
Abnormal Lipoproteins Trigger Oxidative Stress-Mediated Apoptosis of Renal Cells in LCAT Deficiency

 , , , ,
, , , ,  ,
,
Abstract
:1. Introduction
2. Methods
2.1. Subjects
2.2. Biochemical Analysis
2.3. Lipoprotein Isolation for In Vitro Studies
2.4. Analysis of Sphingolipids in HDL
2.5. Cell Cultures
2.6. Evaluation of Apoptosis
2.7. Production of Reactive Oxygen Species (ROS)
2.8. Gene Expression
2.9. Statistical Analysis
3. Results
3.1. Clinical and Biochemical Features of LCAT Deficient Carriers and Controls
3.2. Lipoprotein Composition
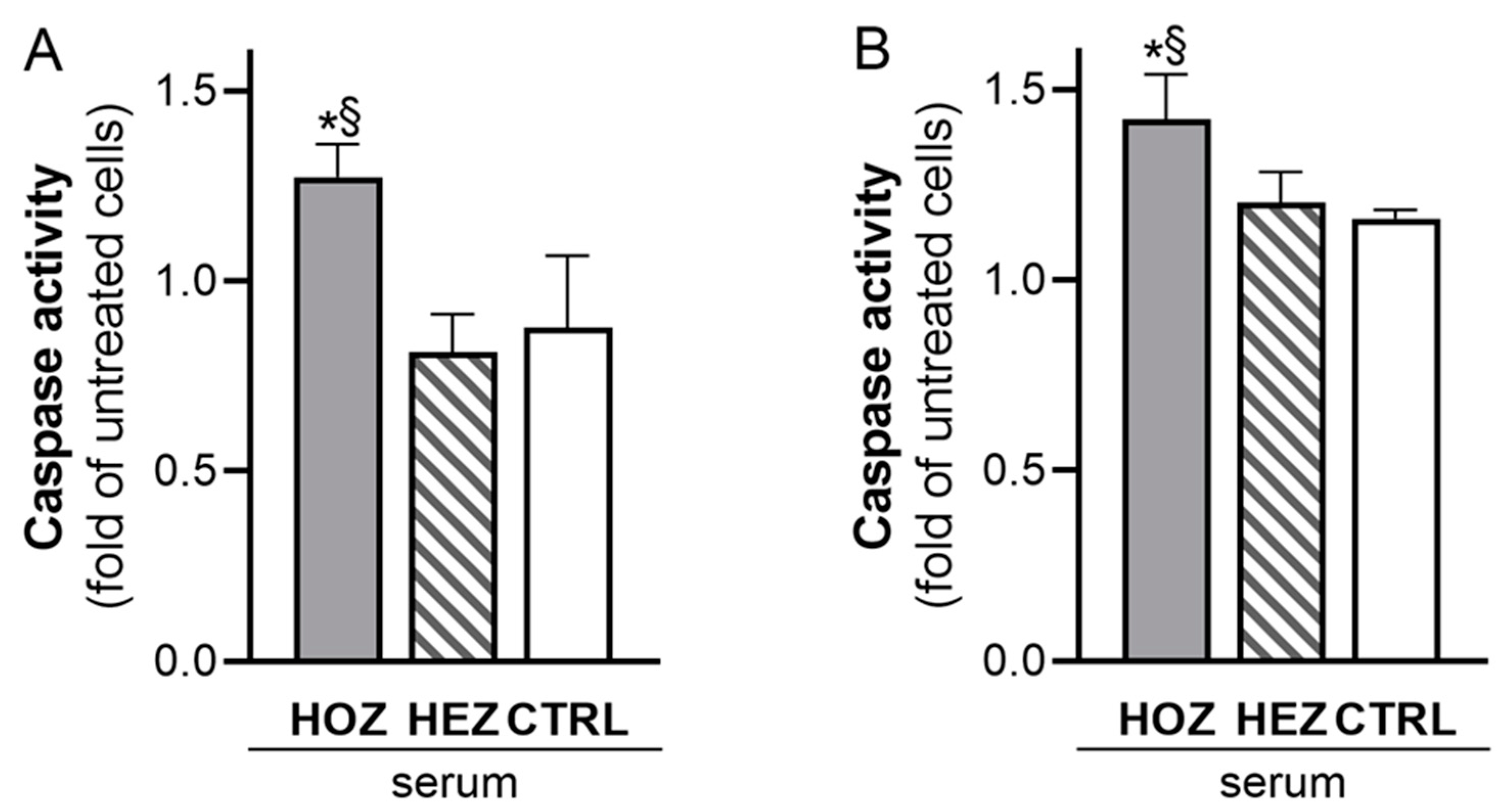
3.3. Induction of Renal Cell Apoptosis by Sera from LCAT Deficient Carriers
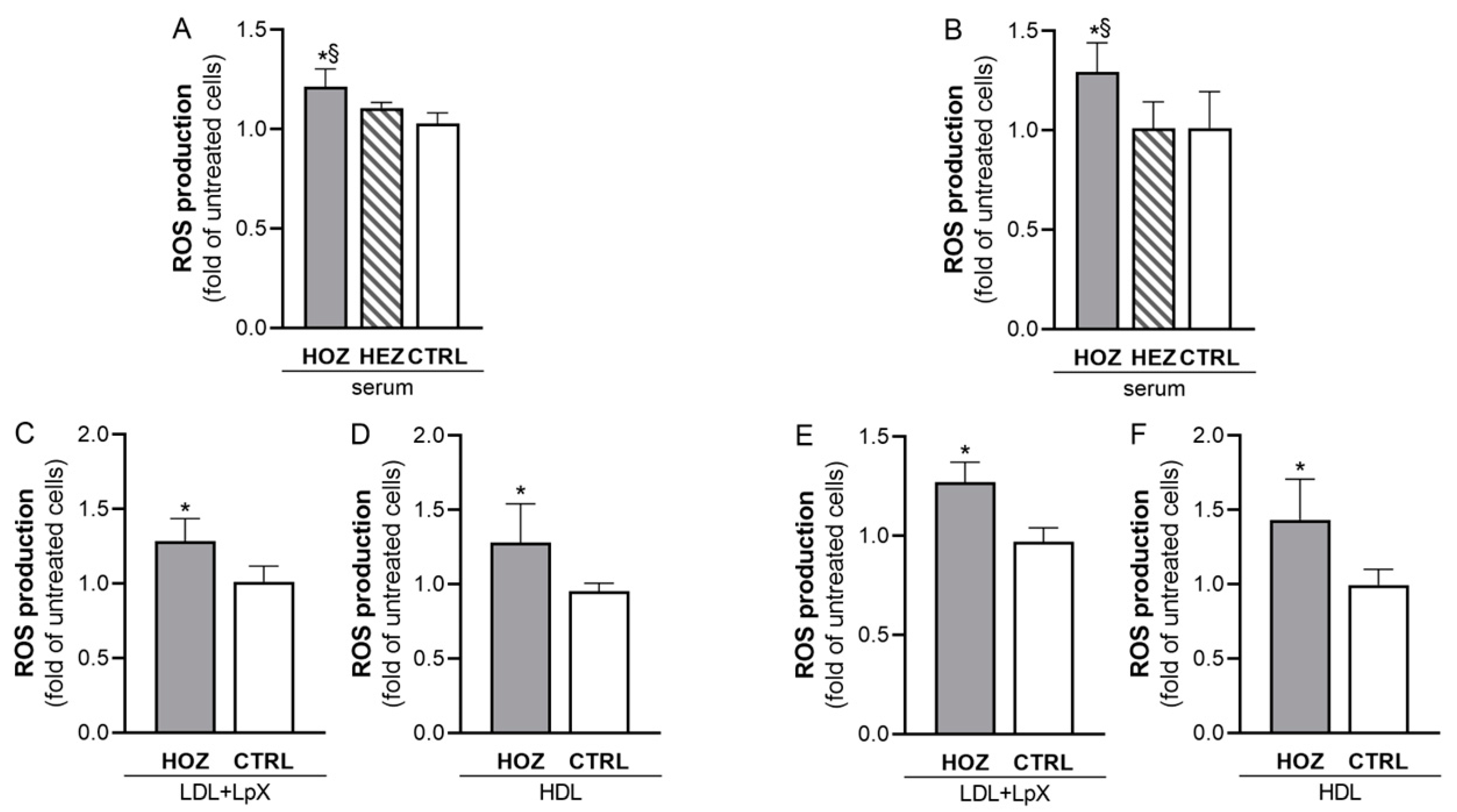
3.4. Induction of ROS by Sera and Lipoproteins from LCAT Deficient Carriers
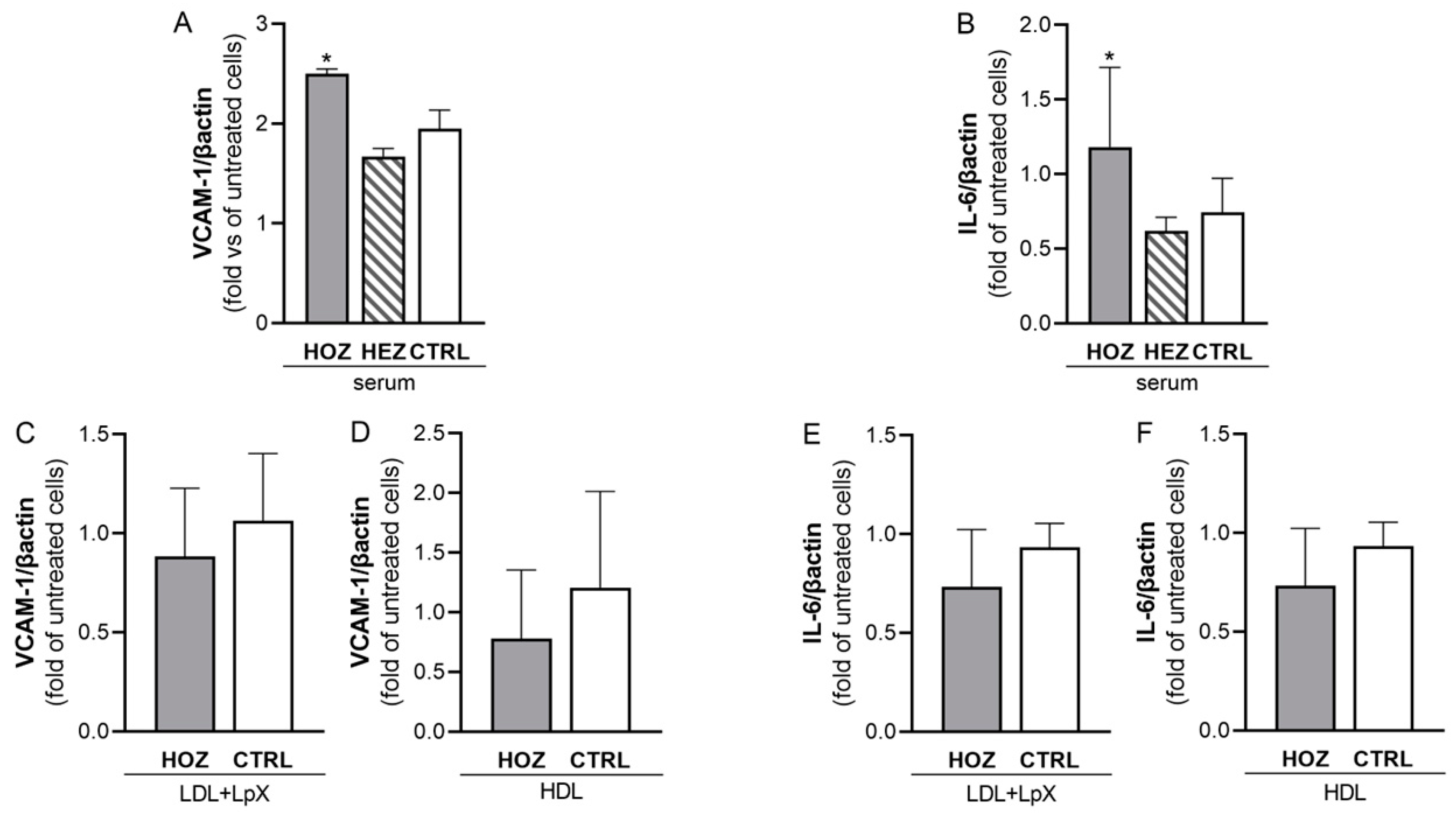
3.5. Lipoprotein-Independent Induction of Inflammation by Sera from LCAT Deficient Carriers
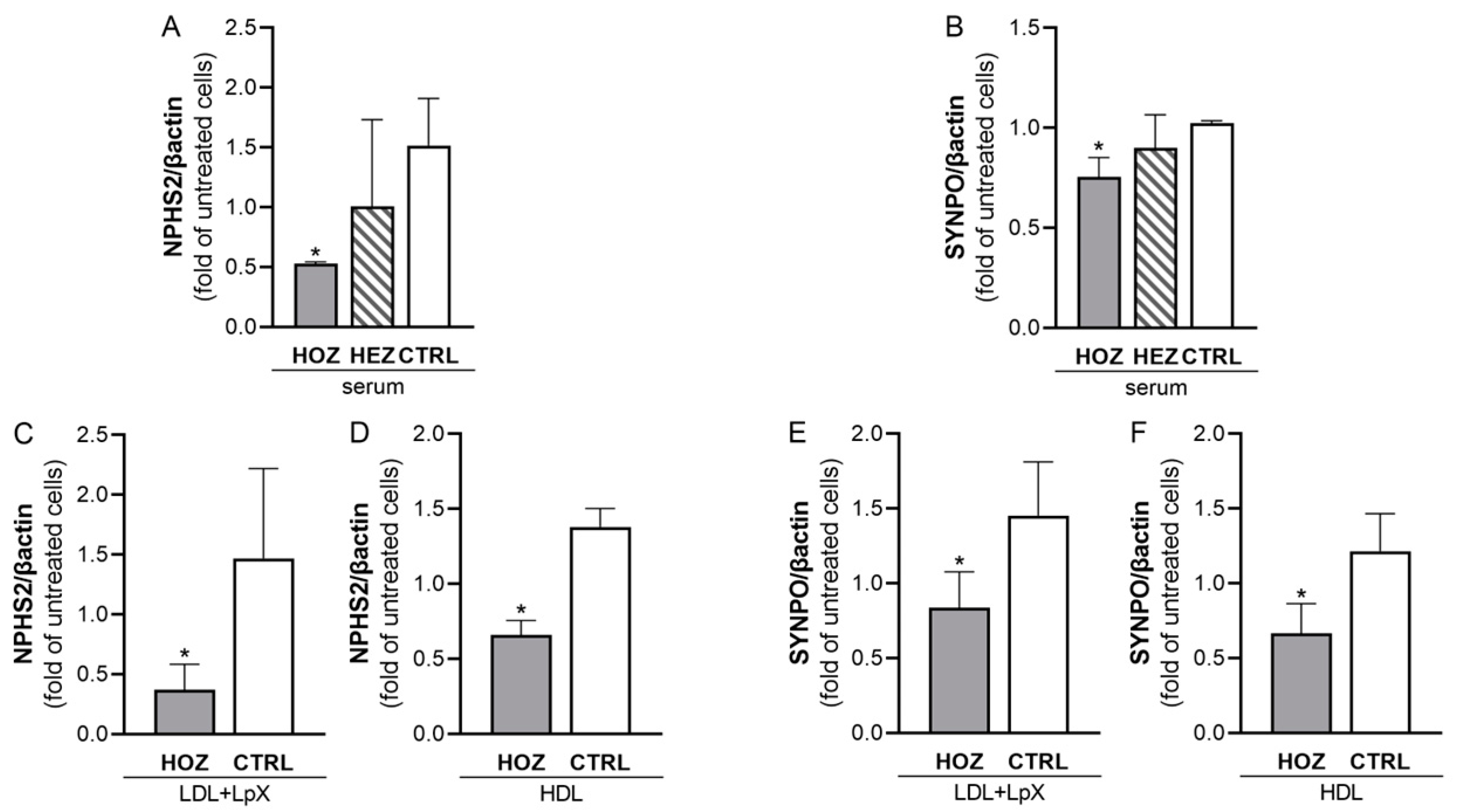
3.6. Inhibition of Podocin and Synaptopodin Expression by Sera and Lipoproteins from LCAT Deficient Carriers
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Santamarina-Fojo, S.; Hoeg, J.M.; Assmann, G.; Brewer, H.B., Jr. Lecithin cholesterol acyltransferase deficiency and fish eye disease. In The Metabolic and Molecular Bases of Inherited Diseases; Scriver, C.R., Beaudet, A.L., Sly, W.S., Valle, D., Eds.; McGraw-Hill: New York, NY, USA, 2001; pp. 2817–2833. [Google Scholar]
- Boscutti, G.; Calabresi, L.; Pizzolitto, S.; Boer, E.; Bosco, M.; Mattei, P.L.; Milutinovic, N.; Berbecar, D.; Beltram, E.; Franceschini, G. LCAT deficiency: A nephrological diagnosis. G. Ital. Nefrol. 2011, 28, 369–382. [Google Scholar]
- Narayanan, S. Biochemistry and clinical relevance of lipoprotein X. Ann. Clin. Lab. Sci. 1984, 14, 371–374. [Google Scholar] [PubMed]
