
Insights on the Multifaceted Roles of Wild-Type and Mutated Superoxide Dismutase 1 in Amyotrophic Lateral Sclerosis Pathogenesis

 , , , and
, , , and 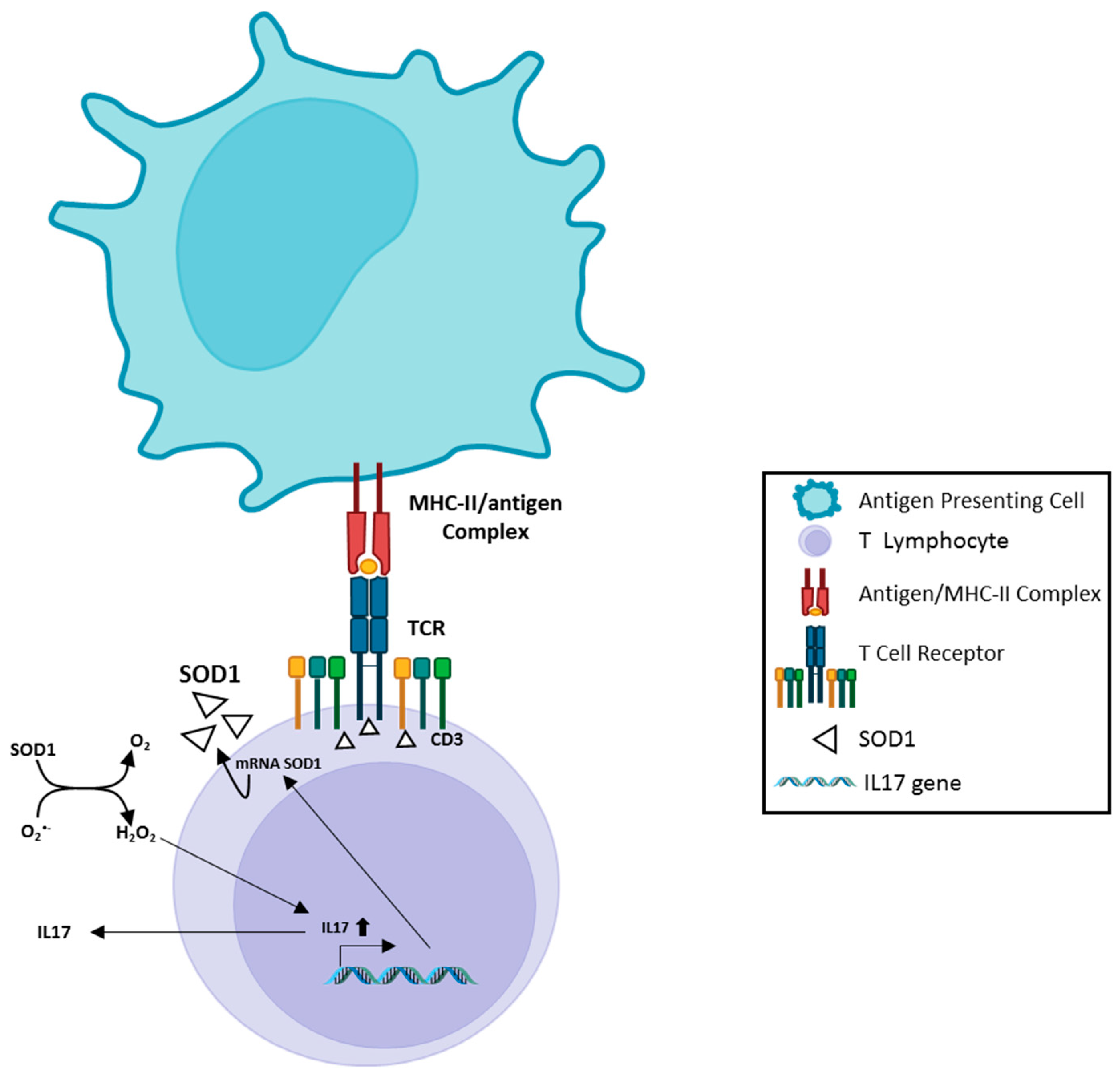{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Pathogenic Mechanisms of Mutant SOD1 in ALS
3. ALS and Mitochondrial Damage
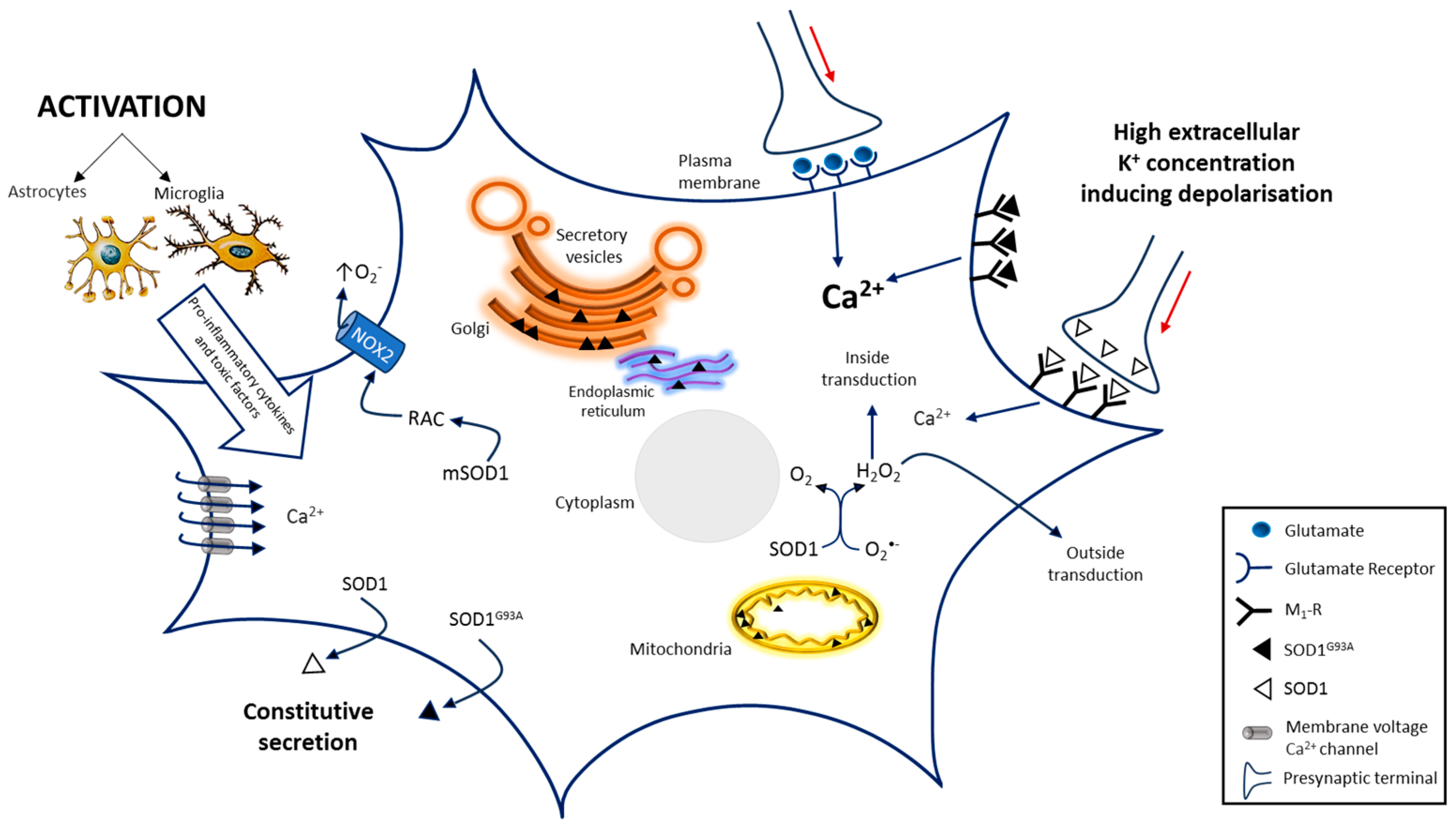
4. Microglia Activation, NADPH Oxidase and SOD1 in ALS
5. SOD1, Immunity and Neuroinflammation Processes in ALS
6. Constitutive and Inducible SOD1 Secretion
7. Role of SOD1 Interaction with the Muscarinic M1 Receptor
8. Discussion
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Phukan, J.; Pender, N.P.; Hardiman, O. Cognitive impairment in amyotrophic lateral sclerosis. Lancet Neurol. 2007, 6, 994–1003. [Google Scholar] [CrossRef]
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; van den Berg, L.H. Amyotrophic lateral sclerosis. Nat. Rev. Dis. Primers 2017, 3, 17071. [Google Scholar] [CrossRef] [PubMed]
- Rowland, L.P.; Shneider, N.A. Amyotrophic lateral sclerosis. N. Engl. J. Med. 2001, 344, 1688–1700. [Google Scholar] [CrossRef]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Renton, A.E.; Chiò, A.; Traynor, B.J. State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 2014, 17, 17–23. [Google Scholar] [CrossRef]
- Brown, R.H.; Al-Chalabi, A. Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2017, 377, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Mejzini, R.; Flynn, L.L.; Pitout, I.L.; Fletcher, S.; Wilton, S.D.; Akkari, P.A. ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now? Front. Neurosci. 2019, 13, 1310. [Google Scholar] [CrossRef]
- Cozzolino, M.; Pesaresi, M.G.; Gerbino, V.; Grosskreutz, J.; Carrì, M.T. Amyotrophic lateral sclerosis: New insights into underlying molecular mechanisms and opportunities for therapeutic intervention. Antioxid. Redox Signal 2012, 17, 1277–1330. [Google Scholar] [CrossRef] [PubMed]
- Ferraiuolo, L.; Kirby, J.; Grierson, A.J.; Sendtner, M.; Shaw, P.J. Molecular pathways of motor neuron injury in amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2011, 7, 616–630. [Google Scholar] [CrossRef]
- Halliwell, B.; Gutteridge, J.M.C. Free Radicals in Biology and Medicine; Clarendon Press: Oxford, UK, 1989. [Google Scholar]
- McCord, J.M.; Fridovich, I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J. Biol. Chem. 1969, 244, 6049–6055. [Google Scholar] [CrossRef]
- Cimini, V.; Ruggiero, G.; Buonomo, T.; Seru, R.; Sciorio, S.; Zanzi, C.; Santangelo, F.; Mondola, P. CuZn-superoxide dismutase in human thymus: Immunocytochemical localisation and secretion in thymus-derived epithelial and fibroblast cell lines. Histochem. Cell Biol. 2002, 118, 163–169. [Google Scholar] [CrossRef]
- Mondola, P.; Ruggiero, G.; Serù, R.; Damiano, S.; Grimaldi, S.; Garbi, C.; Monda, M.; Greco, D.; Santillo, M. The Cu,Zn superoxide dismutase in neuroblastoma SK-N-BE cells is exported by a microvesicles dependent pathway. Brain Res. Mol. Brain Res. 2003, 110, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Gomes, C.; Keller, S.; Altevogt, P.; Costa, J. Evidence for secretion of Cu,Zn superoxide dismutase via exosomes from a cell model of amyotrophic lateral sclerosis. Neurosci. Lett. 2007, 428, 43–46. [Google Scholar] [CrossRef]
- Polazzi, E.; Mengoni, I.; Caprini, M.; Peña-Altamira, E.; Kurtys, E.; Monti, B. Copper-zinc superoxide dismutase (SOD1) is released by microglial cells and confers neuroprotection against 6-OHDA neurotoxicity. NeuroSignals 2013, 21, 112–128. [Google Scholar] [CrossRef] [PubMed]
- Urushitani, M.; Sik, A.; Sakurai, T.; Nukina, N.; Takahashi, R.; Julien, J.P. Chromogranin-mediated secretion of mutant superoxide dismutase proteins linked to amyotrophic lateral sclerosis. Nat. Neurosci. 2006, 9, 108–118. [Google Scholar] [CrossRef]
- Mondola, P.; Santillo, M.; Serù, R.; Damiano, S.; Alvino, C.; Ruggiero, G.; Formisano, P.; Terrazzano, G.; Secondo, A.; Annunziato, L. Cu,Zn superoxide dismutase increases intracellular calcium levels via a phospholipase C-protein kinase C pathway in SK-N-BE neuroblastoma cells. Biochem. Biophys. Res. Commun. 2004, 324, 887–892. [Google Scholar] [CrossRef] [PubMed]
