
Lactobacillus sp. Facilitate the Repair of DNA Damage Caused by Bile-Induced Reactive Oxygen Species in Experimental Models of Gastroesophageal Reflux Disease




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Bacterial Cultures
2.3. Adaptation Assay
2.4. Bacterial Growth Curves
2.5. Biofilm
2.6. Hydrophobicity
2.7. Esophageal Epithelial Cell Treatment with Bile and/or Lactobacillus
2.8. COMET Assay
2.9. Immunofluorescence
2.10. Statistical Analysis
3. Results
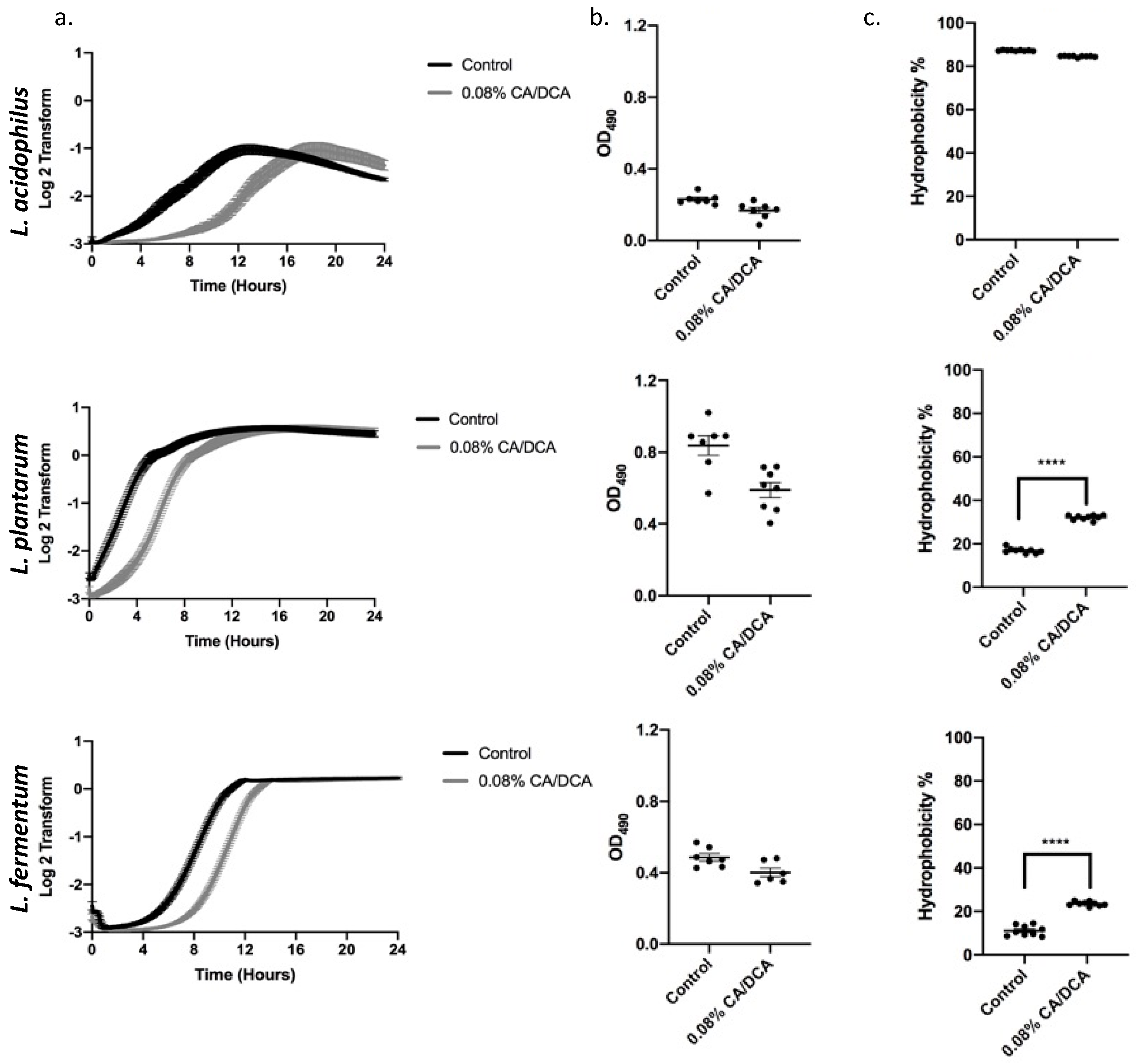
3.1. Lactobacilli Adapt to Cholate/Deoxycholate Bile Salt Challenge
3.2. Bile Exposure Causes a Delay in Exponential Growth without the Induction of a Stress Response
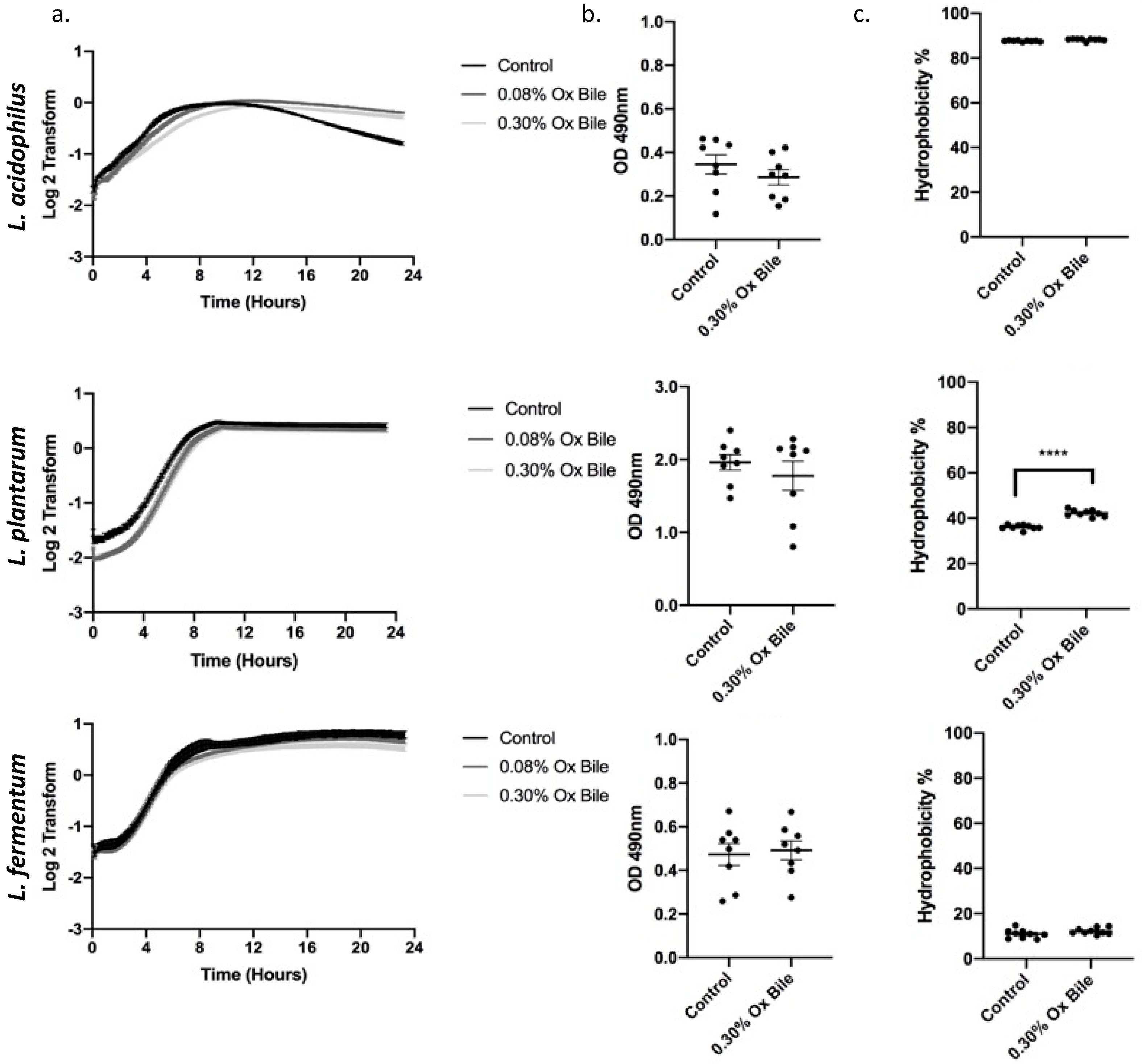
3.3. Ox Bile as a Physiological Model for Refluxate Shows a Dose Dependent Effect in Lactobacilli Growth with Complete Recovery and No Change in Biofilm Formation
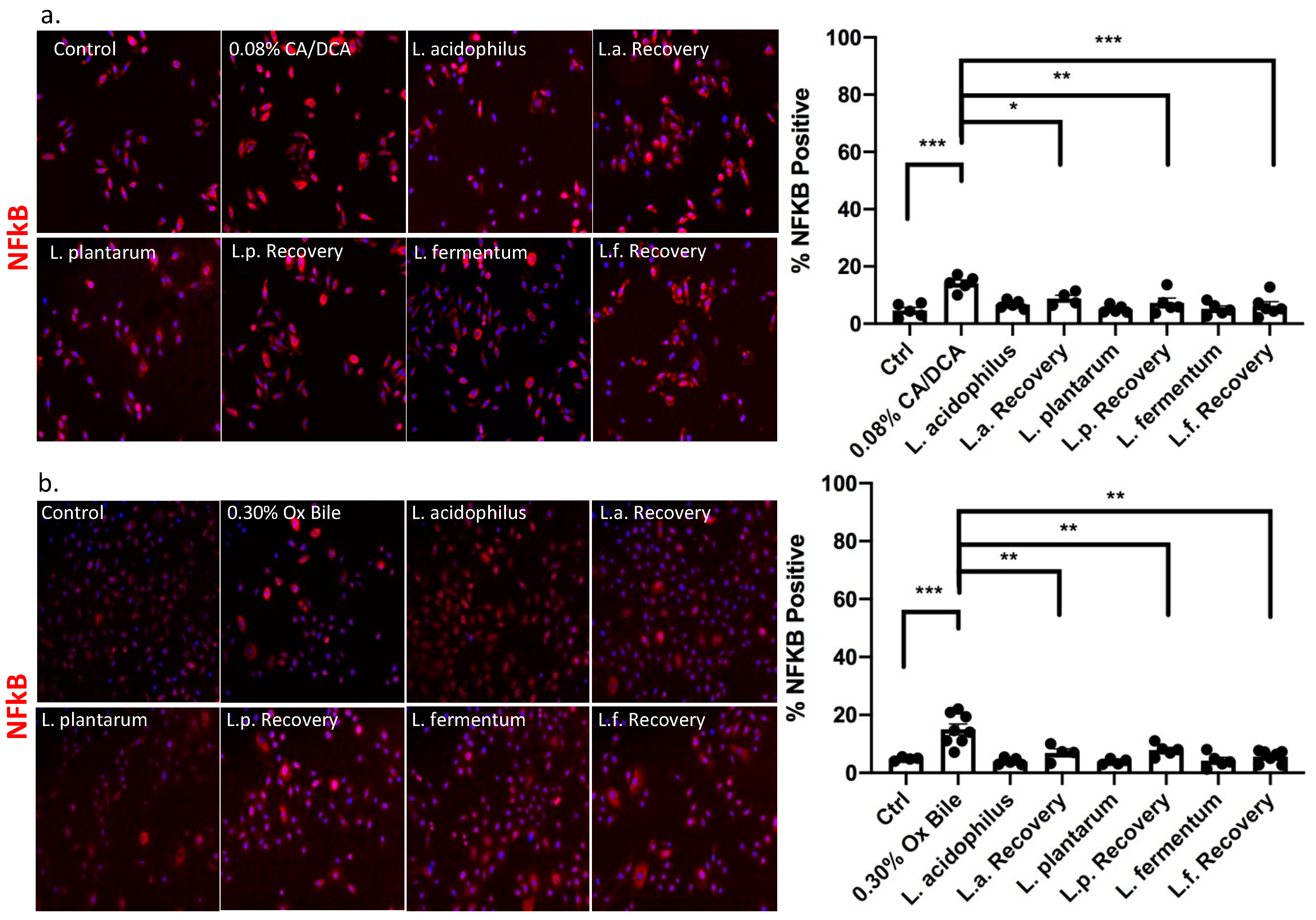
3.4. Lactobacilli Show Anti-Inflammatory Capabilities in the Context of Bile-Induced NFκB Signaling
3.5. Lactobacilli Show an Antioxidant Effect
3.6. Addition of Lactobacilli to Esophageal Cells after Exposure to Bile Reduces DNA Damage
3.7. DNA Double Strand Break Repair Is Accelerated upon Addition of Lactobacilli
4. Discussion
4.1. Antioxidative Function of Lactobacilli during ROS-Induced Inflammation and DNA Damage
4.2. DNA Damage Repair
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, D.; Liu, S.; Li, Z.; Wang, R. Global, regional and national burden of gastroesophageal reflux disease, 1990–2019: Update from the GBD 2019 study. Ann. Med. 2022, 54, 1372–1384. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Liu, X.; Cao, S.; Dong, X.; Rao, S.; Cai, K. Understanding Esophageal Cancer: The Challenges and Opportunities for the Next Decade. Front. Oncol. 2020, 10, 1727. [Google Scholar] [CrossRef] [PubMed]
- Miwa, K.; Sahara, H.; Segawa, M.; Kinami, S.; Sato, T.; Miyazaki, I.; Hattori, T. Reflux of duodenal or gastro-duodenal contents induces esophageal carcinoma in rats. Int. J. Cancer 1996, 67, 269–274. [Google Scholar] [CrossRef]
- Fein, M.; Fuchs, K.H.; Stopper, H.; Diem, S.; Herderich, M. Duodenogastric reflux and foregut carcinogenesis: Analysis of duodenal juice in a rodent model of cancer. Carcinogenesis 2000, 21, 2079–2084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dvorak, K.; Payne, C.M.; Chavarria, M.; Ramsey, L.; Dvorakova, B.; Bernstein, H.; Holubec, H.; Sampliner, R.E.; Guy, N.; Condon, A.; et al. Bile acids in combination with low pH induce oxidative stress and oxidative DNA damage: Relevance to the pathogenesis of Barrett’s oesophagus. Gut 2007, 56, 763–771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernstein, H.; Bernstein, C.; Payne, C.M.; Dvorakova, K.; Garewal, H. Bile acids as carcinogens in human gastrointestinal cancers. Mutat. Res. 2005, 589, 47–65. [Google Scholar] [CrossRef]
- Zhong, L.; Zhang, X.; Covasa, M. Emerging roles of lactic acid bacteria in protection against colorectal cancer. World J. Gastroenterol. 2014, 20, 7878–7886. [Google Scholar] [CrossRef]
- Liu, C.T.; Chu, F.J.; Chou, C.C.; Yu, R.C. Antiproliferative and anticytotoxic effects of cell fractions and exopolysaccharides from Lactobacillus casei 01. Mutat. Res. 2011, 721, 157–162. [Google Scholar] [CrossRef]
