


Vutiglabridin Alleviates Cellular Senescence with Metabolic Regulation and Circadian Clock in Human Dermal Fibroblasts

 , , , , ,
, , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Cell Growth Assay
2.3. Senescence Associated-β-Galactosidase (SA-β-gal) Assay
2.4. RNA Extraction and cDNA Synthesis
2.5. Quantitative Real-Time PCR
2.6. Extracellular Flux Assays
2.7. Confocal Microscopy and Electron Microscopy
2.8. Western Blot
2.9. NAD+ and NADH Measurement
2.10. Lentivirus Production
2.11. BMAL1Luc HDFs Stable Cell Line Production
2.12. Circadian Clock Synchronization and Real-Time Luciferase Monitoring Assay
2.13. Cosinor Analysis
3. Results
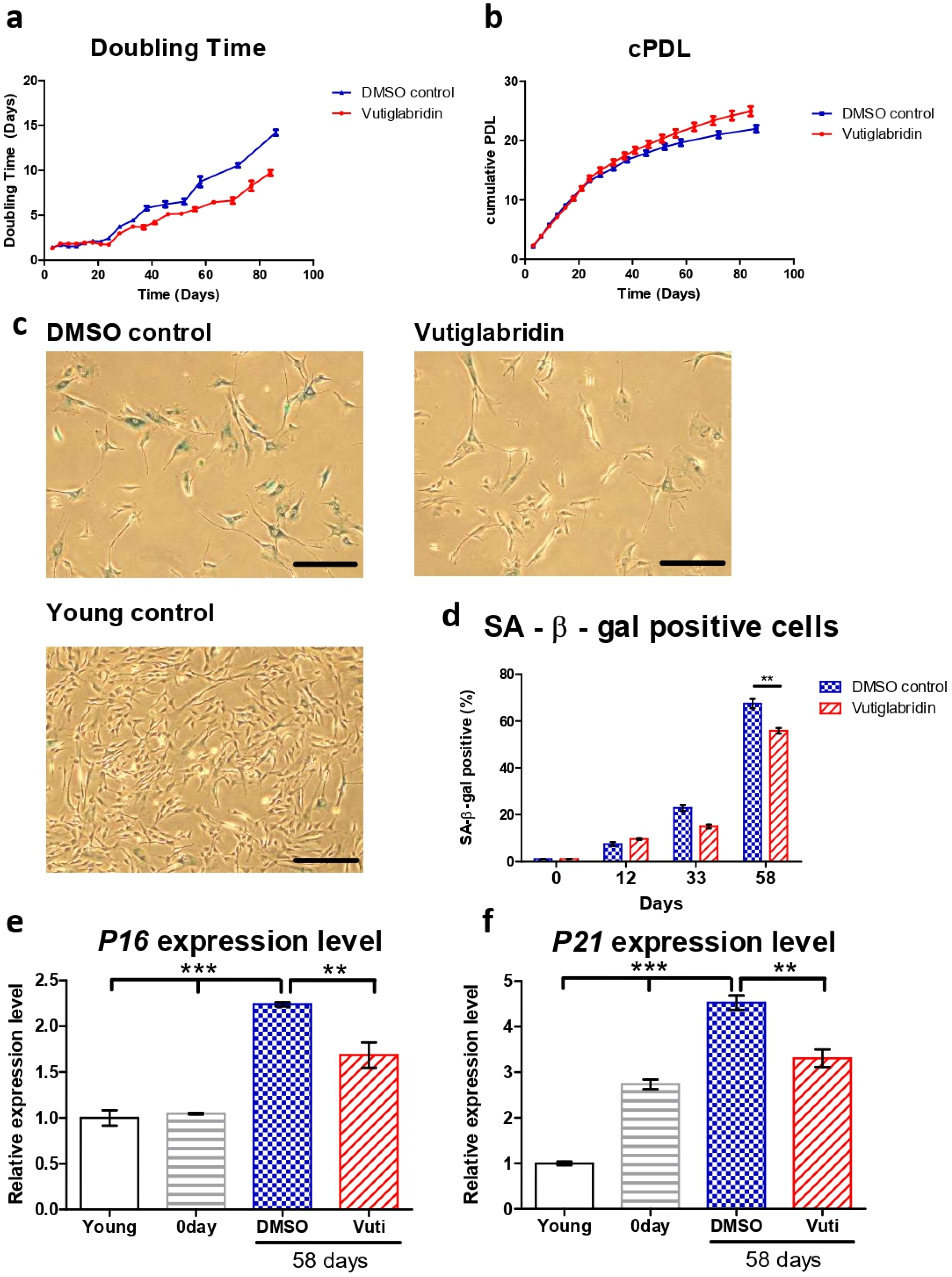
3.1. Vutiglabridin Treatment Alleviates Replicative Senescence of HDFs
3.2. Long-Term Culture with Vutiglabridin Reduces Mitochondrial Dysfunction in High-Passage HDFs
3.3. Long-Term Culture with Vutiglabridin Modulates the Expression of the Metabolic Regulatory Protein in High-Passage HDFs
3.4. Long-Term Culture with Vutiglabridin Alleviates the Senescent Phenotype of the Circadian Clock in High-Passage HDFs
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Childs, B.G.; Durik, M.; Baker, D.J.; van Deursen, J.M. Cellular senescence in aging and age-related disease: From mechanisms to therapy. Nat. Med. 2015, 21, 1424–1435. [Google Scholar] [CrossRef] [PubMed]
- Kudlova, N.; De Sanctis, J.B.; Hajduch, M. Cellular Senescence: Molecular Targets, Biomarkers, and Senolytic Drugs. Int. J. Mol. Sci. 2022, 23, 4168. [Google Scholar] [CrossRef]
- Kumari, R.; Jat, P. Mechanisms of Cellular Senescence: Cell Cycle Arrest and Senescence Associated Secretory Phenotype. Front. Cell Dev. Biol. 2021, 9, 485. [Google Scholar] [CrossRef] [PubMed]
- Di Micco, R.; Krizhanovsky, V.; Baker, D.; di Fagagna, F.D. Cellular senescence in ageing: From mechanisms to therapeutic opportunities. Nat. Rev. Mol. Cell Biol. 2021, 22, 75–95. [Google Scholar] [CrossRef]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O.; et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef]
- Stein, G.H.; Drullinger, L.F.; Soulard, A.; Dulić, V. Differential Roles for Cyclin-Dependent Kinase Inhibitors p21 and p16 in the Mechanisms of Senescence and Differentiation in Human Fibroblasts. Mol. Cell. Biol. 1999, 19, 2109–2117. [Google Scholar] [CrossRef] [PubMed]
- Lago, J.C.; Puzzi, M.B. The effect of aging in primary human dermal fibroblasts. PLoS ONE 2019, 14, e0219165. [Google Scholar] [CrossRef] [PubMed]
