
Cellular Pre-Adaptation to the High O2 Concentration Used in Standard Cell Culture Confers Resistance to Subsequent H2O2-Induced Cell Death



Abstract
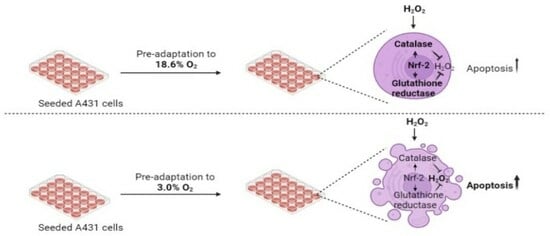
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Methods
2.1. In Vitro Culture of A431 Cells under Standard Cell Culture Oxygen Conditions Compared to Physioxia
2.2. Measurement of the Oxygen Concentration in Experimental Reagents
2.3. Cell Death Analysis by Flow Cytometry
2.4. Detection of Lipid Peroxidation
2.5. Measurement of H2O2 by Amplex Red
2.6. Whole Cell and Nuclear Lysis
2.7. Catalase Activity
2.8. Superoxide Dismutase Activity
2.9. Glutathione Reductase Activity
2.10. Glutathione Peroxidase Activity
2.11. Detection of Transcription Factors (Hypoxia-Inducible Factor-1α and Nrf-2) and Catalase Protein by Western Blotting
2.12. Statistical Analysis
3. Results
3.1. Cells Pre-Adapted to 18.6% O2 Are Resistant to H2O2-Induced Cell Death Compared to Cells Pre-Adapted to 3.0% O2


3.2. Cells Pre-Adapted to 18.6% O2 Are Resistant to H2O2- and Cumene Hydroperoxide-Induced Lipid Peroxidation Compared to Cells Pre-Adapted to 3.0% O2
3.3. Cells Pre-Adapted to 18.6% O2 Exhibit Higher Antioxidant Enzymatic Activities of Catalase and Glutathione Reductase Compared to Cells Pre-Adapted to 3.0% O2
3.4. Cells Pre-Adapted to 18.6% O2 Exhibit a Higher Level of Catalase Protein, and a Higher H2O2 Concentration, Compared to Cells Pre-Adapted to 3.0% O2 for 96 h
3.5. Cells Pre-Adapted to 18.6% O2 Exhibit a Higher Level of Nuclear Nrf-2 Protein Compared to Cells Pre-Adapted to 3.0% O2 for 96 h
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sies, H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biol. 2017, 11, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Konyalioglu, S.; Armagan, G.; Yalcin, A.; Atalayin, C.; Dagci, T. Effects of resveratrol on hydrogen peroxide-induced oxidative stress in embryonic neural stem cells. Neural Regen. Res. 2013, 8, 485. [Google Scholar] [PubMed]
- Gardner, A.M.; Xu, F.H.; Fady, C.; Jacoby, F.J.; Duffey, D.C.; Tu, Y.; Lichtenstein, A. Apoptotic vs. Nonapoptotic Cytotoxicity Induced by Hydrogen Peroxide. Free Radic. Biol. Med. 1997, 22, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, L.; Lu, H. Preservation of hydrogen peroxide-induced oxidative damage in HepG-2 cells by rice protein hydrolysates pretreated with electron beams. Sci. Rep. 2020, 10, 8415. [Google Scholar] [CrossRef]
- Lee, D.; Park, S.; Bae, S. Hydrogen peroxide-activatable antioxidant prodrug as a targeted therapeutic agent for ischemia-reperfusion injury. Sci. Rep. 2015, 5, 16592. [Google Scholar] [CrossRef]
- Roy, P.; Reavey, E.; Rayne, M.; Roy, S.; Abed El Baky, M.; Ishii, Y.; Bartholomew, C. Enhanced sensitivity to hydrogen peroxide-induced apoptosis in Evi1 transformed Rat1 fibroblasts due to repression of carbonic anhydrase III. FEBS J. 2010, 277, 441–452. [Google Scholar] [CrossRef]