- Even, S.; Lindley, N.D.; Loubière, P.; Cocaign-Bousquet, M. Dynamic response of catabolic pathways to autoacidification in Lactococcus lactis: Transcript profiling and stability in relation to metabolic and energetic constraints. Mol. Microbiol. 2002, 45, 1143–1152. [Google Scholar] [CrossRef]
- Reyes-Nava, L.A.; Garduño-Siciliano, L.; Santos, E.L.P.; Hernández-Sánchez, H.; A Arauz, J.; Muriel, P.; Rivera-Espinoza, Y. Use of bile acids as a selection strategy for lactobacillus strains with probiotic potential. J Food Nutr. Disor. 2016, 5, 1000187. [Google Scholar] [CrossRef]
- D’Souza, S.M.; Houston, K.; Keenan, L.; Yoo, B.S.; Parekh, P.J.; Johnson, D.A. Role of microbial dysbiosis in the pathogenesis of esophageal mucosal disease: A paradigm shift from acid to bacteria? World J. Gastroenterol. 2021, 27, 2054–2072. [Google Scholar] [CrossRef]
- Walter, J. Ecological role of lactobacilli in the gastrointestinal tract: Implications for fundamental and biomedical research. Appl. Environ. Microbiol. 2008, 74, 4985–4996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elliott, D.R.F.; Walker, A.W.; O’Donovan, M.; Parkhill, J.; Fitzgerald, R.C. A non-endoscopic device to sample the oesophageal microbiota: A case-control study. Lancet Gastroenterol. Hepatol. 2017, 2, 32–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Dhabi, N.A.; Valan Arasu, M.; Vijayaraghavan, P.; Esmail, G.A.; Duraipandiyan, V.; Kim, Y.O.; Kim, H.; Kim, H.J. Probiotic and Antioxidant Potential of Lactobacillus reuteriLR12 and Lactobacillus lactisLL10 Isolated from Pineapple Puree and Quality Analysis of Pineapple-Flavored Goat Milk Yoghurt during Storage. Microorganisms 2020, 8, 1461. [Google Scholar] [CrossRef]
- Harada, H.; Nakagawa, H.; Oyama, K.; Takaoka, M.; Andl, C.D.; Jacobmeier, B.; von Werder, A.; Enders, G.H.; Opitz, O.G.; Rustgi, A.K. Telomerase induces immortalization of human esophageal keratinocytes without p16INK4a inactivation. Mol. Cancer Res. MCR 2003, 1, 729–738. [Google Scholar]
- Begley, M.; Gahan, C.G.; Hill, C. Bile stress response in Listeria monocytogenes LO28: Adaptation, cross-protection, and identification of genetic loci involved in bile resistance. Appl. Environ. Microbiol. 2002, 68, 6005–6012. [Google Scholar] [CrossRef] [Green Version]
- Serebriakova, E.V.; Darmov, I.V.; Medvedev, N.P.; Alekseev, S.A.; Rybak, S.I. Otsenka gidrofobnykh svoĭstv bakterial’nykh kletok po adsorbtsii na poverkhnosti kapel’ khloroforma [Evaluation of the hydrophobicity of bacterial cells by measuring their adherence to chloroform drops]. Mikrobiologiia 2002, 71, 237–239. [Google Scholar]
- Merritt, M.E.; Donaldson, J.R. Effect of bile salts on the DNA and membrane integrity of enteric bacteria. J. Med. Microbiol. 2009, 58 Pt 12, 1533–1541. [Google Scholar] [CrossRef] [Green Version]
- Bridson, E.Y. (Ed.) The Oxoid Manual; Unipath Ltd.: Basingstoke, UK, 1995. [Google Scholar]
- Rode, D.K.H.; Singh, P.K.; Drescher, K. Multicellular and unicellular responses of microbial biofilms to stress. Biol. Chem. 2020, 401, 1365–1374. [Google Scholar] [CrossRef] [PubMed]
- Heilmann, C. Adhesion mechanisms of staphylococci. Adv. Exp. Med. Biol. 2011, 715, 105–123. [Google Scholar] [CrossRef]