- Viggiano, A.; Serù, R.; Damiano, S.; De Luca, B.; Santillo, M.; Mondola, P. Inhibition of long-term potentiation by CuZn superoxide dismutase injection in rat dentate gyrus: Involvement of muscarinic M1 receptor. J. Cell Physiol. 2012, 227, 3111–3115. [Google Scholar] [CrossRef]
- Becher, B.; Prat, A.; Antel, J.P. Brain-immune connection: Immuno-regulatory properties of CNS-resident cells. Glia 2000, 29, 293–304. [Google Scholar] [CrossRef]
- Norris, G.T.; Kipnis, J. Immune cells and CNS physiology: Microglia and beyond. J. Exp. Med. 2019, 216, 60–70. [Google Scholar] [CrossRef]
- Castellani, G.; Croese, T.; Peralta Ramos, J.M.; Schwartz, M. Transforming the understanding of brain immunity. Science 2023, 380, eabo7649. [Google Scholar] [CrossRef]
- Schnell, A.; Littman, D.R.; Kuchroo, V.K. T(H)17 cell heterogeneity and its role in tissue inflammation. Nat. Immunol. 2023, 24, 19–29. [Google Scholar] [CrossRef]
- Terrazzano, G.; Rubino, V.; Damiano, S.; Sasso, A.; Petrozziello, T.; Ucci, V.; Palatucci, A.T.; Giovazzino, A.; Santillo, M.; De Felice, B.; et al. T cell activation induces CuZn superoxide dismutase (SOD)-1 intracellular re-localization, production and secretion. Biochim. Biophys. Acta 2014, 1843, 265–274. [Google Scholar] [CrossRef]
- Rubino, V.; Palatucci, A.T.; La Rosa, G.; Giovazzino, A.; Aruta, F.; Damiano, S.; Carriero, F.; Santillo, M.; Iodice, R.; Mondola, P.; et al. Superoxide Dismutase-1 Intracellular Content in T Lymphocytes Associates with Increased Regulatory T Cell Level in Multiple Sclerosis Subjects Undergoing Immune-Modulating Treatment. Antioxidants 2021, 10, 1940. [Google Scholar] [CrossRef]
- Becher, B.; Spath, S.; Goverman, J. Cytokine networks in neuroinflammation. Nat. Rev. Immunol. 2017, 17, 49–59. [Google Scholar] [CrossRef]
- Jin, M.; Akgün, K.; Ziemssen, T.; Kipp, M.; Günther, R.; Hermann, A. Interleukin-17 and Th17 Lymphocytes Directly Impair Motoneuron Survival of Wildtype and FUS-ALS Mutant Human iPSCs. Int. J. Mol. Sci. 2021, 22, 8042. [Google Scholar] [CrossRef] [PubMed]
- Atkin, J.D.; Farg, M.A.; Walker, A.K.; McLean, C.; Tomas, D.; Horne, M.K. Endoplasmic reticulum stress and induction of the unfolded protein response in human sporadic amyotrophic lateral sclerosis. Neurobiol. Dis. 2008, 30, 400–407. [Google Scholar] [CrossRef]
- Atkin, J.D.; Farg, M.A.; Soo, K.Y.; Walker, A.K.; Halloran, M.; Turner, B.J.; Nagley, P.; Horne, M.K. Mutant SOD1 inhibits ER-Golgi transport in amyotrophic lateral sclerosis. J. Neurochem. 2014, 129, 190–204. [Google Scholar] [CrossRef] [PubMed]
- Andrus, P.K.; Fleck, T.J.; Gurney, M.E.; Hall, E.D. Protein oxidative damage in a transgenic mouse model of familial amyotrophic lateral sclerosis. J. Neurochem. 1998, 71, 2041–2048. [Google Scholar] [CrossRef] [PubMed]
- Ezzi, S.A.; Urushitani, M.; Julien, J.P. Wild-type superoxide dismutase acquires binding and toxic properties of ALS-linked mutant forms through oxidation. J. Neurochem. 2007, 102, 170–178. [Google Scholar] [CrossRef]
- Elam, J.S.; Taylor, A.B.; Strange, R.; Antonyuk, S.; Doucette, P.A.; Rodriguez, J.A.; Hasnain, S.S.; Hayward, L.J.; Valentine, J.S.; Yeates, T.O.; et al. Amyloid-like filaments and water-filled nanotubes formed by SOD1 mutant proteins linked to familial ALS. Nat. Struct. Biol. 2003, 10, 461–467. [Google Scholar] [CrossRef]
- Stathopulos, P.B.; Rumfeldt, J.A.O.; Scholz, G.A.; Irani, R.A.; Frey, H.E.; Hallewell, R.A.; Lepock, J.R.; Meiering, E.M. Cu/Zn superoxide dismutase mutants associated with amyotrophic lateral sclerosis show enhanced formation of aggregates in vitro. Proc. Natl. Acad. Sci. USA 2003, 100, 7021–7026. [Google Scholar] [CrossRef]
- Rodriguez, J.A.; Valentine, J.S.; Eggers, D.K.; Roe, J.A.; Tiwari, A.; Brown, R.H., Jr.; Hayward, L.J. Familial amyotrophic lateral sclerosis-associated mutations decrease the thermal stability of distinctly metallated species of human copper/zinc superoxide dismutase. J. Biol. Chem. 2002, 277, 15932–15937. [Google Scholar] [CrossRef]