- Miarka, P.; Idzior-Waluś, B.; Kuźniewski, M.; Waluś-Miarka, M.; Klupa, T.; Sułowicz, W. Corticosteroid treatment of kidney disease in a patient with familial lecithin-cholesterol acyltransferase deficiency. Clin. Exp. Nephrol. 2011, 15, 424–429. [Google Scholar] [CrossRef] [PubMed]
- Pavanello, C.; Ossoli, A.; Arca, M.; D’Erasmo, L.; Boscutti, G.; Gesualdo, L.; Lucchi, T.; Sampietro, T.; Veglia, F.; Calabresi, L. Progression of chronic kidney disease in familial LCAT deficiency: A follow-up of the Italian cohort. J. Lipid Res. 2020, 61, 1784–1788. [Google Scholar] [CrossRef]
- Stoekenbroek, R.M.; van den Bergh Weerman, M.A.; Hovingh, G.K.; Potter van Loon, B.J.; Siegert, C.E.; Holleboom, A.G. Familial LCAT deficiency: From renal replacement to enzyme replacement. Neth. J. Med. 2013, 71, 29–31. [Google Scholar] [PubMed]
- Ossoli, A.; Neufeld, E.B.; Thacker, S.G.; Vaisman, B.; Pryor, M.; Freeman, L.A.; Brantner, C.A.; Baranova, I.; Francone, N.O.; Demosky, S.J., Jr.; et al. Lipoprotein X Causes Renal Disease in LCAT Deficiency. PLoS ONE 2016, 11, e0150083. [Google Scholar] [CrossRef] [Green Version]
- Calabresi, L.; Pisciotta, L.; Costantin, A.; Frigerio, I.; Eberini, I.; Alessandrini, P.; Arca, M.; Bon, G.B.; Boscutti, G.; Busnach, G.; et al. The molecular basis of lecithin:cholesterol acyltransferase deficiency syndromes: A comprehensive study of molecular and biochemical findings in 13 unrelated Italian families. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1972–1978. [Google Scholar] [CrossRef] [Green Version]
- Simonelli, S.; Tinti, C.; Salvini, L.; Tinti, L.; Ossoli, A.; Vitali, C.; Sousa, V.; Orsini, G.; Nolli, M.L.; Franceschini, G.; et al. Recombinant human LCAT normalizes plasma lipoprotein profile in LCAT deficiency. Biologicals 2013, 41, 446–449. [Google Scholar] [CrossRef]
- Gomaraschi, M.; Ossoli, A.; Castelnuovo, S.; Simonelli, S.; Pavanello, C.; Balzarotti, G.; Arca, M.; Di Costanzo, A.; Sampietro, T.; Vaudo, G.; et al. Depletion in LpA-I:A-II particles enhances HDL-mediated endothelial protection in familial LCAT deficiency. J. Lipid Res. 2017, 58, 994–1001. [Google Scholar] [CrossRef] [Green Version]