- Colavitti, R.; Finkel, T. Reactive Oxygen Species as Mediators of Cellular Senescence. IUBMB Life 2005, 57, 277–281. [Google Scholar] [CrossRef]
- Vinogradov, A.D.; Grivennikova, V.G. Oxidation of NADH and ROS production by respiratory complex I. Biochim. Biophys. Acta (BBA) Bioenerg. 2016, 1857, 863–871. [Google Scholar] [CrossRef]
- Gallage, S.; Gil, J. Mitochondrial Dysfunction Meets Senescence. Trends Biochem. Sci. 2016, 41, 207–209. [Google Scholar] [CrossRef]
- Alam, F.; Syed, H.; Amjad, S.; Baig, M.; Khan, T.A.; Rehman, R. Interplay between oxidative stress, SIRT1, reproductive and metabolic functions. Curr. Res. Physiol. 2021, 4, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, R.; Reza, H.M.; Shinohara, K.; Nakahata, Y. Cellular senescence and its impact on the circadian clock. J. Biochem. 2021, 171, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, R.; Nakahata, Y.; Shinohara, K.; Bessho, Y. Cellular Senescence Triggers Altered Circadian Clocks With a Prolonged Period and Delayed Phases. Front. Neurosci. 2021, 15, 638122. [Google Scholar] [CrossRef] [PubMed]
- Farhud, D.; Aryan, Z. Circadian Rhythm, Lifestyle and Health: A Narrative Review. Iran. J. Public Health 2018, 47, 1068–1076. [Google Scholar] [PubMed]
- Wilking, M.; Mukhtar, H.; Ahmad, N.; Haddadi, M.; Jahromi, S.R.; Nongthomba, U.; Shivanandappa, T.; Ramesh, S.; Mahasneh, A.A.; Zhao, H.; et al. Circadian Rhythm Connections to Oxidative Stress: Implications for Human Health. Antioxid. Redox Signal. 2013, 19, 192–208. [Google Scholar] [CrossRef]
- Jacobi, D.; Liu, S.; Burkewitz, K.; Kory, N.; Knudsen, N.H.; Alexander, R.K.; Unluturk, U.; Li, X.; Kong, X.; Hyde, A.L.; et al. Hepatic Bmal1 Regulates Rhythmic Mitochondrial Dynamics and Promotes Metabolic Fitness. Cell Metab. 2015, 22, 709–720. [Google Scholar] [CrossRef]
- Greco, C.M.; Sassone–Corsi, P. Circadian blueprint of metabolic pathways in the brain. Nat. Rev. Neurosci. 2019, 20, 71–82. [Google Scholar] [CrossRef]
- Lee, J.; Moulik, M.; Fang, Z.; Saha, P.; Zou, F.; Xu, Y.; Nelson, D.L.; Ma, K.; Moore, D.D.; Yechoor, V.K. Bmal1 and β-Cell Clock Are Required for Adaptation to Circadian Disruption, and Their Loss of Function Leads to Oxidative Stress-Induced β-Cell Failure in Mice. Mol. Cell. Biol. 2013, 33, 2327–2338. [Google Scholar] [CrossRef]
- Kolinjivadi, A.M.; Chong, S.T.; Ngeow, J. Molecular connections between circadian rhythm and genome maintenance pathways. Endocr.-Relat. Cancer 2021, 28, R55–R66. [Google Scholar] [CrossRef]