- Li, Z.; Zhao, J.; Li, Q.; Yang, W.; Song, Q.; Li, W.; Liu, J. KLF4 promotes hydrogen-peroxide-induced apoptosis of chronic myeloid leukemia cells involving the bcl-2/bax pathway. Cell Stress Chaperones 2010, 15, 905–912. [Google Scholar] [CrossRef] [PubMed]
- Pallepati, P.; Averill-Bates, D.A. Mild thermotolerance induced at 40 °C protects HeLa cells against activation of death receptor-mediated apoptosis by hydrogen peroxide. Free Radic. Biol. Med. 2011, 50, 667–679. [Google Scholar] [CrossRef]
- Bejarano, I.; Espino, J.; Marchena, A.M.; Barriga, C.; Paredes, S.D.; Rodríguez, A.B.; Pariente, J.A. Melatonin enhances hydrogen peroxide-induced apoptosis in human promyelocytic leukaemia HL-60 cells. Mol. Cell. Biochem. 2011, 353, 167–176. [Google Scholar] [CrossRef]
- Tochigi, M.; Inoue, T.; Suzuki-Karasaki, M.; Ochiai, T.; Ra, C.; Suzuki-Karasaki, Y. Hydrogen peroxide induces cell death in human TRAIL-resistant melanoma through intracellular superoxide generation. Int. J. Oncol. 2013, 42, 863–872. [Google Scholar] [CrossRef]
- Lin, K.Y. Molecular damage and responses of oral keratinocyte to hydrogen peroxide. BMC Oral Health 2019, 19, 10. [Google Scholar] [CrossRef] [PubMed]
- Armeni, T.; Battino, M.; Stronati, A.; Pugnaloni, A.; Tomassini, G.; Rosi, G.; Biagini, G.; Principato, G. Total antioxidant capacity and nuclear DNA damage in keratinocytes after exposure to H2O2. Biol. Chem. 2001, 382, 1697–1705. [Google Scholar] [CrossRef]
- Wenger, R.; Kurtcuoglu, V.; Scholz, C.; Marti, H.; Hoogewijs, D. Frequently asked questions in hypoxia research. Hypoxia 2015, 3, 35. [Google Scholar] [CrossRef] [PubMed]
- Keeley, T.P.; Mann, G.E. Defining physiological normoxia for improved translation of cell physiology to animal models and humans. Physiol. Rev. 2019, 99, 161–234. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, D.C.J.; Smerdon, G.R.; Harries, L.W.; Dodd, N.J.F.; Murphy, M.P.; Curnow, A.; Winyard, P.G. Altered cellular redox homeostasis and redox responses under standard oxygen cell culture conditions versus physioxia. Free Radic. Biol. Med. 2018, 126, 322–333. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.; Chrusciel, S.; Fayad-Kobeissi, S.; Dubois-Randé, J.L.; Azuaje, F.; Boczkowski, J.; Motterlini, R.; Foresti, R. Permanent culture of macrophages at physiological oxygen attenuates the antioxidant and immunomodulatory properties of dimethyl fumarate. J. Cell Physiol. 2014, 230, 1128–1138. [Google Scholar] [CrossRef] [PubMed]
- Chapple, S.J.; Keeley, T.P.; Mastronicola, D.; Arno, M.; Vizcay-Barrena, G.; Fleck, R.; Siow, R.C.M.; Mann, G.E. Bach1 differentially regulates distinct Nrf2-dependent genes in human venous and coronary artery endothelial cells adapted to physiological oxygen levels. Free Radic. Biol. Med. 2016, 92, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Dailey, L.A.; Swedrowska, M.; Siow, R.; Mann, G.E.; Vizcay-Barrena, G.; Arno, M.; Mudway, I.S.; Forbes, B. Quantifying the magnitude of the oxygen artefact inherent in culturing airway cells under atmospheric oxygen versus physiological levels. FEBS Lett. 2016, 590, 258–269. [Google Scholar] [CrossRef]