- Hanna, E.V.; Simonelli, S.; Chamney, S.; Ossoli, A.; Mullan, R.N. Paradoxical fall in proteinuria during pregnancy in an LCAT-deficient patient-A case report. J. Clin. Lipidol. 2018, 12, 1151–1156. [Google Scholar] [CrossRef]
- Havel, R.J.; Eder, H.A.; Bragdon, J.H. The distribution and chemical composition of ultracentrifugally separated lipoproteins in human serum. J. Clin. Investig. 1955, 34, 1345–1353. [Google Scholar] [CrossRef] [Green Version]
- Arnaboldi, L.; Ossoli, A.; Giorgio, E.; Pisciotta, L.; Lucchi, T.; Grigore, L.; Pavanello, C.; Granata, A.; Pasta, A.; Arosio, B.; et al. LIPA gene mutations affect the composition of lipoproteins: Enrichment in ACAT-derived cholesteryl esters. Atherosclerosis 2020, 297, 8–15. [Google Scholar] [CrossRef]
- Ejsing, C.S.; Duchoslav, E.; Sampaio, J.; Simons, K.; Bonner, R.; Thiele, C.; Ekroos, K.; Shevchenko, A. Automated Identification and Quantification of Glycerophospholipid Molecular Species by Multiple Precursor Ion Scanning. Anal. Chem. 2006, 78, 6202–6214. [Google Scholar] [CrossRef]
- Camont, L.; Lhomme, M.; Rached, F.; Le Goff, W.; Nègre-Salvayre, A.; Salvayre, R.; Calzada, C.; Lagarde, M.; Chapman, M.J.; Kontush, A. Small, dense high-density lipoprotein-3 particles are enriched in negatively charged phospholipids: Relevance to cellular cholesterol efflux, antioxidative, antithrombotic, anti-inflammatory, and antiapoptotic functionalities. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2715–2723. [Google Scholar] [CrossRef] [Green Version]
- Kayser, B.D.; Prifti, E.; Lhomme, M.; Belda, E.; Dao, M.C.; Aron-Wisnewsky, J.; Kontush, A.; Zucker, J.D.; Rizkalla, S.W.; Dugail, I.; et al. Elevated serum ceramides are linked with obesity-associated gut dysbiosis and impaired glucose metabolism. Metabolomics 2019, 15, 140. [Google Scholar] [CrossRef]