- Krasowska, A.; Sigler, K. How microorganisms use hydrophobicity and what does this mean for human needs? Front. Cell. Infect. Microbiol. 2014, 4, 112. [Google Scholar] [CrossRef] [Green Version]
- McQuaid, K.R.; Laine, L.; Fennerty, M.B.; Souza, R.; Spechler, S.J. Systematic review: The role of bile acids in the pathogenesis of gastro-oesophageal reflux disease and related neoplasia. Aliment. Pharmacol. Ther. 2011, 34, 146–165. [Google Scholar] [CrossRef] [PubMed]
- Zhao, A.; Wang, S.; Chen, W.; Zheng, X.; Huang, F.; Han, X.; Ge, K.; Rajani, C.; Huang, Y.; Yu, H.; et al. Increased levels of conjugated bile acids are associated with human bile reflux gastritis. Sci. Rep. 2020, 10, 11601. [Google Scholar] [CrossRef]
- Hu, P.L.; Yuan, Y.H.; Yue, T.L.; Guo, C.F. Bile acid patterns in commercially available oxgall powders used for the evaluation of the bile tolerance ability of potential probiotics. PLoS ONE 2018, 13, e0192964. [Google Scholar] [CrossRef] [Green Version]
- Gunn, J.S. Mechanism of bacterial resistance and response to bile. Microbes Infect 2000, 2, 907–913. [Google Scholar] [CrossRef]
- Colleypriest, B.J.; Ward, S.G.; Tosh, D. How does inflammation cause Barrett’s metaplasia? Curr. Opin. Pharmacol. 2009, 9, 721–726. [Google Scholar] [CrossRef] [PubMed]
- Souza, R.F. From Reflux Esophagitis to Esophageal Adenocarcinoma. Dig. Dis. 2016, 34, 483–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, Y.; Chen, X.; Bajpai, M.; Verma, A.; Das, K.M.; Souza, R.F.; Garman, K.S.; Donohoe, C.L.; O’Farrell, N.J.; Reynolds, J.V.; et al. Cellular origins and molecular mechanisms of Barrett’s esophagus and esophageal adenocarcinoma. Ann. N. Y. Acad. Sci. 2013, 1300, 187–199. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, K.E.; Phelan, J.J.; O’Hanlon, C.; Lysaght, J.; O’Sullivan, J.N.; Reynolds, J.V. The role of inflammation in cancer of the esophagus. Expert Rev. Gastroenterol. Hepatol. 2014, 8, 749–760. [Google Scholar] [CrossRef] [PubMed]
- O’Riordan, J.M.; Abdel-latif, M.M.; Ravi, N.; McNamara, D.; Byrne, P.J.; McDonald, G.S.; Keeling, P.W.; Kelleher, D.; Reynolds, J.V. Proinflammatory cytokine and nuclear factor kappa-B expression along the inflammation-metaplasia-dysplasia-adenocarcinoma sequence in the esophagus. Am. J. Gastroenterol. 2005, 100, 1257–1264. [Google Scholar] [CrossRef]
- Huo, X.; Juergens, S.; Zhang, X.; Rezaei, D.; Yu, C.; Strauch, E.D.; Wang, J.Y.; Cheng, E.; Meyer, F.; Wang, D.H.; et al. Deoxycholic acid causes DNA damage while inducing apoptotic resistance through NF-κB activation in benign Barrett’s epithelial cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 301, G278–G286. [Google Scholar] [CrossRef]
- Peng, S.; Huo, X.; Rezaei, D.; Zhang, Q.; Zhang, X.; Yu, C.; Asanuma, K.; Cheng, E.; Pham, T.H.; Wang, D.H.; et al. In Barrett’s esophagus patients and Barrett’s cell lines, ursodeoxycholic acid increases antioxidant expression and prevents DNA damage by bile acids. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 307, G129–G139. [Google Scholar] [CrossRef] [PubMed]
- Hahm, J.Y.; Park, J.; Jang, E.S.; Chi, S.W. 8-Oxoguanine: From oxidative damage to epigenetic and epitranscriptional modification. Exp. Mol. Med. 2022, 54, 1626–1642. [Google Scholar] [CrossRef]