- Jakubiak, G.K.; Osadnik, K.; Lejawa, M.; Kasperczyk, S.; Osadnik, T.; Pawlas, N. Oxidative Stress in Association with Metabolic Health and Obesity in Young Adults. Oxidative Med. Cell. Longev. 2021, 2021, 9987352. [Google Scholar] [CrossRef]
- Tam, B.T.; Morais, J.A.; Santosa, S. Obesity and ageing: Two sides of the same coin. Obes. Rev. 2020, 21, e12991. [Google Scholar] [CrossRef]
- Lefranc, C.; Friederich-Persson, M.; Palacios-Ramirez, R.; Cat, A.N.D. Mitochondrial oxidative stress in obesity: Role of the mineralocorticoid receptor. J. Endocrinol. 2018, 238, R143–R159. [Google Scholar] [CrossRef]
- Youn, J.-Y.; Siu, K.L.; Lob, H.E.; Itani, H.; Harrison, D.G.; Cai, H. Role of Vascular Oxidative Stress in Obesity and Metabolic Syndrome. Diabetes 2014, 63, 2344–2355. [Google Scholar] [CrossRef] [PubMed]
- Yakes, F.M.; Van Houten, B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc. Natl. Acad. Sci. USA 1997, 94, 514–519. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. A Mitochondrial Paradigm of Metabolic and Degenerative Diseases, Aging, and Cancer: A Dawn for Evolutionary Medicine. Annu. Rev. Genet. 2005, 39, 359–407. [Google Scholar] [CrossRef] [PubMed]
- Rafelski, S.M. Mitochondrial network morphology: Building an integrative, geometrical view. BMC Biol. 2013, 11, 71. [Google Scholar] [CrossRef]
- Chen, H.; Vermulst, M.; Wang, Y.E.; Chomyn, A.; Prolla, T.A.; McCaffery, J.M.; Chan, D.C. Mitochondrial Fusion Is Required for mtDNA Stability in Skeletal Muscle and Tolerance of mtDNA Mutations. Cell 2010, 141, 280–289. [Google Scholar] [CrossRef]
- Vaya, J.; Belinky, P.A.; Aviram, M. Antioxidant Constituents from Licorice Roots: Isolation, Structure Elucidation and Antioxidative Capacity Toward LDL Oxidation. Free Radic. Biol. Med. 1997, 23, 302–313. [Google Scholar] [CrossRef]
- Lee, J.-W.; Choe, S.S.; Jang, H.; Kim, J.; Jeong, H.W.; Jo, H.; Jeong, K.-H.; Tadi, S.; Park, M.G.; Kwak, T.H.; et al. AMPK activation with glabridin ameliorates adiposity and lipid dysregulation in obesity. J. Lipid Res. 2012, 53, 1277–1286. [Google Scholar] [CrossRef]
- Choi, L.S.; Jo, I.G.; Kang, K.S.; Im, J.H.; Kim, J.; Chung, J.W.; Yoo, S.-K. Discovery and preclinical efficacy of HSG4112, a synthetic structural analog of glabridin, for the treatment of obesity. Int. J. Obes. 2020, 45, 130–142. [Google Scholar] [CrossRef]