- Stallone, G.; Infante, B.; Pontrelli, P.; Gigante, M.; Montemurno, E.; Loverre, A.; Rossini, M.; Schena, F.P.; Grandaliano, G.; Gesualdo, L. Sirolimus and proteinuria in renal transplant patients: Evidence for a dose-dependent effect on slit diaphragm-associated proteins. Transplantation 2011, 91, 997–1004. [Google Scholar] [CrossRef]
- Baragetti, A.; Ossoli, A.; Strazzella, A.; Simonelli, S.; Baragetti, I.; Grigore, L.; Pellegatta, F.; Catapano, A.L.; Norata, G.D.; Calabresi, L. Low plasma lecithin: Cholesterol acyltransferase (LCAT) concentration predicts chronic kidney disease. J. Clin. Med. 2020, 9, 2289. [Google Scholar] [CrossRef]
- Oldoni, F.; Baldassarre, D.; Castelnuovo, S.; Ossoli, A.; Amato, M.; van Capelleveen, J.; Hovingh, G.K.; De Groot, E.; Bochem, A.; Simonelli, S.; et al. Complete and Partial Lecithin:Cholesterol Acyltransferase Deficiency Is Differentially Associated With Atherosclerosis. Circulation 2018, 138, 1000–1007. [Google Scholar] [CrossRef]
- Pavanello, C.; Ossoli, A.; Strazzella, A.; Risè, P.; Veglia, F.; Lhomme, M.; Parini, P.; Calabresi, L. Plasma FA composition in familial LCAT deficiency indicates SOAT2-derived cholesteryl ester formation in humans. J. Lipid Res. 2022, 63, 100232. [Google Scholar] [CrossRef]
- Duni, A.; Liakopoulos, V.; Roumeliotis, S.; Peschos, D.; Dounousi, E. Oxidative stress in the pathogenesis and evolution of chronic kidney disease: Untangling Ariadne’s thread. Int. J. Mol. Sci. 2019, 20, 3711. [Google Scholar] [CrossRef] [Green Version]
- Cantaluppi, V.; Quercia, A.D.; Dellepiane, S.; Ferrario, S.; Camussi, G.; Biancone, L. Interaction between systemic inflammation and renal tubular epithelial cells. Nephrol. Dial. Transplant. 2014, 29, 2004–2011. [Google Scholar] [CrossRef] [Green Version]
- Haraldsson, B.; Nyström, J.; Deen, W.M. Properties of the glomerular barrier and mechanisms of proteinuria. Physiol. Rev. 2008, 88, 451–487. [Google Scholar] [CrossRef] [Green Version]