- Ock, C.Y.; Kim, E.H.; Choi, D.J.; Lee, H.J.; Hahm, K.B.; Chung, M.H. 8-Hydroxydeoxyguanosine: Not mere biomarker for oxidative stress, but remedy for oxidative stress-implicated gastrointestinal diseases. World J. Gastroenterol. 2012, 18, 302–308. [Google Scholar] [CrossRef]
- Peng, D.; Zaika, A.; Que, J.; El-Rifai, W. The antioxidant response in Barrett’s tumorigenesis: A double-edged sword. Redox Biol. 2021, 41, 101894. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, V.; Horvat, A.; Korolkova, O.; Washington, M.K.; El-Rifai, W.; Dikalov, S.I.; Zaika, A.I. Prevention of DNA damage in Barrett’s esophageal cells exposed to acidic bile salts. Carcinogenesis 2016, 37, 1161–1169. [Google Scholar] [CrossRef] [Green Version]
- Olive, P.L.; Banáth, J.P. The COMET assay: A method to measure DNA damage in individual cells. Nat. Protoc. 2006, 1, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Boiteux, S.; Coste, F.; Castaing, B. Repair of 8-oxo-7,8-dihydroguanine in prokaryotic and eukaryotic cells: Properties and biological roles of the Fpg and OGG1 DNA N-glycosylases. Free. Radic. Biol. Med. 2017, 107, 179–201. [Google Scholar] [CrossRef]
- Jiang, B.; Zhao, S.; Tao, Z.; Wen, J.; Yang, Y.; Zheng, Y.; Yan, H.; Sheng, Y.; Gao, A. Controlled bile acid exposure to oesophageal mucosa causes up-regulation of nuclear γ-H2AX possibly via iNOS induction. Biosci. Rep. 2016, 36, e00357. [Google Scholar] [CrossRef] [Green Version]
- Shen, W.; Ma, Y.; Qi, H.; Wang, W.; He, J.; Xiao, F.; Zhu, H.; He, S. Kinetics model of DNA double-strand break repair in eukaryotes. DNA Repair 2021, 100, 103035. [Google Scholar] [CrossRef]
- Kang, M.A.; So, E.Y.; Simons, A.L.; Spitz, D.R.; Ouchi, T. DNA damage induces reactive oxygen species generation through the H2AX-Nox1/Rac1 pathway. Cell Death Dis. 2012, 3, e249. [Google Scholar] [CrossRef] [Green Version]
- Huo, X.; Dunbar, K.B.; Zhang, X.; Zhang, Q.; Spechler, S.J.; Souza, R.F. In Barrett’s epithelial cells, weakly acidic bile salt solutions cause oxidative DNA damage with response and repair mediated by p38. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 318, G464–G478. [Google Scholar] [CrossRef]
- Spechler, S.J. Does Barrett’s esophagus regress after surgery (or proton pump inhibitors)? Dig. Dis. 2014, 32, 156–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alsalahi, O.; Dobrian, A.D. Proton Pump Inhibitors: The Culprit for Barrett’s Esophagus? Front. Oncol. 2015, 4, 373. [Google Scholar] [CrossRef]
- Sifrim, D. Management of bile reflux. Gastroenterol. Hepatol. 2013, 9, 179–180. [Google Scholar]
- Sarela, A.I.; Hick, D.G.; Verbeke, C.S.; Casey, J.F.; Guillou, P.J.; Clark, G.W. Persistent acid and bile reflux in asymptomatic patients with Barrett esophagus receiving proton pump inhibitor therapy. Arch. Surg. 2004, 139, 547–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofmann, A.F.; Mysels, K.J. Bile acid solubility and precipitation in vitro and in vivo: The role of conjugation, pH, and Ca2+ ions. J. Lipid Res. 1992, 33, 617–626. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Oh, T.Y.; Ahn, B.O.; Cho, H.; Kim, W.B.; Kim, Y.B.; Surh, Y.J.; Kim, H.J.; Hahm, K.B. Involvement of oxidative stress in experimentally induced reflux esophagitis and Barrett’s esophagus: Clue for the chemoprevention of esophageal carcinoma by antioxidants. Mutat. Res. 2001, 480–481, 189–200. [Google Scholar] [CrossRef]