- Villeneuve, L.; Tiede, L.M.; Morsey, B.; Fox, H.S. Quantitative proteomics reveals oxygen-dependent changes in neuronal mitochondria affecting function and sensitivity to rotenone. J. Proteome Res. 2013, 12, 4599–4606. [Google Scholar] [CrossRef]
- Tiede, L.M.; Cook, E.A.; Morsey, B.; Fox, H.S. Oxygen matters: Tissue culture oxygen levels affect mitochondrial function and structure as well as responses to HIV viroproteins. Cell Death Dis. 2011, 2, 246. [Google Scholar] [CrossRef]
- Lee, K.H.; Kim, D.Y.; Kim, W. Cultivation of human skin cells under physiological oxygen concentration modulates expression of skin significant genes and response to hydroxy acids. Biochem. Biophys. Res. Commun. 2021, 551, 161–167. [Google Scholar] [CrossRef]
- Gatenby, R.A.; Kessler, H.B.; Rosenblum, J.S.; Coia, L.R.; Moldofsky, P.J.; Hartz, W.H.; Broder, G.J. Oxygen distribution in squamous cell carcinoma metastases and its relationship to outcome of radiation therapy. Int. J. Radiat. Oncol. Biol. Phys. 1988, 14, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Stücker, M.; Struk, A.; Altmeyer, P.; Herde, M.; Baumgärtl, H.; Lübbers, D.W. The cutaneous uptake of atmospheric oxygen contributes significantly to the oxygen supply of human dermis and epidermis. J. Physiol. 2002, 538, 985–994. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Kang, R.; Tang, D. Ferroptosis by Lipid Peroxidation: The Tip of the Iceberg? Front. Cell Dev. Biol. 2021, 9, 647. [Google Scholar] [CrossRef] [PubMed]
- Weiss, R.H.; Estabrook, R.W. The mechanism of cumene hydroperoxide-dependent lipid peroxidation: The function of cytochrome P-450. Arch. Biochem. Biophys. 1986, 251, 348–360. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.D.; Marshall, T.S.; Kwit, A.D.; Miller Jenkins, L.M.; Dulcey, A.E.; Madigan, J.P.; Pluchino, K.M.; Goldsborough, A.S.; Brimacombe, K.R.; Griffiths, G.L.; et al. Inhibition of glutathione peroxidase mediates the collateral sensitivity of multidrug-resistant cells to tiopronin. J. Biol. Chem. 2014, 289, 21473–21489. [Google Scholar] [CrossRef] [PubMed]
- Behnisch-Cornwell, S.; Laubenstein, G.; Bednarski, P.J. Studies of the inhibitory activities of tiopronin and mercaptosuccinic acid on glutathione peroxidase and their cytotoxic and antioxidant properties. Pharmazie 2019, 74, 536–542. [Google Scholar] [PubMed]
- Tarr, J.M.; Winyard, P.G.; Ryan, B.; Harries, L.W.; Haigh, R.; Viner, N.; Eggleton, P. Extracellular calreticulin is present in the joints of patients with rheumatoid arthritis and inhibits FasL (CD95L)-mediated apoptosis of T cells. Arthritis Rheum. 2010, 62, 2919–2929. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Chu, J.; Chen, H.; Cheng, H.; Su, J.; Wang, X.; Cao, Y.; Tian, S.; Li, Q. Gastrodin Inhibits H2O2-Induced Ferroptosis through Its Antioxidative Effect in Rat Glioma Cell Line C6. Biol. Pharm. Bull. 2020, 43, 480–487. [Google Scholar] [CrossRef]