- Shin, G.-C.; Lee, H.M.; Kim, N.Y.; Yoo, S.-K.; Park, Y.S.; Park, H.S.; Ryu, D.; Kim, K.P.; Kim, K.-H. Synthetic glabridin derivatives mitigate steatohepatitis in a diet-induced biopsy-confirmed non-alcoholic steatosis hepatitis mouse model through paraoxonase-2. BioRxiv 2021, 462722. [Google Scholar] [CrossRef]
- van Deursen, J.M. The role of senescent cells in ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Itahana, K.; Itahana, Y.; Dimri, G.P. Colorimetric detection of senescence-associated β galactosidase. Methods Mol. Biol. 2013, 965, 143–156. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.A.; Fleury-Olela, F.; Nagoshi, E.; Hauser, C.; Juge, C.; Meier, C.A.; Chicheportiche, R.; Dayer, J.-M.; Albrecht, U.; Schibler, U. The Period Length of Fibroblast Circadian Gene Expression Varies Widely among Human Individuals. PLoS Biol. 2005, 3, e338. [Google Scholar] [CrossRef] [PubMed]
- Mookerjee, S.A.; Brand, M.D. Measurement and Analysis of Extracellular Acid Production to Determine Glycolytic Rate. J. Vis. Exp. 2015, 106, e53464. [Google Scholar] [CrossRef]
- Divakaruni, A.S.; Paradyse, A.; Ferrick, D.A.; Murphy, A.N.; Jastroch, M. Analysis and Interpretation of Microplate-Based Oxygen Consumption and pH Data. Methods Enzymol. 2014, 547, 309–354. [Google Scholar] [CrossRef] [PubMed]
- James, E.L.; Michalek, R.D.; Pitiyage, G.N.; de Castro, A.M.; Vignola, K.S.; Jones, J.; Mohney, R.P.; Karoly, E.D.; Prime, S.S.; Parkinson, E.K. Senescent Human Fibroblasts Show Increased Glycolysis and Redox Homeostasis with Extracellular Metabolomes That Overlap with Those of Irreparable DNA Damage, Aging, and Disease. J. Proteome Res. 2015, 14, 1854–1871. [Google Scholar] [CrossRef]
- Tanaka, H.; Igata, T.; Etoh, K.; Koga, T.; Takebayashi, S.; Nakao, M. The NSD2/WHSC1/MMSET methyltransferase prevents cellular senescence-associated epigenomic remodeling. Aging Cell 2020, 19, e13173. [Google Scholar] [CrossRef]
- Son, J.M.; Sarsour, E.H.; Balaraju, A.K.; Fussell, J.; Kalen, A.L.; Wagner, B.A.; Buettner, G.R.; Goswami, P.C. Mitofusin 1 and optic atrophy 1 shift metabolism to mitochondrial respiration during aging. Aging Cell 2017, 16, 1136–1145. [Google Scholar] [CrossRef]
- Valente, A.J.; Maddalena, L.A.; Robb, E.L.; Moradi, F.; Stuart, J.A. A simple ImageJ macro tool for analyzing mitochondrial network morphology in mammalian cell culture. Acta Histochem. 2017, 119, 315–326. [Google Scholar] [CrossRef]
- Riahi, Y.; Cohen, G.; Shamni, O.; Sasson, S. Signaling and cytotoxic functions of 4-hydroxyalkenals. Am. J. Physiol. Metab. 2010, 299, E879–E886. [Google Scholar] [CrossRef] [PubMed]