- Peng, D.; Belkhiri, A.; Hu, T.; Chaturvedi, R.; Asim, M.; Wilson, K.T.; Zaika, A.; El-Rifai, W. Glutathione peroxidase 7 protects against oxidative DNA damage in oesophageal cells. Gut 2012, 61, 1250–1260. [Google Scholar] [CrossRef] [Green Version]
- Nehra, D.; Howell, P.; Williams, C.P.; Pye, J.K.; Beynon, J. Toxic bile acids in gastro-oesophageal reflux disease: Influence of gastric acidity. Gut 1999, 44, 598–602. [Google Scholar] [CrossRef]
- Thanan, R.; Ma, N.; Hiraku, Y.; Iijima, K.; Koike, T.; Shimosegawa, T.; Murata, M.; Kawanishi, S. DNA Damage in CD133-Positive Cells in Barrett’s Esophagus and Esophageal Adenocarcinoma. Mediat. Inflamm. 2016, 7937814. [Google Scholar] [CrossRef] [Green Version]
- Thanan, R.; Ma, N.; Iijima, K.; Abe, Y.; Koike, T.; Shimosegawa, T.; Pinlaor, S.; Hiraku, Y.; Oikawa, S.; Murata, M.; et al. Proton pump inhibitors suppress iNOS-dependent DNA damage in Barrett’s esophagus by increasing Mn-SOD expression. Biochem. Biophys. Res. Commun. 2012, 421, 280–285. [Google Scholar] [CrossRef] [PubMed]
- Kameoka, M.; Ota, K.; Tetsuka, T.; Tanaka, Y.; Itaya, A.; Okamoto, T.; Yoshihara, K. Evidence for regulation of NF-kappaB by poly(ADP-ribose) polymerase. Biochem. J. 2000, 346 Pt 3, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Ba, X.; Bacsi, A.; Luo, J.; Aguilera-Aguirre, L.; Zeng, X.; Radak, Z.; Brasier, A.R.; Boldogh, I. 8-oxoguanine DNA glycosylase-1 augments proinflammatory gene expression by facilitating the recruitment of site-specific transcription factors. J. Immunol. 2014, 192, 2384–2394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguilera-Aguirre, L.; Bacsi, A.; Radak, Z.; Hazra, T.K.; Mitra, S.; Sur, S.; Brasier, A.R.; Ba, X.; Boldogh, I. Innate inflammation induced by the 8-oxoguanine DNA glycosylase-1-KRAS-NF-κB pathway. J. Immunol. 2014, 193, 4643–4653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Visnes, T.; Cázares-Körner, A.; Hao, W.; Wallner, O.; Masuyer, G.; Loseva, O.; Mortusewicz, O.; Wiita, E.; Sarno, A.; Manoilov, A.; et al. Small-molecule inhibitor of OGG1 suppresses proinflammatory gene expression and inflammation. Science 2018, 362, 834–839. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Latif, M.M.; Duggan, S.; Reynolds, J.V.; Kelleher, D. Inflammation and esophageal carcinogenesis. Curr. Opin. Pharmacol. 2009, 9, 396–404. [Google Scholar] [CrossRef]
- Souza, R.F.; Huo, X.; Mittal, V.; Schuler, C.M.; Carmack, S.W.; Zhang, H.Y.; Zhang, X.; Yu, C.; Hormi-Carver, K.; Genta, R.M.; et al. Gastroesophageal reflux might cause esophagitis through a cytokine-mediated mechanism rather than caustic acid injury. Gastroenterology 2009, 137, 1776–1784. [Google Scholar] [CrossRef]
- Fitzgerald, R.C.; Abdalla, S.; Onwuegbusi, B.A.; Sirieix, P.; Saeed, I.T.; Burnham, W.R.; Farthing, M.J. Inflammatory gradient in Barrett’s oesophagus: Implications for disease complications. Gut 2002, 51, 316–322. [Google Scholar] [CrossRef] [Green Version]
- Greten, F.R.; Eckmann, L.; Greten, T.F.; Park, J.M.; Li, Z.W.; Egan, L.J.; Kagnoff, M.F.; Karin, M. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell 2004, 118, 285–296. [Google Scholar] [CrossRef] [Green Version]