- Bendotti, C.; Carrì, M.T. Lessons from models of SOD1-linked familial ALS. Trends Mol. Med. 2004, 10, 393–400. [Google Scholar] [CrossRef]
- Rakhit, R.; Cunningham, P.; Furtos-Matei, A.; Dahan, S.; Qi, X.F.; Crow, J.P.; Cashman, N.R.; Kondejewski, L.H.; Chakrabartty, A. Oxidation-induced misfolding and aggregation of superoxide dismutase and its implications for amyotrophic lateral sclerosis. J. Biol. Chem. 2002, 277, 47551–47556. [Google Scholar] [CrossRef]
- Alexander, G.M.; Erwin, K.L.; Byers, N.; Deitch, J.S.; Augelli, B.J.; Blankenhorn, E.P.; Heiman-Patterson, T.D. Effect of transgene copy number on survival in the G93A SOD1 transgenic mouse model of ALS. Brain Res. Mol. Brain Res. 2004, 130, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Andersen, P.M.; Nilsson, P.; Ala-Hurula, V.; Keränen, M.L.; Tarvainen, I.; Haltia, T.; Nilsson, L.; Binzer, M.; Forsgren, L.; Marklund, S.L. Amyotrophic lateral sclerosis associated with homozygosity for an Asp90Ala mutation in CuZn-superoxide dismutase. Nat. Genet. 1995, 10, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Alexianu, M.E.; Ho, B.K.; Mohamed, A.H.; La Bella, V.; Smith, R.G.; Appel, S.H. The role of calcium-binding proteins in selective motoneuron vulnerability in amyotrophic lateral sclerosis. Ann. Neurol. 1994, 36, 846–858. [Google Scholar] [CrossRef]
- Beckman, J.S.; Estévez, A.G.; Crow, J.P.; Barbeito, L. Superoxide dismutase and the death of motoneurons in ALS. Trends Neurosci. 2001, 24, S15–S20. [Google Scholar] [CrossRef] [PubMed]
- Howland, D.S.; Liu, J.; She, Y.; Goad, B.; Maragakis, N.J.; Kim, B.; Erickson, J.; Kulik, J.; DeVito, L.; Psaltis, G.; et al. Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS). Proc. Natl. Acad. Sci. USA 2002, 99, 1604–1609. [Google Scholar] [CrossRef]
- Beers, D.R.; Henkel, J.S.; Xiao, Q.; Zhao, W.; Wang, J.; Yen, A.A.; Siklos, L.; McKercher, S.R.; Appel, S.H. Wild-type microglia extend survival in PU.1 knockout mice with familial amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2006, 103, 16021–16026. [Google Scholar] [CrossRef]
- Gurney, M.E.; Pu, H.; Chiu, A.Y.; Dal Canto, M.C.; Polchow, C.Y.; Alexander, D.D.; Caliendo, J.; Hentati, A.; Kwon, Y.W.; Deng, H.X.; et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 1994, 264, 1772–1775. [Google Scholar] [CrossRef]
- Sun, H.; Bénardais, K.; Stanslowsky, N.; Thau-Habermann, N.; Hensel, N.; Huang, D.; Claus, P.; Dengler, R.; Stangel, M.; Petri, S. Therapeutic potential of mesenchymal stromal cells and MSC conditioned medium in Amyotrophic Lateral Sclerosis (ALS)--in vitro evidence from primary motor neuron cultures, NSC-34 cells, astrocytes and microglia. PLoS ONE 2013, 8, e72926. [Google Scholar] [CrossRef]
- Atkin, J.D.; Farg, M.A.; Turner, B.J.; Tomas, D.; Lysaght, J.A.; Nunan, J.; Rembach, A.; Nagley, P.; Beart, P.M.; Cheema, S.S.; et al. Induction of the unfolded protein response in familial amyotrophic lateral sclerosis and association of protein-disulfide isomerase with superoxide dismutase 1. J. Biol. Chem. 2006, 281, 30152–30165. [Google Scholar] [CrossRef] [PubMed]
- Evans, C.S.; Holzbaur, E.L.F. Autophagy and mitophagy in ALS. Neurobiol. Dis. 2019, 122, 35–40. [Google Scholar] [CrossRef]
- Taylor, J.P.; Brown Jr, R.H.; Cleveland, D.W. Decoding ALS: From genes to mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Maday, S.; Wallace, K.E.; Holzbaur, E.L.F. Autophagosomes initiate distally and mature during transport toward the cell soma in primary neurons. J. Cell Biol. 2012, 196, 407–417. [Google Scholar] [CrossRef]
- Rudnick, N.D.; Griffey, C.J.; Guarnieri, P.; Gerbino, V.; Wang, X.; Piersaint, J.A.; Tapia, J.C.; Rich, M.M.; Maniatis, T. Distinct roles for motor neuron autophagy early and late in the SOD1G93A mouse model of ALS. Proc. Natl. Acad. Sci. USA 2017, 114, E8294–E8303. [Google Scholar] [CrossRef]
- Johnston, J.A.; Dalton, M.J.; Gurney, M.E.; Kopito, R.R. Formation of high molecular weight complexes of mutant Cu,Zn-superoxide dismutase in a mouse model for familial amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2000, 97, 12571–12576. [Google Scholar] [CrossRef] [PubMed]