- Pavanello, C.; Calabresi, L. Genetic, biochemical, and clinical features of LCAT deficiency: Update for 2020. Curr. Opin. Lipidol. 2020, 31, 232–237. [Google Scholar] [CrossRef]
- Tabas, I. Consequences of cellular cholesterol accumulation: Basic concepts and physiological implications. J. Clin. Investig. 2002, 110, 905–911. [Google Scholar] [CrossRef]
- Chen, S.; Meng, X.F.; Zhang, C. Role of NADPH oxidase-mediated reactive oxygen species in podocyte injury. Biomed. Res. Int. 2013, 2013, 839761. [Google Scholar] [CrossRef] [Green Version]
- Duann, P.; Lianos, E.A.; Ma, J.; Lin, P.H. Autophagy, innate immunity and tissue repair in acute kidney injury. Int. J. Mol. Sci. 2016, 17, 662. [Google Scholar] [CrossRef] [Green Version]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta. 2016, 1863, 2977–2992. [Google Scholar] [CrossRef]
- Brites, F.; Martin, M.; Guillas, I.; Kontush, A. Antioxidative activity of high-density lipoprotein (HDL): Mechanistic insights into potential clinical benefit. BBA Clin. 2017, 8, 66–77. [Google Scholar] [CrossRef]
- Li, P.L.; Zhang, Y. Cross talk between ceramide and redox signaling: Implications for endothelial dysfunction and renal disease. Handb. Exp. Pharmacol. 2013, 216, 171–197. [Google Scholar] [CrossRef] [Green Version]
- Hannun, Y.A.; Obeid, L.M. Sphingolipids and their metabolism in physiology and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 175–191. [Google Scholar] [CrossRef]
- Zamzami, N.; Marchetti, P.; Castedo, M.; Decaudin, D.; Macho, A.; Hirsch, T.; Susin, S.A.; Petit, P.X.; Mignotte, B.; Kroemer, G. Sequential reduction of mitochondrial transmembrane potential and generation of reactive oxygen species in early programmed cell death. J. Exp. Med. 1995, 182, 367–377. [Google Scholar] [CrossRef]
- Kroemer, G.; Zamzami, N.; Susin, S.A. Mitochondrial control of apoptosis. Immunol. Today 1997, 18, 44–51. [Google Scholar] [CrossRef]
- Caridi, G.; Perfumo, F.; Ghiggeri, G.M. NPHS2 (Podocin) mutations in nephrotic syndrome. Clinical spectrum and fine mechanisms. Pediatr. Res. 2005, 57 5 Pt 2, 54R–61R. [Google Scholar] [CrossRef]