- Glinghammar, B.; Inoue, H.; Rafter, J.J. Deoxycholic acid causes DNA damage in colonic cells with subsequent induction of caspases, COX-2 promoter activity and the transcription factors NF-kB and AP-1. Carcinogenesis 2002, 23, 839–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef] [PubMed]
- Rains, J.L.; Jain, S.K. Oxidative stress, insulin signaling, and diabetes. Free. Radic. Biol. Med. 2011, 50, 567–575. [Google Scholar] [CrossRef] [Green Version]
- Zerrouki, M.; Benkaci-Ali, F. DFT study of the mechanisms of nonenzymatic DNA repair by phytophenolic antioxidants. J. Mol. Model. 2018, 24, 78. [Google Scholar] [CrossRef] [PubMed]
- Peng, D.F.; Hu, T.L.; Soutto, M.; Belkhiri, A.; El-Rifai, W. Loss of glutathione peroxidase 7 promotes TNF-α-induced NF-κB activation in Barrett’s carcinogenesis. Carcinogenesis 2014, 35, 1620–1628. [Google Scholar] [CrossRef]
- Peng, D.F.; Hu, T.L.; Soutto, M.; Belkhiri, A.; El-Rifai, W. Glutathione Peroxidase 7 Suppresses Bile Salt-Induced Expression of Pro-Inflammatory Cytokines in Barrett’s Carcinogenesis. J. Cancer 2014, 5, 510–517. [Google Scholar] [CrossRef] [Green Version]
- Hong, J.; Chen, Z.; Peng, D.; Zaika, A.; Revetta, F.; Washington, M.K.; Belkhiri, A.; El-Rifai, W. APE1-mediated DNA damage repair provides survival advantage for esophageal adenocarcinoma cells in response to acidic bile salts. Oncotarget 2016, 7, 16688–16702. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Lu, H.; Peng, D.; Cao, L.L.; Ballout, F.; Srirmajayam, K.; Chen, Z.; Bhat, A.; Wang, T.C.; Capobianco, A.; et al. Activation of NOTCH signaling via DLL1 is mediated by APE1-redox-dependent NF-κB activation in oesophageal adenocarcinoma. Gut 2023, 72, 421–432. [Google Scholar] [CrossRef]
- Wojcik, M.; Burzynska-Pedziwiatr, I.; Wozniak, L.A. A review of natural and synthetic antioxidants important for health and longevity. Curr. Med. Chem. 2010, 17, 3262–3288. [Google Scholar] [CrossRef]
- Arasu, M.V.; Al-Dhabi, N.A. In vitro antifungal, probiotic, and antioxidant functional properties of a novel Lactobacillus paraplantarum isolated from fermented dates in Saudi Arabia. J. Sci. Food Agric. 2017, 97, 5287–5295. [Google Scholar] [CrossRef]
- Mishra, V.; Shah, C.; Mokashe, N.; Chavan, R.; Yadav, H.; Prajapati, J. Probiotics as potential antioxidants: A systematic review. J. Agric. Food Chem. 2015, 63, 3615–3626. [Google Scholar] [CrossRef]
- Songisepp, E.; Kals, J.; Kullisaar, T.; Mändar, R.; Hütt, P.; Zilmer, M.; Mikelsaar, M. Evaluation of the functional efficacy of an antioxidative probiotic in healthy volunteers. Nutr. J. 2005, 4, 22. [Google Scholar] [CrossRef] [Green Version]
- Kairane, C.; Mahlapuu, R.; Ehrlich, K.; Kilk, K.; Zilmer, M.; Soomets, U. Diverse Effects of Glutathione and UPF Peptides on Antioxidant Defense System in Human Erythroleukemia Cells K562. Int. J. Pept. 2012, 124163. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.; Gui, W.; Koo, I.; Smith, P.B.; Allman, E.L.; Nichols, R.G.; Rimal, B.; Cai, J.; Liu, Q.; Patterson, A.D. The microbiome modulating activity of bile acids. Gut Microbes 2020, 11, 979–996. [Google Scholar] [CrossRef] [PubMed]