- Beal, M.F. Mitochondria and the pathogenesis of ALS. Brain 2000, 123, 1291–1292. [Google Scholar] [CrossRef]
- Siklós, L.; Engelhardt, J.; Harati, Y.; Smith, R.G.; Joó, F.; Appel, S.H. Ultrastructural evidence for altered calcium in motor nerve terminals in amyotrophic lateral sclerosis. Ann. Neurol. 1996, 39, 203–216. [Google Scholar] [CrossRef]
- Okado-Matsumoto, A.; Fridovich, I. Amyotrophic lateral sclerosis: A proposed mechanism. Proc. Natl. Acad. Sci. USA 2002, 99, 9010–9014. [Google Scholar] [CrossRef] [PubMed]
- Bruijn, L.I.; Miller, T.M.; Cleveland, D.W. Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annu. Rev. Neurosci. 2004, 27, 723–749. [Google Scholar] [CrossRef]
- Martin, L.J.; Gertz, B.; Pan, V.; Price, A.C.; Molkentin, J.D.; Chang, Q. The mitochondrial permeability transition pore in motor neurons: Involvement in the pathobiology of ALS mice. Exp. Neurol. 2009, 218, 333–346. [Google Scholar] [CrossRef]
- Martin, L.J. The mitochondrial permeability transition pore: A molecular target for amyotrophic lateral sclerosis therapy. Biochim. Biophys. Acta 2010, 1802, 186–197. [Google Scholar] [CrossRef]
- Higgins, C.M.J.; Jung, C.; Ding, H.; Xu, Z. Mutant Cu, Zn superoxide dismutase that causes motoneuron degeneration is present in mitochondria in the CNS. J. Neurosci. 2002, 22, RC215. [Google Scholar] [CrossRef] [PubMed]
- Guareschi, S.; Cova, E.; Cereda, C.; Ceroni, M.; Donetti, E.; Bosco, D.A.; Trotti, D.; Pasinelli, P. An over-oxidized form of superoxide dismutase found in sporadic amyotrophic lateral sclerosis with bulbar onset shares a toxic mechanism with mutant SOD1. Proc. Natl. Acad. Sci. USA 2012, 109, 5074–5079. [Google Scholar] [CrossRef] [PubMed]
- Bogaert, E.; Van Damme, P.; Van Den Bosch, L.; Robberecht, W. Vascular endothelial growth factor in amyotrophic lateral sclerosis and other neurodegenerative diseases. Muscle Nerve 2006, 34, 391–405. [Google Scholar] [CrossRef]
- Bomsztyk, K.; Denisenko, O.; Ostrowski, J. hnRNP K: One protein multiple processes. Bioessays 2004, 26, 629–638. [Google Scholar] [CrossRef] [PubMed]
- Borchelt, D.R.; Lee, M.K.; Slunt, H.S.; Guarnieri, M.; Xu, Z.S.; Wong, P.C.; Brown, R.H., Jr.; Price, D.L.; Sisodia, S.S.; Cleveland, D.W. Superoxide dismutase 1 with mutations linked to familial amyotrophic lateral sclerosis possesses significant activity. Proc. Natl. Acad. Sci. USA 1994, 91, 8292–8296. [Google Scholar] [CrossRef] [PubMed]
- Borroni, B.; Bonvicini, C.; Alberici, A.; Buratti, E.; Agosti, C.; Archetti, S.; Papetti, A.; Stuani, C.; Di Luca, M.; Gennarelli, M.; et al. Mutation within TARDBP leads to frontotemporal dementia without motor neuron disease. Hum. Mutat. 2009, 30, E974–E983. [Google Scholar] [CrossRef]
- McGeer, P.L.; Itagaki, S.; McGeer, E.G. Expression of the histocompatibility glycoprotein HLA-DR in neurological disease. Acta Neuropathol. 1988, 76, 550–557. [Google Scholar] [CrossRef]
- McGeer, P.L.; McGeer, E.G. History of innate immunity in neurodegenerative disorders. Front. Pharmacol. 2011, 2, 77. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.L.; Itagaki, S.; Tago, H.; McGeer, E.G. Reactive microglia in patients with senile dementia of the Alzheimer type are positive for the histocompatibility glycoprotein HLA-DR. Neurosci. Lett. 1987, 79, 195–200. [Google Scholar] [CrossRef]
- Alexianu, M.E.; Kozovska, M.; Appel, S.H. Immune reactivity in a mouse model of familial ALS correlates with disease progression. Neurology 2001, 57, 1282–1289. [Google Scholar] [CrossRef] [PubMed]
- Pramatarova, A.; Laganière, J.; Roussel, J.; Brisebois, K.; Rouleau, G.A. Neuron-specific expression of mutant superoxide dismutase 1 in transgenic mice does not lead to motor impairment. J. Neurosci. 2001, 21, 3369–3374. [Google Scholar] [CrossRef]
- Qin, L.; Liu, Y.; Wang, T.; Wei, S.J.; Block, M.L.; Wilson, B.; Liu, B.; Hong, J.S. NADPH oxidase mediates lipopolysaccharide-induced neurotoxicity and proinflammatory gene expression in activated microglia. J. Biol. Chem. 2004, 279, 1415–1421. [Google Scholar] [CrossRef] [PubMed]