- Yu, H.; Kistler, A.; Faridi, M.H.; Meyer, J.O.; Tryniszewska, B.; Mehta, D.; Yue, L.; Dryer, S.; Reiser, J. Synaptopodin limits TRPC6 podocyte surface expression and attenuates proteinuria. J. Am. Soc. Nephrol. 2016, 27, 3308–3319. [Google Scholar] [CrossRef] [Green Version]
- Ossoli, A.; Strazzella, A.; Rottoli, D.; Zanchi, C.; Locatelli, M.; Zoja, C.; Simonelli, S.; Veglia, F.; Barbaras, R.; Tupin, C.; et al. CER-001 ameliorates lipid profile and kidney disease in a mouse model of familial LCAT deficiency. Metabolism 2021, 116, 154464. [Google Scholar] [CrossRef]
- Pavanello, C.; Turri, M.; Strazzella, A.; Tulissi, P.; Pizzolitto, S.; De Maglio, G.; Nappi, R.; Calabresi, L.; Boscutti, G. The HDL mimetic CER-001 remodels plasma lipoproteins and reduces kidney lipid deposits in inherited lecithin:cholesterol acyltransferase deficiency. J. Intern. Med. 2022, 291, 364–370. [Google Scholar] [CrossRef] [PubMed]
- Pavanello, C.; Ossoli, A.; Turri, M.; Strazzella, A.; Simonelli, S.; Laurenzi, T.; Kono, K.; Yamada, K.; Kiyosawa, N.; Eberini, I.; et al. Activation of naturally occurring lecithin:cholesterol acyltransferase mutants by a novel activator compound. J. Pharmacol. Exp. Ther. 2020, 375, 463–468. [Google Scholar] [CrossRef]
- Manthei, K.A.; Yang, S.M.; Baljinnyam, B.; Chang, L.; Glukhova, A.; Yuan, W.; Freeman, L.A.; Maloney, D.J.; Schwendeman, A.; Remaley, A.T.; et al. Molecular basis for activation of lecithin:cholesterol acyltransferase by a compound that increases HDL cholesterol. Elife 2018, 7, e41604. [Google Scholar] [CrossRef] [PubMed]
- Freeman, L.A.; Demosky, S.J., Jr.; Konaklieva, M.; Kuskovsky, R.; Aponte, A.; Ossoli, A.F.; Gordon, S.M.; Koby, R.F.; Manthei, K.A.; Shen, M.; et al. Lecithin:cholesterol acyltransferase activation by sulfhydryl-reactive small molecules: Role of cysteine-31. J. Pharmacol. Exp. Ther. 2017, 362, 