
Toward Ameliorating Insulin Resistance: Targeting a Novel PAK1 Signaling Pathway Required for Skeletal Muscle Mitochondrial Function

 ,
, 
Abstract
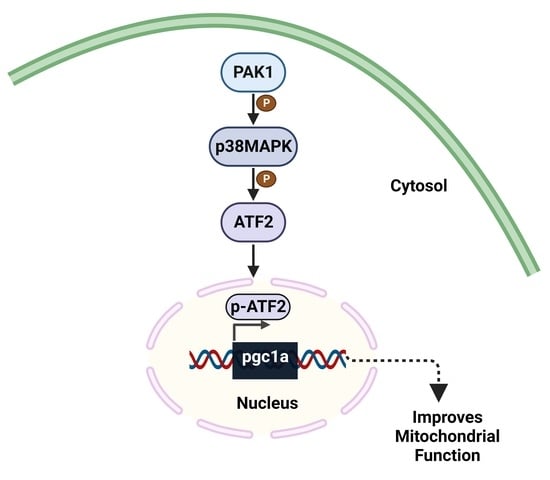
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Human Skeletal Muscle
2.2. Cell Culture
2.3. Immunoblot Analysis
2.4. Mitochondrial Fragmentation and Superoxide Radical Detection
2.5. Adenoviral Transduction
2.6. Mitochondrial Copy Number Evaluation
2.7. Oxygen Consumption Rate (OCR) Measurements
2.8. RNA Isolation and qPCR
2.9. Immunostaining
2.10. Statistics
3. Results
3.1. PAK1 Suppression in Skeletal Muscle Impairs Mitochondrial Structure/Function in L6.GLUT4myc Myotubes
3.2. PAK1 Enrichment in Insulin-Resistant L6 Myotubes Protects from Mitochondrial Damage
3.3. PAK1 Regulates PGC1α Gene Expression
3.4. PAK1 Regulates p38MAPK/ATF2/PGC1α-Axis Signaling
3.5. PAK1 Regulates the Nuclear Translocation of ATF2
3.6. T2D Human Muscle and PAK1-Deficient Myotubes Exhibit Impaired Mitochondrial Function and PGC1α Expression
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zheng, Y.; Ley, S.H.; Hu, F.B. Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat. Rev. Endocrinol. 2018, 14, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Unnikrishnan, R.; Pradeepa, R.; Joshi, S.R.; Mohan, V. Type 2 Diabetes: Demystifying the Global Epidemic. Diabetes 2017, 66, 1432–1442. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Jacot, E.; Jequier, E.; Maeder, E.; Wahren, J.; Felber, J.P. The effect of insulin on the disposal of intravenous glucose. Results from indirect calorimetry and hepatic and femoral venous catheterization. Diabetes 1981, 30, 1000–1007. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.A.; Gunnarsson, R.; Bjorkman, O.; Olsson, M.; Wahren, J. Effects of insulin on peripheral and splanchnic glucose metabolism in noninsulin-dependent (type II) diabetes mellitus. J. Clin. Investig. 1985, 76, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Balakrishnan, R.; Thurmond, D.C. Mechanisms by Which Skeletal Muscle Myokines Ameliorate Insulin Resistance. Int. J. Mol. Sci. 2022, 23, 4636. [Google Scholar] [CrossRef] [PubMed]
- Ahn, M.; Oh, E.; McCown, E.M.; Wang, X.; Veluthakal, R.; Thurmond, D.C. A requirement for PAK1 to support mitochondrial function and maintain cellular redox balance via electron transport chain proteins to prevent beta-cell apoptosis. Metabolism 2021, 115, 154431. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. National Diabetes Statistics Report; Centers for Disease Control and Prevention: Atlanta, GA, USA, 2017. Available online: https://dev.diabetes.org/sites/default/files/2019-06/cdc-statistics-report-2017.pdf (accessed on 12 June 2023).