- Burns, P.; Sanchez, B.; Vinderola, G.; Ruas-Madiedo, P.; Ruiz, L.; Margolles, A.; Reinheimer, J.; de los Reyes-Gavilán, C.G. Inside the adaptation process of Lactobacillus delbrueckii subsp. lactis to bile. Int. J. Food Microbiol. 2010, 142, 132–141. [Google Scholar] [CrossRef] [Green Version]
- Kay, J.; Thadhani, E.; Samson, L.; Engelward, B. Inflammation-induced DNA damage, mutation and cancer. DNA Repair 2019, 83, 102673. [Google Scholar] [CrossRef] [PubMed]
- Scully, R.; Panday, A.; Elango, R.; Willis, N.A. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell Biol. 2019, 20, 698–714. [Google Scholar] [CrossRef] [PubMed]
- Shibata, A. Regulation of repair pathway choice at two-ended DNA double-strand breaks. Mutat. Res. 2017, 803–805, 51–55. [Google Scholar] [CrossRef]
- Huertas, P. DNA resection in eukaryotes: Deciding how to fix the break. Nat. Struct. Mol. Biol. 2010, 17, 11–16. [Google Scholar] [CrossRef] [Green Version]
- Johnson, R.D.; Jasin, M. Sister chromatid gene conversion is a prominent double-strand break repair pathway in mammalian cells. EMBO J. 2000, 19, 3398–3407. [Google Scholar] [CrossRef]
- Bonner, W.M.; Redon, C.E.; Dickey, J.S.; Nakamura, A.J.; Sedelnikova, O.A.; Solier, S.; Pommier, Y. GammaH2AX and cancer. Nat. Rev. Cancer 2008, 8, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Rogakou, E.P.; Pilch, D.R.; Orr, A.H.; Ivanova, V.S.; Bonner, W.M. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998, 273, 5858–5868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scully, R.; Xie, A. Double strand break repair functions of histone H2AX. Mutat. Res. 2013, 750, 5–14. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.R.; Quinet, A.; Byrum, A.K.; Jackson, J.; Berti, M.; Thangavel, S.; Bredemeyer, A.L.; Hindi, I.; Mosammaparast, N.; Tyler, J.K.; et al. XLF and H2AX function in series to promote replication fork stability. J. Cell Biol. 2019, 218, 2113–2123. [Google Scholar] [CrossRef] [Green Version]
- Paull, T.T.; Rogakou, E.P.; Yamazaki, V.; Kirchgessner, C.U.; Gellert, M.; Bonner, W.M. A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr. Biol. CB 2000, 10, 886–895. [Google Scholar] [CrossRef] [Green Version]
- Oizumi, T.; Ohno, R.; Yamabe, S.; Funayama, T.; Nakamura, A.J. Repair Kinetics of DNA Double Strand Breaks Induced by Simulated Space Radiation. Life 2020, 10, 341. [Google Scholar] [CrossRef]
- Krishna, S.; Wagener, B.M.; Liu, H.P.; Lo, Y.C.; Sterk, R.; Petrini, J.H.; Nickoloff, J.A. Mre11 and Ku regulation of double-strand break repair by gene conversion and break-induced replication. DNA Repair 2007, 6, 797–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhardwaj, V.; Gokulan, R.C.; Horvat, A.; Yermalitskaya, L.; Korolkova, O.; Washington, K.M.; El-Rifai, W.; Dikalov, S.I.; Zaika, A.I. Activation of NADPH oxidases leads to DNA damage in esophageal cells. Sci. Rep. 2017, 7, 9956. [Google Scholar] [CrossRef] [Green Version]
- Arthur, J.C.; Perez-Chanona, E.; Mühlbauer, M.; Tomkovich, S.; Uronis, J.M.; Fan, T.J.; Campbell, B.J.; Abujamel, T.; Dogan, B.; Rogers, A.B.; et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science 2012, 338, 120–123. [Google Scholar] [CrossRef] [Green Version]
- Cuevas-Ramos, G.; Petit, C.R.; Marcq, I.; Boury, M.; Oswald, E.; Nougayrède, J.P. Escherichia coli induces DNA damage in vivo and triggers genomic instability in mammalian cells. Proc. Natl. Acad. Sci. USA 2010, 107, 11537–11542. [Google Scholar] [CrossRef] [Green Version]