- Lambeth, J.D. NOX enzymes and the biology of reactive oxygen. Nat. Rev. Immunol. 2004, 4, 181–189. [Google Scholar] [CrossRef]
- Marden, J.J.; Harraz, M.M.; Williams, A.J.; Nelson, K.; Luo, M.; Paulson, H.; Engelhardt, J.F. Redox modifier genes in amyotrophic lateral sclerosis in mice. J. Clin. Investig. 2007, 117, 2913–2919. [Google Scholar] [CrossRef]
- Pasinelli, P.; Brown, R.H. Molecular biology of amyotrophic lateral sclerosis: Insights from genetics. Nat. Rev. Neurosci. 2006, 7, 710–723. [Google Scholar] [CrossRef]
- Barber, S.C.; Mead, R.J.; Shaw, P.J. Oxidative stress in ALS: A mechanism of neurodegeneration and a therapeutic target. Biochim. Biophys. Acta 2006, 1762, 1051–1067. [Google Scholar] [CrossRef]
- Wu, D.C.; Ré, D.B.; Nagai, M.; Ischiropoulos, H.; Przedborski, S. The inflammatory NADPH oxidase enzyme modulates motor neuron degeneration in amyotrophic lateral sclerosis mice. Proc. Natl. Acad. Sci. USA 2006, 103, 12132–12137. [Google Scholar] [CrossRef]
- Harraz, M.M.; Marden, J.J.; Zhou, W.; Zhang, Y.; Williams, A.; Sharov, V.S.; Nelson, K.; Luo, M.; Paulson, H.; Schöneich, C.; et al. SOD1 mutations disrupt redox-sensitive Rac regulation of NADPH oxidase in a familial ALS model. J. Clin. Investig. 2008, 118, 659–670. [Google Scholar] [CrossRef]
- Harraz, M.M.; Park, A.; Abbott, D.; Zhou, W.; Zhang, Y.; Engelhardt, J.F. MKK6 phosphorylation regulates production of superoxide by enhancing Rac GTPase activity. Antioxid. Redox Signal 2007, 9, 1803–1813. [Google Scholar] [CrossRef]
- Ransohoff, R.M. How neuroinflammation contributes to neurodegeneration. Science 2016, 353, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Cirulli, E.T.; Lasseigne, B.N.; Petrovski, S.; Sapp, P.C.; Dion, P.A.; Leblond, C.S.; Couthouis, J.; Lu, Y.F.; Wang, Q.; Krueger, B.J.; et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 2015, 347, 1436–1441. [Google Scholar] [CrossRef]
- Freischmidt, A.; Wieland, T.; Richter, B.; Ruf, W.; Schaeffer, V.; Müller, K.; Marroquin, N.; Nordin, F.; Hübers, A.; Weydt, P.; et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat. Neurosci. 2015, 18, 631–636. [Google Scholar] [CrossRef]
- Maruyama, H.; Morino, H.; Ito, H.; Izumi, Y.; Kato, H.; Watanabe, Y.; Kinoshita, Y.; Kamada, M.; Nodera, H.; Suzuki, H.; et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature 2010, 465, 223–226. [Google Scholar] [CrossRef] [PubMed]
- Dobson-Stone, C.; Hallupp, M.; Bartley, L.; Shepherd, C.E.; Halliday, G.M.; Schofield, P.R.; Hodges, J.R.; Kwok, J.B. C9ORF72 repeat expansion in clinical and neuropathologic frontotemporal dementia cohorts. Neurology 2012, 79, 995–1001. [Google Scholar] [CrossRef]
- Ono, S.; Hu, J.; Shimizu, N.; Imai, T.; Nakagawa, H. Increased interleukin-6 of skin and serum in amyotrophic lateral sclerosis. J. Neurol. Sci. 2001, 187, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Henkel, J.S.; Beers, D.R.; Wen, S.; Rivera, A.L.; Toennis, K.M.; Appel, J.E.; Zhao, W.; Moore, D.H.; Powell, S.Z.; Appel, S.H. Regulatory T-lymphocytes mediate amyotrophic lateral sclerosis progression and survival. EMBO Mol. Med. 2013, 5, 64–79. [Google Scholar] [CrossRef]
- Chiu, I.M.; Phatnani, H.; Kuligowski, M.; Tapia, J.C.; Carrasco, M.A.; Zhang, M.; Maniatis, T.; Carroll, M.C. Activation of innate and humoral immunity in the peripheral nervous system of ALS transgenic mice. Proc. Natl. Acad. Sci. USA 2009, 106, 20960–20965. [Google Scholar] [CrossRef] [PubMed]
- Gasco, S.; Zaragoza, P.; García-Redondo, A.; Calvo, A.C.; Osta, R. Inflammatory and non-inflammatory monocytes as novel prognostic biomarkers of survival in SOD1G93A mouse model of Amyotrophic Lateral Sclerosis. PLoS ONE 2017, 12, e0184626. [Google Scholar] [CrossRef] [PubMed]
- Paré, B.; Gros-Louis, F. Potential skin involvement in ALS: Revisiting Charcot’s observation—A review of skin abnormalities in ALS. Rev. Neurosci. 2017, 28, 551–572. [Google Scholar] [CrossRef] [PubMed]
- Nardo, G.; Trolese, M.C.; de Vito, G.; Cecchi, R.; Riva, N.; Dina, G.; Heath, P.R.; Quattrini, A.; Shaw, P.J.; Piazza, V.; et al. Immune response in peripheral axons delays disease progression in SOD1(G93A) mice. J. Neuroinflammation 2016, 13, 261. [Google Scholar] [CrossRef]