- Kelley, D.E.; He, J.; Menshikova, E.V.; Ritov, V.B. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes 2002, 51, 2944–2950. [Google Scholar] [CrossRef]
- Gerbitz, K.D.; Gempel, K.; Brdiczka, D. Mitochondria and diabetes. Genetic, biochemical, and clinical implications of the cellular energy circuit. Diabetes 1996, 45, 113–126. [Google Scholar] [CrossRef]
- Ruegsegger, G.N.; Creo, A.L.; Cortes, T.M.; Dasari, S.; Nair, K.S. Altered mitochondrial function in insulin-deficient and insulin-resistant states. J. Clin. Investig. 2018, 128, 3671–3681. [Google Scholar] [CrossRef]
- Sergi, D.; Naumovski, N.; Heilbronn, L.K.; Abeywardena, M.; O’Callaghan, N.; Lionetti, L.; Luscombe-Marsh, N. Mitochondrial (Dys)function and Insulin Resistance: From Pathophysiological Molecular Mechanisms to the Impact of Diet. Front. Physiol. 2019, 10, 532. [Google Scholar] [CrossRef]
- Scott, L.J.; Erdos, M.R.; Huyghe, J.R.; Welch, R.P.; Beck, A.T.; Wolford, B.N.; Chines, P.S.; Didion, J.P.; Narisu, N.; Stringham, H.M.; et al. The genetic regulatory signature of type 2 diabetes in human skeletal muscle. Nat. Commun. 2016, 7, 11764. [Google Scholar] [CrossRef] [PubMed]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstrale, M.; Laurila, E.; et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in man diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Patti, M.E.; Butte, A.J.; Crunkhorn, S.; Cusi, K.; Berria, R.; Kashyap, S.; Miyazaki, Y.; Kohane, I.; Costello, M.; Saccone, R.; et al. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: Potential role of PGC1 and NRF1. Proc. Natl. Acad. Sci. USA 2003, 100, 8466–8471. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Puigserver, P.; Andersson, U.; Zhang, C.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.; Scarpulla, R.C.; et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 1999, 98, 115–124. [Google Scholar] [CrossRef]
- Li, L.; Pan, R.; Li, R.; Niemann, B.; Aurich, A.C.; Chen, Y.; Rohrbach, S. Mitochondrial biogenesis and peroxisome proliferator-activated receptor-gamma coactivator-1alpha (PGC-1alpha) deacetylation by physical activity: Intact adipocytokine signaling is required. Diabetes 2011, 60, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Stump, C.S.; Short, K.R.; Bigelow, M.L.; Schimke, J.M.; Nair, K.S. Effect of insulin on human skeletal muscle mitochondrial ATP production, protein synthesis, and mRNA transcripts. Proc. Natl. Acad. Sci. USA 2003, 100, 7996–8001. [Google Scholar] [CrossRef]
- Yu, T.; Robotham, J.L.; Yoon, Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc. Natl. Acad. Sci. USA 2006, 103, 2653–2658. [Google Scholar] [CrossRef]
- Merz, K.E.; Tunduguru, R.; Ahn, M.; Salunkhe, V.A.; Veluthakal, R.; Hwang, J.; Bhattacharya, S.; McCown, E.M.; Garcia, P.A.; Zhou, C.; et al. Changes in skeletal muscle pak1 levels regulate tissue crosstalk to impact whole body glucose homeostasis. FASEB J. 2022, 13, 821849. [Google Scholar] [CrossRef]
- Tunduguru, R.; Zhang, J.; Aslamy, A.; Salunkhe, V.A.; Brozinick, J.T.; Elmendorf, J.S.; Thurmond, D.C. The actin-related p41ARC subunit contributes to p21-activated kinase-1 (PAK1)-mediated glucose uptake into skeletal muscle cells. J. Biol. Chem. 2017, 292, 19034–19043. [Google Scholar] [CrossRef]
- Kalwat, M.A.; Yoder, S.M.; Wang, Z.; Thurmond, D.C. A p21-activated kinase (PAK1) signaling cascade coordinately regulates F-actin remodeling and insulin granule exocytosis in pancreatic beta cells. Biochem. Pharmacol. 2013, 85, 808–816. [Google Scholar] [CrossRef]
- Wang, Z.; Oh, E.; Clapp, D.W.; Chernoff, J.; Thurmond, D.C. Inhibition or ablation of p21-activated kinase (PAK1) disrupts glucose homeostatic mechanisms in vivo. J. Biol. Chem. 2011, 286, 41359–41367. [Google Scholar] [CrossRef] [PubMed]