- Lundin, C.; Schultz, N.; Arnaudeau, C.; Mohindra, A.; Hansen, L.T.; Helleday, T. RAD51 is involved in repair of damage associated with DNA replication in mammalian cells. J. Mol. Biol. 2003, 328, 521–535. [Google Scholar] [CrossRef] [PubMed]
- Lopetuso, L.R.; Severgnini, M.; Pecere, S.; Ponziani, F.R.; Boskoski, I.; Larghi, A.; Quaranta, G.; Masucci, L.; Ianiro, G.; Camboni, T.; et al. Esophageal microbiome signature in patients with Barrett’s esophagus and esophageal adenocarcinoma. PLoS ONE 2020, 15, e0231789. [Google Scholar] [CrossRef] [PubMed]
- Gomi, A.; Yamaji, K.; Watanabe, O.; Yoshioka, M.; Miyazaki, K.; Iwama, Y.; Urita, Y. Bifidobacterium bifidum YIT 10347 fermented milk exerts beneficial effects on gastrointestinal discomfort and symptoms in healthy adults: A double-blind, randomized, placebo-controlled study. J. Dairy Sci. 2018, 101, 4830–4841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, C.B.; Odamaki, T.; Xiao, J.-Z. Insights into the reason of human-residential bifidobacterial (HR) being the natural inhibitants of the human gut and their potential health promoting benefits. FEMS Microbiol. Rev. 2020, 44, 369–385. [Google Scholar] [CrossRef] [PubMed]
- Nakae, H.; Tsuda, A.; Matsuoka, T.; Mine, T.; Koga, Y. Gastric microbiota in the functional dyspepsia patients treated with probiotic yogurt. BMJ Open Gastroenterol. 2016, 3, e000109. [Google Scholar] [CrossRef] [Green Version]
- Ohtsu, T.; Takagi, A.; Uemura, N.; Inoue, K.; Sekino, H.; Kawashima, A.; Uchida, M.; Koga, Y. The Ameliorating Effect of Lactobacillus gasseri OLL2716 on Functional Dyspepsia in Helicobacter pylori-Uninfected Individuals: A Randomized Controlled Study. Digestion 2017, 96, 92–102. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Dai, H.; Liang, W.; Zhang, L.; Deng, Z. Fermented dairy foods intake and risk of cancer. Int. J. Cancer 2019, 144, 2099–2108. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bernard, J.N.; Chinnaiyan, V.; Almeda, J.; Catala-Valentin, A.; Andl, C.D. Lactobacillus sp. Facilitate the Repair of DNA Damage Caused by Bile-Induced Reactive Oxygen Species in Experimental Models of Gastroesophageal Reflux Disease. Antioxidants 2023, 12, 1314. https://doi.org/10.3390/antiox12071314
Bernard JN, Chinnaiyan V, Almeda J, Catala-Valentin A, Andl CD. Lactobacillus sp. Facilitate the Repair of DNA Damage Caused by Bile-Induced Reactive Oxygen Species in Experimental Models of Gastroesophageal Reflux Disease. Antioxidants. 2023; 12(7):1314. https://doi.org/10.3390/antiox12071314
Chicago/Turabian StyleBernard, Joshua N., Vikram Chinnaiyan, Jasmine Almeda, Alma Catala-Valentin, and Claudia D. Andl. 2023. "Lactobacillus sp. Facilitate the Repair of DNA Damage Caused by Bile-Induced Reactive Oxygen Species in Experimental Models of Gastroesophageal Reflux Disease" Antioxidants 12, no. 7: 1314. https://doi.org/10.3390/antiox12071314
APA StyleBernard, J. N., Chinnaiyan, V., Almeda, J., Catala-Valentin, A., & Andl, C. D. (2023). Lactobacillus sp. Facilitate the Repair of DNA Damage Caused by Bile-Induced Reactive Oxygen Species in Experimental Models of Gastroesophageal Reflux Disease. Antioxidants, 12(7), 1314. https://doi.org/10.3390/antiox12071314