- Baruch, K.; Deczkowska, A.; David, E.; Castellano, J.M.; Miller, O.; Kertser, A.; Berkutzki, T.; Barnett-Itzhaki, Z.; Bezalel, D.; Wyss-Coray, T.; et al. Aging. Aging-induced type I interferon response at the choroid plexus negatively affects brain function. Science 2014, 346, 89–93. [Google Scholar] [CrossRef]
- Coque, E.; Salsac, C.; Espinosa-Carrasco, G.; Varga, B.; Degauque, N.; Cadoux, M.; Crabé, R.; Virenque, A.; Soulard, C.; Fierle, J.K.; et al. Cytotoxic CD8(+) T lymphocytes expressing ALS-causing SOD1 mutant selectively trigger death of spinal motoneurons. Proc. Natl. Acad. Sci. USA 2019, 116, 2312–2317. [Google Scholar] [CrossRef]
- Sakaguchi, S.; Ono, M.; Setoguchi, R.; Yagi, H.; Hori, S.; Fehervari, Z.; Shimizu, J.; Takahashi, T.; Nomura, T. Foxp3+ CD25+ CD4+ natural regulatory T cells in dominant self-tolerance and autoimmune disease. Immunol. Rev. 2006, 212, 8–27. [Google Scholar] [CrossRef]
- Sheean, R.K.; McKay, F.C.; Cretney, E.; Bye, C.R.; Perera, N.D.; Tomas, D.; Weston, R.A.; Scheller, K.J.; Djouma, E.; Menon, P.; et al. Association of Regulatory T-Cell Expansion With Progression of Amyotrophic Lateral Sclerosis: A Study of Humans and a Transgenic Mouse Model. JAMA Neurol. 2018, 75, 681–689. [Google Scholar] [CrossRef]
- de Candia, P.; Procaccini, C.; Russo, C.; Lepore, M.T.; Matarese, G. Regulatory T cells as metabolic sensors. Immunity 2022, 55, 1981–1992. [Google Scholar] [CrossRef]
- Procaccini, C.; Matarese, G. Regulatory T cells, mTOR kinase, and metabolic activity. Cell Mol. Life Sci. 2012, 69, 3975–3987. [Google Scholar] [CrossRef]
- Tsang, C.K.; Zheng, X. F S. A balancing act: mTOR integrates nutrient signals to regulate redox-dependent growth and survival through SOD1. Mol. Cell Oncol. 2018, 5, e1488372. [Google Scholar] [CrossRef]
- Tsang, C.K.; Chen, M.; Cheng, X.; Qi, Y.; Chen, Y.; Das, I.; Li, X.; Vallat, B.; Fu, L.W.; Qian, C.N.; et al. SOD1 Phosphorylation by mTORC1 Couples Nutrient Sensing and Redox Regulation. Mol. Cell 2018, 70, 502–515.e508. [Google Scholar] [CrossRef]
- Damiano, S.; Sozio, C.; La Rosa, G.; Guida, B.; Faraonio, R.; Santillo, M.; Mondola, P. Metabolism Regulation and Redox State: Insight into the Role of Superoxide Dismutase 1. Int. J. Mol. Sci. 2020, 21, 6606. [Google Scholar] [CrossRef] [PubMed]
- Mondola, P.; Annella, T.; Santillo, M.; Santangelo, F. Evidence for secretion of cytosolic CuZn superoxide dismutase by Hep G2 cells and human fibroblasts. Int. J. Biochem. Cell Biol. 1996, 28, 677–681. [Google Scholar] [CrossRef]
- Mondola, P.; Annella, T.; Serù, R.; Santangelo, F.; Iossa, S.; Gioielli, A.; Santillo, M. Secretion and increase of intracellular CuZn superoxide dismutase content in human neuroblastoma SK-N-BE cells subjected to oxidative stress. Brain Res. Bull. 1998, 45, 517–520. [Google Scholar] [CrossRef] [PubMed]
- Ghaemmaghami, S.; Huh, W.K.; Bower, K.; Howson, R.W.; Belle, A.; Dephoure, N.; O’Shea, E.K.; Weissman, J.S. Global analysis of protein expression in yeast. Nature 2003, 425, 737–741. [Google Scholar] [CrossRef] [PubMed]
- Turner, B.J.; Atkin, J.D.; Farg, M.A.; Zang, D.W.; Rembach, A.; Lopes, E.C.; Patch, J.D.; Hill, A.F.; Cheema, S.S. Impaired extracellular secretion of mutant superoxide dismutase 1 associates with neurotoxicity in familial amyotrophic lateral sclerosis. J. Neurosci. 2005, 25, 108–117. [Google Scholar] [CrossRef]
- Zhao, W.; Beers, D.R.; Henkel, J.S.; Zhang, W.; Urushitani, M.; Julien, J.P.; Appel, S.H. Extracellular mutant SOD1 induces microglial-mediated motoneuron injury. Glia 2010, 58, 231–243. [Google Scholar] [CrossRef]
- Fetrow, J.S.; Siew, N.; Skolnick, J. Structure-based functional motif identifies a potential disulfide oxidoreductase active site in the serine/threonine protein phosphatase-1 subfamily. FASEB J. 1999, 13, 1866–1874. [Google Scholar] [CrossRef]
- Juarez, J.C.; Manuia, M.; Burnett, M.E.; Betancourt, O.; Boivin, B.; Shaw, D.E.; Tonks, N.K.; Mazar, A.P.; Doñate, F. Superoxide dismutase 1 (SOD1) is essential for H2O2-mediated oxidation and inactivation of phosphatases in growth factor signaling. Proc. Natl. Acad. Sci. USA 2008, 105, 7147–7152. [Google Scholar] [CrossRef]