- Tunduguru, R.; Chiu, T.T.; Ramalingam, L.; Elmendorf, J.S.; Klip, A.; Thurmond, D.C. Signaling of the p21-activated kinase (PAK1) coordinates insulin-stimulated actin remodeling and glucose uptake in skeletal muscle cells. Biochem. Pharmacol. 2014, 92, 380–388. [Google Scholar] [CrossRef] [PubMed]
- Niemann, B.; Haufs-Brusberg, S.; Puetz, L.; Feickert, M.; Jaeckstein, M.Y.; Hoffmann, A.; Zurkovic, J.; Heine, M.; Trautmann, E.M.; Muller, C.E.; et al. Apoptotic brown adipocytes enhance energy expenditure via extracellular inosine. Nature 2022, 609, 361–368. [Google Scholar] [CrossRef]
- McCarthy, A.M.; Spisak, K.O.; Brozinick, J.T.; Elmendorf, J.S. Loss of cortical actin filaments in insulin-resistant skeletal muscle cells impairs GLUT4 vesicle trafficking and glucose transport. Am. J. Physiol. Cell Physiol. 2006, 291, C860–C868. [Google Scholar] [CrossRef]
- Zhu, C.H.; Mouly, V.; Cooper, R.N.; Mamchaoui, K.; Bigot, A.; Shay, J.W.; Di Santo, J.P.; Butler-Browne, G.S.; Wright, W.E. Cellular senescence in human myoblasts is overcome by human telomerase reverse transcriptase and cyclin-dependent kinase 4: Consequences in aging muscle and therapeutic strategies for muscular dystrophies. Aging Cell 2007, 6, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Gan, D.; Hu, F.; Cheng, Y.; Yu, J.; Lei, B.; Shu, Q.; Gu, R.; Xu, G. Intravitreal injection of mitochondrial DNA induces cell damage and retinal dysfunction in rats. Biol. Res. 2022, 55, 22. [Google Scholar] [CrossRef] [PubMed]
- Aswani, A.; Manson, J.; Itagaki, K.; Chiazza, F.; Collino, M.; Wupeng, W.L.; Chan, T.K.; Wong, W.S.F.; Hauser, C.J.; Thiemermann, C.; et al. Scavenging Circulating Mitochondrial DNA as a Potential Therapeutic Option for Multiple Organ Dysfunction in Trauma Hemorrhage. Front. Immunol. 2018, 9, 891. [Google Scholar] [CrossRef]
- Nisr, R.B.; Shah, D.S.; Ganley, I.G.; Hundal, H.S. Proinflammatory NFkB signalling promotes mitochondrial dysfunction in skeletal muscle in response to cellular fuel overloading. Cell. Mol. Life Sci. 2019, 76, 4887–4904. [Google Scholar] [CrossRef]
- Ueyama, A.; Yaworsky, K.L.; Wang, Q.; Ebina, Y.; Klip, A. GLUT-4myc ectopic expression in L6 myoblasts generates a GLUT-4-specific pool conferring insulin sensitivity. Am. J. Physiol. 1999, 277, E572–E578. [Google Scholar] [CrossRef]
- Mouchiroud, L.; Eichner, L.J.; Shaw, R.J.; Auwerx, J. Transcriptional coregulators: Fine-tuning metabolism. Cell Metab. 2014, 20, 26–40. [Google Scholar] [CrossRef]
- Liang, H.; Ward, W.F. PGC-1alpha: A key regulator of energy metabolism. Adv. Physiol. Educ. 2006, 30, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Chiang, Y.A.; Shao, W.; Xu, X.X.; Chernoff, J.; Jin, T. P21-activated protein kinase 1 (Pak1) mediates the cross talk between insulin and beta-catenin on proglucagon gene expression and its ablation affects glucose homeostasis in male C57BL/6 mice. Endocrinology 2013, 154, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Oladimeji, P.; Diakonova, M. PAK1 translocates into nucleus in response to prolactin but not to estrogen. Biochem. Biophys. Res. Commun. 2016, 473, 206–211. [Google Scholar] [CrossRef]
- Kelly, M.L.; Chernoff, J. Mouse models of PAK function. Cell. Logist. 2012, 2, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Joseph, G.A.; Lu, M.; Radu, M.; Lee, J.K.; Burden, S.J.; Chernoff, J.; Krauss, R.S. Group I Paks Promote Skeletal Myoblast Differentiation In Vivo and In Vitro. Mol. Cell. Biol. 2017, 37, e00222-16. [Google Scholar] [CrossRef] [PubMed]