306–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shamburek, R.D.; Bakker-Arkema, R.; Auerbach, B.J.; Krause, B.R.; Homan, R.; Amar, M.J.; Freeman, L.A.; Remaley, A.T. Familial lecithin:cholesterol acyltransferase deficiency: First-in-human treatment with enzyme replacement. J. Clin. Lipidol. 2016, 10, 356–367. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HOZ | HEZ | CTRL | p-Value | |
|---|---|---|---|---|
| N | 6 | 8 | 8 | |
| Sex (m/f) | 5/1 | 5/3 | 4/4 | 0.201 |
| Age | 37.8 ± 9.3 | 57.0 ± 15.1 | 36.3 ± 7.7 | 0.111 |
| Total cholesterol (mg/dL) | 154.3 ± 71.0 | 185.9 ± 48.7 | 194 ± 20.3 | 0.663 |
| Unesterified cholesterol (mg/dL) | 122.0 ± 58.1 §* | 53.5 ± 14.9 | 26.4 ± 16.9 | 0.002 |
| UC/TC (%) | 79.2 ± 15.3 §* | 29.7 ± 8.0 | 14.3 ± 9.9 | <0.001 |
| LDL-cholesterol (mg/dL) | 83.2 ± 43.7.0 | 119.0 ± 45.7 | 115.7 ± 26.4 | 0.228 |
| HDL-cholesterol (mg/dL) | 7.0 ± 1.4 §* | 38.9 ± 9.9 | 64.7 ± 20.8 | <0.001 |
| Triglycerides (mg/dL) | 321.0 ± 266.9 §* | 139.6 ± 51.8 * | 73.6 ± 27.0 | 0.007 |
| Phospholipids (mg/dL) | 330.4 ± 88.3 §* | 226.4 ± 39.5 | 239.7 ± 42.7 | 0.017 |
| apoA-I (mg/dL) | 37.0 ± 10.8 §* | 116.1 ± 24.3 | 120.8 ± 25.2 | <0.001 |
| apoA-II (mg/dL) | 4.9 ± 1.3 §* | 32.6 ± 3.8 | 29.5 ± 2.1 | <0.001 |
| apoB (mg/dL) | 62.3 ± 33.1 §* | 107.3 ± 29.9 | 93.5 ± 15.1 | 0.035 |
| Preβ-HDL (%) | 51.0 ± 11.5 §* | 23.9 ± 3.2 | 12.1 ± 1.1 | <0.001 |
| LpX (+/−) | 6/0 §* | 0/8 | 0/8 | <0.001 |
| LDL+LpX | ||||
|---|---|---|---|---|
| HOZ | HEZ | CTRL | p-Value | |
| UC (%) | 19.6 ± 3.9 §* | 9.0 ± 1.6 | 9.0 ± 2.3 | <0.001 |
| CE (%) | 4.1 ± 4.2 §* | 30.3 ± 10.0 | 37.1 ± 5.7 | <0.001 |
| PL (%) | 42.0 ± 6.4 §* | 25.4 ± 4.4 | 22.5 ± 5.7 | <0.001 |
| TG (%) | 23.0 ± 5.1 §* | 14.6 ± 3.3 * | 9.0 ± 2.3 | 0.016 |
| PT (%) | 11.3 ± 4.8 §* | 21.0 ± 4.4 | 22.2 ± 6.0 | <0.001 |
| HDL | ||||
| HOZ | HEZ | CTRL | p-Value | |
| UC (%) | 17.7 ± 2.8 §* | 3.9 ± 1.2 | 3.8 ± 0.9 | <0.001 |
| CE (%) | 0.3 ± 0.8 §* | 16.0 ± 3.1 * | 21.6 ± 3.9 | <0.001 |