- Flor, A.C.; Kron, S.J. Lipid-derived reactive aldehydes link oxidative stress to cell senescence. Cell Death Dis. 2016, 7, e2366. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.H.; Zou, Y.; Kim, D.H.; Kim, N.D.; Yu, B.P.; Chung, H.Y. Proteomic Analysis of Nitrated and 4-Hydroxy-2-Nonenal-Modified Serum Proteins During Aging. J. Gerontol. Ser. A 2006, 61, 332–338. [Google Scholar] [CrossRef] [PubMed]
- Alers, S.; Löffler, A.S.; Wesselborg, S.; Stork, B. Role of AMPK-mTOR-Ulk1/2 in the Regulation of Autophagy: Cross Talk, Shortcuts, and Feedbacks. Mol. Cell. Biol. 2012, 32, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Onken, B.; Driscoll, M. Metformin Induces a Dietary Restriction–Like State and the Oxidative Stress Response to Extend C. elegans Healthspan via AMPK, LKB1, and SKN-1. PLoS ONE 2010, 5, e8758. [Google Scholar] [CrossRef] [PubMed]
- Manco, G.; Porzio, E.; Carusone, T.M. Human Paraoxonase-2 (PON2): Protein Functions and Modulation. Antioxidants 2021, 10, 256. [Google Scholar] [CrossRef]
- Wiley, C.D.; Velarde, M.C.; Lecot, P.; Liu, S.; Sarnoski, E.A.; Freund, A.; Shirakawa, K.; Lim, H.W.; Davis, S.S.; Ramanathan, A.; et al. Mitochondrial Dysfunction Induces Senescence with a Distinct Secretory Phenotype. Cell Metab. 2016, 23, 303–314. [Google Scholar] [CrossRef]
- Correia-Melo, C.; Birch, J.; Fielder, E.; Rahmatika, D.; Taylor, J.; Chapman, J.; Lagnado, A.; Carroll, B.M.; Miwa, S.; Richardson, G.; et al. Rapamycin improves healthspan but not inflammaging in nfκb1−/− mice. Aging Cell 2018, 18, e12882. [Google Scholar] [CrossRef]
- Algire, C.; Moiseeva, O.; Deschênes-Simard, X.; Amrein, L.; Petruccelli, L.; Birman, E.; Viollet, B.; Ferbeyre, G.; Pollak, M.N. Metformin Reduces Endogenous Reactive Oxygen Species and Associated DNA Damage. Cancer Prev. Res. 2012, 5, 536–543. [Google Scholar] [CrossRef]
- Cheng, X.-Y.; Li, Y.-Y.; Huang, C.; Li, J.; Yao, H.-W. AMP-activated protein kinase reduces inflammatory responses and cellular senescence in pulmonary emphysema. Oncotarget 2017, 8, 22513–22523. [Google Scholar] [CrossRef]
- Um, J.H.; Yang, S.; Yamazaki, S.; Kang, H.; Viollet, B.; Foretz, M.; Chung, J.H. Activation of 5′-AMP-activated Kinase with Diabetes Drug Metformin Induces Casein Kinase Iɛ (CKIɛ)-dependent Degradation of Clock Protein mPer2. J. Biol. Chem. 2007, 282, 20794–20798. [Google Scholar] [CrossRef]