- Park, S.J.; Kim, H.Y.; Kim, H.; Park, S.M.; Joe, E.H.; Jou, I.; Choi, Y.H. Oxidative stress induces lipid-raft-mediated activation of Src homology 2 domain-containing protein-tyrosine phosphatase 2 in astrocytes. Free Radic. Biol. Med. 2009, 46, 1694–1702. [Google Scholar] [CrossRef] [PubMed]
- Santillo, M.; Secondo, A.; Serù, R.; Damiano, S.; Garbi, C.; Taverna, E.; Rosa, P.; Giovedì, S.; Benfenati, F.; Mondola, P. Evidence of calcium- and SNARE-dependent release of CuZn superoxide dismutase from rat pituitary GH3 cells and synaptosomes in response to depolarization. J. Neurochem. 2007, 102, 679–685. [Google Scholar] [CrossRef] [PubMed]
- Floyd, C.L.; Gorin, F.A.; Lyeth, B.G. Mechanical strain injury increases intracellular sodium and reverses Na+/Ca2+ exchange in cortical astrocytes. Glia 2005, 51, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Xiao, B.; Chen, D.; Zhou, Q.; Hang, J.; Zhang, W.; Kuang, Z.; Sun, Z.; Li, L. Glutamate metabotropic receptor 4 (GRM4) inhibits cell proliferation, migration and invasion in breast cancer and is regulated by miR-328-3p and miR-370-3p. BMC Cancer 2019, 19, 891. [Google Scholar] [CrossRef]
- Lewerenz, J.; Hewett, S.J.; Huang, Y.; Lambros, M.; Gout, P.W.; Kalivas, P.W.; Massie, A.; Smolders, I.; Methner, A.; Pergande, M.; et al. The cystine/glutamate antiporter system x(c)(-) in health and disease: From molecular mechanisms to novel therapeutic opportunities. Antioxid. Redox Signal 2013, 18, 522–555. [Google Scholar] [CrossRef]
- Luo, T.; Wu, W.H.; Chen, B.S. NMDA receptor signaling: Death or survival? Front. Biol. 2011, 6, 468–476. [Google Scholar] [CrossRef] [PubMed]
- Rothstein, J.D.; Martin, L.J.; Kuncl, R.W. Decreased Glutamate Transport by the Brain and Spinal Cord in Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 1992, 326, 1464–1468. [Google Scholar] [CrossRef]
- Damiano, S.; Sasso, A.; Accetta, R.; Monda, M.; De Luca, B.; Pavone, L.M.; Belfiore, A.; Santillo, M.; Mondola, P. Effect of Mutated Cu, Zn Superoxide Dismutase (SOD1(G93A)) on Modulation of Transductional Pathway Mediated by M1 Muscarinic Receptor in SK-N-BE and NSC-34 Cells. Front. Physiol. 2018, 9, 611. [Google Scholar] [CrossRef]
- Peggion, C.; Scalcon, V.; Massimino, M.L.; Nies, K.; Lopreiato, R.; Rigobello, M.P.; Bertoli, A. SOD1 in ALS: Taking Stock in Pathogenic Mechanisms and the Role of Glial and Muscle Cells. Antioxidants 2022, 11, 614. [Google Scholar] [CrossRef]
- Zhou, J.; Yi, J.; Fu, R.; Liu, E.; Siddique, T.; Ríos, E.; Deng, H.-X. Hyperactive intracellular calcium signaling associated with localized mitochondrial defects in skeletal muscle of an animal model of amyotrophic lateral sclerosis. J. Biol. Chem. 2010, 285, 705–712. [Google Scholar] [CrossRef]
- Li, A.; Yi, J.; Li, X.; Zhou, J. Physiological Ca2+ Transients Versus Pathological Steady-State Ca2+ Elevation, Who Flips the ROS Coin in Skeletal Muscle Mitochondria. Front. Physiol. 2020, 11, 595800. [Google Scholar] [CrossRef] [PubMed]
- Bonner, T.I.; Young, A.C.; Brann, M.R.; Buckley, N.J. Cloning and expression of the human and rat m5 muscarinic acetylcholine receptor genes. Neuron 1988, 1, 403–410. [Google Scholar] [CrossRef]
- Bonner, T.I.; Buckley, N.J.; Young, A.C.; Brann, M.R. Identification of a family of muscarinic acetylcholine receptor genes. Science 1987, 237, 527–532. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, K.; Fujii, T. Extraneuronal cholinergic system in lymphocytes. Pharmacol. Ther. 2000, 86, 29–48. [Google Scholar] [CrossRef]
- Strong, M.J.; Kesavapany, S.; Pant, H.C. The pathobiology of amyotrophic lateral sclerosis: A proteinopathy? J. Neuropathol. Exp. Neurol. 2005, 64, 649–664. [Google Scholar] [CrossRef]
- Dion, P.A.; Daoud, H.; Rouleau, G.A. Genetics of motor neuron disorders: New insights into pathogenic mechanisms. Nat. Rev. Genet. 2009, 10, 769–782. [Google Scholar] [CrossRef]