- Deacon, S.W.; Beeser, A.; Fukui, J.A.; Rennefahrt, U.E.; Myers, C.; Chernoff, J.; Peterson, J.R. An isoform-selective, small-molecule inhibitor targets the autoregulatory mechanism of p21-activated kinase. Chem. Biol. 2008, 15, 322–331. [Google Scholar] [CrossRef]
- Akimoto, T.; Pohnert, S.C.; Li, P.; Zhang, M.; Gumbs, C.; Rosenberg, P.B.; Williams, R.S.; Yan, Z. Exercise stimulates Pgc-1alpha transcription in skeletal muscle through activation of the p38 MAPK pathway. J. Biol. Chem. 2005, 280, 19587–19593. [Google Scholar] [CrossRef]
- Vitucci, D.; Imperlini, E.; Arcone, R.; Alfieri, A.; Canciello, A.; Russomando, L.; Martone, D.; Cola, A.; Labruna, G.; Orru, S.; et al. Serum from differently exercised subjects induces myogenic differentiation in LHCN-M2 human myoblasts. J. Sports Sci. 2018, 36, 1630–1639. [Google Scholar] [CrossRef]
- Koranova, T.; Dvoracek, L.; Grebenova, D.; Roselova, P.; Obr, A.; Kuzelova, K. PAK1 and PAK2 in cell metabolism regulation. J. Cell. Biochem. 2022, 123, 375–389. [Google Scholar] [CrossRef]
- Gupta, A.; Ajith, A.; Singh, S.; Panday, R.K.; Samaiya, A.; Shukla, S. PAK2-c-Myc-PKM2 axis plays an essential role in head and neck oncogenesis via regulating Warburg effect. Cell Death Dis. 2018, 9, 825. [Google Scholar] [CrossRef]
- Ainbinder, A.; Boncompagni, S.; Protasi, F.; Dirksen, R.T. Role of Mitofusin-2 in mitochondrial localization and calcium uptake in skeletal muscle. Cell Calcium 2015, 57, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Vendelin, M.; Beraud, N.; Guerrero, K.; Andrienko, T.; Kuznetsov, A.V.; Olivares, J.; Kay, L.; Saks, V.A. Mitochondrial regular arrangement in muscle cells: A “crystal-like” pattern. Am. J. Physiol. Cell Physiol. 2005, 288, C757–C767. [Google Scholar] [CrossRef] [PubMed]
- Yaffe, M.P. The machinery of mitochondrial inheritance and behavior. Science 1999, 283, 1493–1497. [Google Scholar] [CrossRef] [PubMed]
- Wagner, O.I.; Lifshitz, J.; Janmey, P.A.; Linden, M.; McIntosh, T.K.; Leterrier, J.F. Mechanisms of mitochondria-neurofilament interactions. J. Neurosci. 2003, 23, 9046–9058. [Google Scholar] [CrossRef] [PubMed]
- Joseph, G.A.; Hung, M.; Goel, A.J.; Hong, M.; Rieder, M.K.; Beckmann, N.D.; Serasinghe, M.N.; Chipuk, J.E.; Devarakonda, P.M.; Goldhamer, D.J.; et al. Late-onset megaconial myopathy in mice lacking group I Paks. Skelet. Muscle 2019, 9, 5. [Google Scholar] [CrossRef]
- Petersen, K.F.; Dufour, S.; Befroy, D.; Garcia, R.; Shulman, G.I. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N. Engl. J. Med. 2004, 350, 664–671. [Google Scholar] [CrossRef]
- Asmann, Y.W.; Stump, C.S.; Short, K.R.; Coenen-Schimke, J.M.; Guo, Z.; Bigelow, M.L.; Nair, K.S. Skeletal muscle mitochondrial functions, mitochondrial DNA copy numbers, and gene transcript profiles in type 2 diabetic and nondiabetic subjects at equal levels of low or high insulin and euglycemia. Diabetes 2006, 55, 3309–3319. [Google Scholar] [CrossRef]
- Ohman, T.; Teppo, J.; Datta, N.; Makinen, S.; Varjosalo, M.; Koistinen, H.A. Skeletal muscle proteomes reveal downregulation of mitochondrial proteins in transition from prediabetes into type 2 diabetes. iScience 2021, 24, 102712. [Google Scholar] [CrossRef]
- Finck, B.N.; Kelly, D.P. PGC-1 coactivators: Inducible regulators of energy metabolism in health and disease. J. Clin. Investig. 2006, 116, 615–622. [Google Scholar] [CrossRef]
- Pagel-Langenickel, I.; Bao, J.; Joseph, J.J.; Schwartz, D.R.; Mantell, B.S.; Xu, X.; Raghavachari, N.; Sack, M.N. PGC-1alpha integrates insulin signaling, mitochondrial regulation, and bioenergetic function in skeletal muscle. J. Biol. Chem. 2008, 283, 22464–22472. [Google Scholar] [CrossRef]