- Massudi, H.; Grant, R.; Braidy, N.; Guest, J.; Farnsworth, B.; Guillemin, G.J. Age-Associated Changes In Oxidative Stress and NAD+ Metabolism In Human Tissue. PLoS ONE 2012, 7, e42357. [Google Scholar] [CrossRef] [PubMed]
- Amorim, J.A.; Coppotelli, G.; Rolo, A.P.; Palmeira, C.M.; Ross, J.M.; Sinclair, D.A. Mitochondrial and metabolic dysfunction in ageing and age-related diseases. Nat. Rev. Endocrinol. 2022, 18, 243–258. [Google Scholar] [CrossRef] [PubMed]
- Nohara, K.; Mallampalli, V.; Nemkov, T.; Wirianto, M.; Yang, J.; Ye, Y.; Sun, Y.; Han, L.; Esser, K.A.; Mileykovskaya, E.; et al. Nobiletin fortifies mitochondrial respiration in skeletal muscle to promote healthy aging against metabolic challenge. Nat. Commun. 2019, 10, 3923. [Google Scholar] [CrossRef] [PubMed]
- Halling, J.F.; Pilegaard, H. PGC-1α-mediated regulation of mitochondrial function and physiological implications. Appl. Physiol. Nutr. Metab. 2020, 45, 927–936. [Google Scholar] [CrossRef]
- Lin, J.D.; Liu, C.; Li, S. Integration of energy metabolism and the mammalian clock. Cell Cycle 2008, 7, 453–457. [Google Scholar] [CrossRef]
- Li, S.; Lin, J.D. Transcriptional control of circadian metabolic rhythms in the liver. Diabetes Obes. Metab. 2015, 17 (Suppl. 1), 33–38. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heo, J.-W.; Lee, H.-E.; Lee, J.; Choi, L.S.; Shin, J.; Mun, J.-Y.; Park, H.-S.; Park, S.-C.; Nam, C.-H. Vutiglabridin Alleviates Cellular Senescence with Metabolic Regulation and Circadian Clock in Human Dermal Fibroblasts. Antioxidants 2024, 13, 109. https://doi.org/10.3390/antiox13010109
Heo J-W, Lee H-E, Lee J, Choi LS, Shin J, Mun J-Y, Park H-S, Park S-C, Nam C-H. Vutiglabridin Alleviates Cellular Senescence with Metabolic Regulation and Circadian Clock in Human Dermal Fibroblasts. Antioxidants. 2024; 13(1):109. https://doi.org/10.3390/antiox13010109
Chicago/Turabian StyleHeo, Jin-Woong, Hye-Eun Lee, Jimin Lee, Leo Sungwong Choi, Jaejin Shin, Ji-Young Mun, Hyung-Soon Park, Sang-Chul Park, and Chang-Hoon Nam. 2024. "Vutiglabridin Alleviates Cellular Senescence with Metabolic Regulation and Circadian Clock in Human Dermal Fibroblasts" Antioxidants 13, no. 1: 109. https://doi.org/10.3390/antiox13010109
APA StyleHeo, J. -W., Lee, H. -E., Lee, J., Choi, L. S., Shin, J., Mun, J. -Y., Park, H. -S., Park, S. -C., & Nam, C. -H. (2024). Vutiglabridin Alleviates Cellular Senescence with Metabolic Regulation and Circadian Clock in Human Dermal Fibroblasts. Antioxidants, 13(1), 109. https://doi.org/10.3390/antiox13010109