- Bosco, D.A.; Morfini, G.; Karabacak, N.M.; Song, Y.; Gros-Louis, F.; Pasinelli, P.; Goolsby, H.; Fontaine, B.A.; Lemay, N.; McKenna-Yasek, D.; et al. Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat. Neurosci. 2010, 13, 1396–1403. [Google Scholar] [CrossRef]
- Forsberg, K.; Jonsson, P.A.; Andersen, P.M.; Bergemalm, D.; Graffmo, K.S.; Hultdin, M.; Jacobsson, J.; Rosquist, R.; Marklund, S.L.; Brännström, T. Novel antibodies reveal inclusions containing non-native SOD1 in sporadic ALS patients. PLoS ONE 2010, 5, e11552. [Google Scholar] [CrossRef] [PubMed]
- Nassif, M.; Matus, S.; Castillo, K.; Hetz, C. Amyotrophic lateral sclerosis pathogenesis: A journey through the secretory pathway. Antioxid. Redox Signal 2010, 13, 1955–1989. [Google Scholar] [CrossRef]
- Turner, B.J.; Atkin, J.D. ER stress and UPR in familial amyotrophic lateral sclerosis. Curr. Mol. Med. 2006, 6, 79–86. [Google Scholar] [CrossRef]
- Damiano, S.; Sasso, A.; De Felice, B.; Terrazzano, G.; Bresciamorra, V.; Carotenuto, A.; Orefice, N.S.; Orefice, G.; Vacca, G.; Belfiore, A.; et al. The IFN-β 1b effect on Cu Zn superoxide dismutase (SOD1) in peripheral mononuclear blood cells of relapsing-remitting multiple sclerosis patients and in neuroblastoma SK-N-BE cells. Brain Res. Bull. 2015, 118, 1–6. [Google Scholar] [CrossRef]
- Lee, A.J.B.; Kittel, T.E.; Kim, R.B.; Bach, T.-N.; Zhang, T.; Mitchell, C.S. Comparing therapeutic modulators of the SOD1 G93A Amyotrophic Lateral Sclerosis mouse pathophysiology. Front. Neurosci. 2023, 16, 1111763. [Google Scholar] [CrossRef] [PubMed]
- Franchina, D.G.; Dostert, C.; Brenner, D. Reactive Oxygen Species: Involvement in T Cell Signaling and Metabolism. Trends Immunol. 2018, 39, 489–502. [Google Scholar] [CrossRef]
- Rigutto, S.; Hoste, C.; Dumont, J.E.; Corvilain, B.; Miot, F.; De Deken, X. Duox1 is the main source of hydrogen peroxide in the rat thyroid cell line PCCl3. Exp. Cell Res. 2007, 313, 3892–3901. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Moalem, G.; Leibowitz-Amit, R.; Yoles, E.; Mor, F.; Cohen, I.R.; Schwartz, M. Autoimmune T cells protect neurons from secondary degeneration after central nervous system axotomy. Nat. Med. 1999, 5, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Ellwardt, E.; Walsh, J.T.; Kipnis, J.; Zipp, F. Understanding the Role of T Cells in CNS Homeostasis. Trends Immunol. 2016, 37, 154–165. [Google Scholar] [CrossRef]
- Xie, L.; Choudhury, G.R.; Winters, A.; Yang, S.H.; Jin, K. Cerebral regulatory T cells restrain microglia/macrophage-mediated inflammatory responses via IL-10. Eur. J. Immunol. 2015, 45, 180–191. [Google Scholar] [CrossRef] [PubMed]
- Korn, T.; Kallies, A. T cell responses in the central nervous system. Nat. Rev. Immunol. 2017, 17, 179–194. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rubino, V.; La Rosa, G.; Pipicelli, L.; Carriero, F.; Damiano, S.; Santillo, M.; Terrazzano, G.; Ruggiero, G.; Mondola, P. Insights on the Multifaceted Roles of Wild-Type and Mutated Superoxide Dismutase 1 in Amyotrophic Lateral Sclerosis Pathogenesis. Antioxidants 2023, 12, 1747. https://doi.org/10.3390/antiox12091747
Rubino V, La Rosa G, Pipicelli L, Carriero F, Damiano S, Santillo M, Terrazzano G, Ruggiero G, Mondola P. Insights on the Multifaceted Roles of Wild-Type and Mutated Superoxide Dismutase 1 in Amyotrophic Lateral Sclerosis Pathogenesis. Antioxidants. 2023; 12(9):1747. https://doi.org/10.3390/antiox12091747
Chicago/Turabian StyleRubino, Valentina, Giuliana La Rosa, Luca Pipicelli, Flavia Carriero, Simona Damiano, Mariarosaria Santillo, Giuseppe Terrazzano, Giuseppina Ruggiero, and Paolo Mondola. 2023. "Insights on the Multifaceted Roles of Wild-Type and Mutated Superoxide Dismutase 1 in Amyotrophic Lateral Sclerosis Pathogenesis" Antioxidants 12, no. 9: 1747. https://doi.org/10.3390/antiox12091747
APA StyleRubino, V., La Rosa, G., Pipicelli, L., Carriero, F., Damiano, S., Santillo, M., Terrazzano, G., Ruggiero, G., & Mondola, P. (2023). Insights on the Multifaceted Roles of Wild-Type and Mutated Superoxide Dismutase 1 in Amyotrophic Lateral Sclerosis Pathogenesis. Antioxidants, 12(9), 1747. https://doi.org/10.3390/antiox12091747