- Ling, C.; Poulsen, P.; Carlsson, E.; Ridderstrale, M.; Almgren, P.; Wojtaszewski, J.; Beck-Nielsen, H.; Groop, L.; Vaag, A. Multiple environmental and genetic factors influence skeletal muscle PGC-1alpha and PGC-1beta gene expression in twins. J. Clin. Investig. 2004, 114, 1518–1526. [Google Scholar] [CrossRef] [PubMed]
- Motwani, M.; Li, D.Q.; Horvath, A.; Kumar, R. Identification of novel gene targets and functions of p21-activated kinase 1 during DNA damage by gene expression profiling. PLoS ONE 2013, 8, e66585. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.R.; Song, C.; Yang, Z.; Kumar, R. Nuclear localization and chromatin targets of p21-activated kinase 1. J. Biol. Chem. 2005, 280, 18130–18137. [Google Scholar] [CrossRef]
- Zhang, N.; Li, X.; Liu, X.; Cao, Y.; Chen, D.; Liu, X.; Wang, Q.; Du, J.; Weng, J.; Ma, W. p21-activated kinase 1 activity is required for histone H3 Ser(10) phosphorylation and chromatin condensation in mouse oocyte meiosis. Reprod. Fertil. Dev. 2017, 29, 1287–1296. [Google Scholar] [CrossRef]
- Cao, W.; Daniel, K.W.; Robidoux, J.; Puigserver, P.; Medvedev, A.V.; Bai, X.; Floering, L.M.; Spiegelman, B.M.; Collins, S. p38 mitogen-activated protein kinase is the central regulator of cyclic AMP-dependent transcription of the brown fat uncoupling protein 1 gene. Mol. Cell. Biol. 2004, 24, 3057–3067. [Google Scholar] [CrossRef]
- Frost, J.A.; Xu, S.; Hutchison, M.R.; Marcus, S.; Cobb, M.H. Actions of Rho family small G proteins and p21-activated protein kinases on mitogen-activated protein kinase family members. Mol. Cell. Biol. 1996, 16, 3707–3713. [Google Scholar] [CrossRef]
- Beeser, A.; Jaffer, Z.M.; Hofmann, C.; Chernoff, J. Role of group A p21-activated kinases in activation of extracellular-regulated kinase by growth factors. J. Biol. Chem. 2005, 280, 36609–36615. [Google Scholar] [CrossRef] [PubMed]
- Ryder, J.W.; Fahlman, R.; Wallberg-Henriksson, H.; Alessi, D.R.; Krook, A.; Zierath, J.R. Effect of contraction on mitogen-activated protein kinase signal transduction in skeletal muscle. Involvement Of the mitogen- and stress-activated protein kinase 1. J. Biol. Chem. 2000, 275, 1457–1462. [Google Scholar] [CrossRef] [PubMed]
- Kerris, J.P.; Betik, A.C.; Li, J.; McConell, G.K. Passive stretch regulates skeletal muscle glucose uptake independent of nitric oxide synthase. J. Appl. Physiol. 2019, 126, 239–245. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balakrishnan, R.; Garcia, P.A.; Veluthakal, R.; Huss, J.M.; Hoolachan, J.M.; Thurmond, D.C. Toward Ameliorating Insulin Resistance: Targeting a Novel PAK1 Signaling Pathway Required for Skeletal Muscle Mitochondrial Function. Antioxidants 2023, 12, 1658. https://doi.org/10.3390/antiox12091658
Balakrishnan R, Garcia PA, Veluthakal R, Huss JM, Hoolachan JM, Thurmond DC. Toward Ameliorating Insulin Resistance: Targeting a Novel PAK1 Signaling Pathway Required for Skeletal Muscle Mitochondrial Function. Antioxidants. 2023; 12(9):1658. https://doi.org/10.3390/antiox12091658
Chicago/Turabian StyleBalakrishnan, Rekha, Pablo A. Garcia, Rajakrishnan Veluthakal, Janice M. Huss, Joseph M. Hoolachan, and Debbie C. Thurmond. 2023. "Toward Ameliorating Insulin Resistance: Targeting a Novel PAK1 Signaling Pathway Required for Skeletal Muscle Mitochondrial Function" Antioxidants 12, no. 9: 1658. https://doi.org/10.3390/antiox12091658
APA StyleBalakrishnan, R., Garcia, P. A., Veluthakal, R., Huss, J. M., Hoolachan, J. M., & Thurmond, D. C. (2023). Toward Ameliorating Insulin Resistance: Targeting a Novel PAK1 Signaling Pathway Required for Skeletal Muscle Mitochondrial Function. Antioxidants, 12(9), 1658. https://doi.org/10.3390/antiox12091658