolism via PGC1alpha and MITF. Cancer Cell 2013, 23, 302–315. [Google Scholar] [CrossRef] [Green Version]
- Cesi, G.; Walbrecq, G.; Zimmer, A.; Kreis, S.; Haan, C. ROS production induced by BRAF inhibitor treatment rewires metabolic processes affecting cell growth of melanoma cells. Mol. Cancer 2017, 16, 102. [Google Scholar] [CrossRef] [Green Version]
- Furfaro, A.L.; Traverso, N.; Domenicotti, C.; Piras, S.; Moretta, L.; Marinari, U.M.; Pronzato, M.A.; Nitti, M. The Nrf2/HO-1 Axis in Cancer Cell Growth and Chemoresistance. Oxid. Med. Cell. Longev. 2016, 2016, 1958174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Was, H.; Cichon, T.; Smolarczyk, R.; Lackowska, B.; Mazur-Bialy, A.; Mazur, M.; Szade, A.; Dominik, P.; Mazan, M.; Kotlinowski, J.; et al. Effect of heme oxygenase-1 on melanoma development in mice—Role of tumor-infiltrating immune cells. Antioxidants 2020, 9, 1223. [Google Scholar] [CrossRef] [PubMed]
- Longhitano, L.; Broggi, G.; Giallongo, S.; Failla, M.; Puzzo, L.; Avitabile, T.; Tibullo, D.; Distefano, A.; Pittalà, V.; Reibaldi, M.; et al. Heme Oxygenase-1 Overexpression Promotes Uveal Melanoma Progression and Is Associated with Poor Clinical Outcomes. Antioxidants 2022, 11, 1997. [Google Scholar] [CrossRef] [PubMed]
- Furfaro, A.L.; Ottonello, S.; Loi, G.; Cossu, I.; Piras, S.; Spagnolo, F.; Queirolo, P.; Marinari, U.M.; Moretta, L.; Pronzato, M.A.; et al. HO-1 downregulation favors BRAFV600 melanoma cell death induced by Vemurafenib/PLX4032 and increases NK recognition. Int. J. Cancer 2020, 146, 1950–1962. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, C.; Yi, C. YAP/TAZ Signaling and Resistance to Cancer Therapy. Trends Cancer 2019, 5, 283–296. [Google Scholar] [CrossRef]
- Takeuchi, H.; Morton, D.L.; Elashoff, D.; Hoon, D.S. Survivin expression by metastatic melanoma predicts poor disease outcome in patients receiving adjuvant polyvalent vaccine. Int. J. Cancer 2005, 117, 1032–1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, T.; Kohashi, K.; Yamada, Y.; Maekawa, A.; Kuda, M.; Furue, M.; Oda, Y. Prognostic significance of forkhead box M1 (FoxM1) expression and antitumour effect of FoxM1 inhibition in melanoma. Histopathology 2016, 69, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.H.; Kim, J.; Hong, H.; Lee, S.; Lee, J.; Jung, E.; Kim, J. Actin remodeling confers BRAF inhibitor resistance to melanoma cells through YAP / TAZ activation. EMBO J. 2016, 35, 462–478. [Google Scholar] [CrossRef] [Green Version]
- Fisher, M.L.; Grun, D.; Adhikary, G.; Xu, W.; Eckert, R.L. Inhibition of YAP function overcomes BRAF inhibitor resistance in melanoma cancer stem cells. Oncotarget 2017, 8, 110257–110272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, B.; Shin, H.J.; Bak, I.S.; Bak, Y.; Jeong, Y.L.; Kwon, T.; Park, Y.H.; Sun, H.N.; Kim, C.H.; Yu, D.Y. Peroxiredoxin I is important for cancer-cell survival in Ras-induced hepatic tumorigenesis. Oncotarget 2016, 7, 68044–68056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–109. [Google Scholar] [CrossRef] [Green Version]
- Baird, L.; Dinkova-Kostova, A.T. The cytoprotective role of the Keap1-Nrf2 pathway. Arch. Toxicol. 2011, 85, 241–272. [Google Scholar] [CrossRef]
- Komander, D.; Clague, M.J.; Urbé, S. Breaking the chains: Structure and function of the deubiquitinases. Nat. Rev. Mol. Cell Biol. 2009, 10, 550–563. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Reverter, D. Molecular Mechanisms of DUBs Regulation in Signaling and Disease. Int. J. Mol. Sci. 2021, 22, 986. [Google Scholar] [CrossRef] [PubMed]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cucci, M.A.; Grattarola, M.; Monge, C.; Roetto, A.; Barrera, G.; Caputo, E.; Dianzani, C.; Pizzimenti, S. Nrf2 as a Therapeutic Target in the Resistance to Targeted Therapies in Melanoma. Antioxidants 2023, 12, 1313. https://doi.org/10.3390/antiox12061313
Cucci MA, Grattarola M, Monge C, Roetto A, Barrera G, Caputo E, Dianzani C, Pizzimenti S. Nrf2 as a Therapeutic Target in the Resistance to Targeted Therapies in Melanoma. Antioxidants. 2023; 12(6):1313. https://doi.org/10.3390/antiox12061313
Chicago/Turabian StyleCucci, Marie Angèle, Margherita Grattarola, Chiara Monge, Antonella Roetto, Giuseppina Barrera, Emilia Caputo, Chiara Dianzani, and Stefania Pizzimenti. 2023. "Nrf2 as a Therapeutic Target in the Resistance to Targeted Therapies in Melanoma" Antioxidants 12, no. 6: 1313. https://doi.org/10.3390/antiox12061313
APA StyleCucci, M. A., Grattarola, M., Monge, C., Roetto, A., Barrera, G., Caputo, E., Dianzani, C., & Pizzimenti, S. (2023). Nrf2 as a Therapeutic Target in the Resistance to Targeted Therapies in Melanoma. Antioxidants, 12(6), 1313. https://doi.org/10.3390/antiox12061313