| PL (%) | 45.9 ± 3.1 §* | 29.4 ± 5.9 | 32.0 ± 5.6 | <0.001 |
| TG (%) | 4.9 ± 2.2 | 5.6 ± 1.1 | 3.6 ± 1.1 | 0.011 |
| PT (%) | 31.3 ± 3.5 §* | 45.4 ± 7.6 | 39.3 ± 7.3 | 0.003 |
| Fold Change HEZ vs. CTRL | Fold Change HOZ vs. CTRL | |||||
|---|---|---|---|---|---|---|
| Mol % (PL + SL) | mol/g prot. | nmol/μL | Mol % (PL + SL) | mol/g prot. | nmol/μL | |
| Total SM | 0.98 | 0.92 | 0.81 | 1.01 | 1.25 | 0.54 |
| Total DHSM | 0.86 | 0.81 | 0.73 | 1.21 | 1.44 | 0.63 |
| Total DHC | 1.50 | 1.40 | 1.23 | 4.96 * | 6.09 * | 2.71 |
| Total Cer 18:1;O2 | 1.23 | 1.14 | 1.00 | 2.33 * | 2.78 | 1.21 |
| Total Cer 18:2;O2 | 0.96 | 0.90 | 0.79 | 1.26 | 1.61 | 0.69 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gomaraschi, M.; Turri, M.; Strazzella, A.; Lhomme, M.; Pavanello, C.; Le Goff, W.; Kontush, A.; Calabresi, L.; Ossoli, A. Abnormal Lipoproteins Trigger Oxidative Stress-Mediated Apoptosis of Renal Cells in LCAT Deficiency. Antioxidants 2023, 12, 1498. https://doi.org/10.3390/antiox12081498
Gomaraschi M, Turri M, Strazzella A, Lhomme M, Pavanello C, Le Goff W, Kontush A, Calabresi L, Ossoli A. Abnormal Lipoproteins Trigger Oxidative Stress-Mediated Apoptosis of Renal Cells in LCAT Deficiency. Antioxidants. 2023; 12(8):1498. https://doi.org/10.3390/antiox12081498
Chicago/Turabian StyleGomaraschi, Monica, Marta Turri, Arianna Strazzella, Marie Lhomme, Chiara Pavanello, Wilfried Le Goff, Anatol Kontush, Laura Calabresi, and Alice Ossoli. 2023. "Abnormal Lipoproteins Trigger Oxidative Stress-Mediated Apoptosis of Renal Cells in LCAT Deficiency" Antioxidants 12, no. 8: 1498. https://doi.org/10.3390/antiox12081498
APA StyleGomaraschi, M., Turri, M., Strazzella, A., Lhomme, M., Pavanello, C., Le Goff, W., Kontush, A., Calabresi, L., & Ossoli, A. (2023). Abnormal Lipoproteins Trigger Oxidative Stress-Mediated Apoptosis of Renal Cells in LCAT Deficiency. Antioxidants, 12(8), 1498. https://doi.org/10.3390/antiox12081498