- Schreiber, E.; Matthias, P.; Müller, M.M.; Schaffner, W. Rapid detection of octamer binding proteins with ‘mini extracts’, prepared from a small number of cells. Nucleic Acids Res. 1989, 17, 6419. [Google Scholar] [CrossRef]
- Li, Y.; Schellhorn, H.E. Rapid kinetic microassay for catalase activity. J. Biomol. Tech. 2007, 18, 185–187. [Google Scholar] [PubMed]
- Winterbourn, C.C. Revisiting the reactions of superoxide with glutathione and other thiols. Arch. Biochem. Biophys. 2016, 595, 68–71. [Google Scholar] [CrossRef] [PubMed]
- Mannervik, B. Chapter 7, unit 7.2: Measurement of glutathione reductase activity. In Current Protocols in Toxicology; Maines, M.D., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 1999. [Google Scholar]
- Ren, Y.; Yeoh, K.W.; Hao, P.; Kon, O.L.; Sze, S.K. Irradiation of Epithelial Carcinoma Cells Upregulates Calcium-Binding Proteins That Promote Survival under Hypoxic Conditions. J. Proteome Res. 2016, 15, 4258–4264. [Google Scholar] [CrossRef] [PubMed]
- Lameijer, L.N.; Ernst, D.; Hopkins, S.L.; Meijer, M.S.; Askes, S.H.C.; Le Dévédec, S.E.; Bonnet, S.A. Red-Light-Activated Ruthenium-Caged NAMPT Inhibitor Remains Phototoxic in Hypoxic Cancer Cells. Angew. Chem. Int. Ed. Engl. 2017, 56, 11549–11553. [Google Scholar] [CrossRef]
- Krymov, S.K.; Scherbakov, A.M.; Dezhenkova, L.G.; Salnikova, D.I.; Solov’eva, S.E.; Sorokin, D.V.; Vullo, D.; De Luca, V.; Capasso, C.; Supuran, C.T.; et al. Indoline-5-Sulfonamides: A Role of the Core in Inhibition of Cancer-Related Carbonic Anhydrases, Antiproliferative Activity and Circumventing of Multidrug Resistance. Pharmaceuticals 2022, 15, 1453. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Hao, P.; Dutta, B.; Cheow, E.S.; Sim, K.H.; Gan, C.S.; Lim, S.K.; Sze, S.K. Hypoxia Modulates A431 Cellular Pathways Association to Tumor Radioresistance and Enhanced Migration Revealed by Comprehensive Proteomic and Functional Studies. Mol. Cell Proteom. 2013, 12, 485. [Google Scholar] [CrossRef]
- Vissers, M.C.M.; Gunningham, S.P.; Morrison, M.J.; Dachs, G.U.; Currie, M.J. Modulation of hypoxia-inducible factor-1 alpha in cultured primary cells by intracellular ascorbate. Free Radic. Biol. Med. 2007, 42, 765–772. [Google Scholar] [CrossRef]
- Mieremet, A.; Vázquez García, A.; Boiten, W. Human skin equivalents cultured under hypoxia display enhanced epidermal morphogenesis and lipid barrier formation. Sci. Rep. 2019, 9, 7811. [Google Scholar] [CrossRef]
- Lardinois, O.M.; Mestdagh, M.M.; Rouxhet, P.G. Reversible inhibition and irreversible inactivation of catalase in presence of hydrogen peroxide. Biochim. Biophys. Acta 1996, 1295, 222–238. [Google Scholar] [CrossRef]
- Ghadermarzi, M.; Moosavi-Movahedi, A.A. Determination of the kinetic parameters for the ‘suicide substrate’ inactivation of bovine liver catalase by hydrogen peroxide. J. Enzym. Inhib. Med. Chem. 1996, 10, 167–175. [Google Scholar] [CrossRef] [PubMed]
- DeLuca, D.C.; Dennis, R.; Smith, W.G. Inactivation of an animal and a fungal catalase by hydrogen peroxide. Arch Biochem. Biophys. 1995, 320, 129–134. [Google Scholar] [CrossRef]
- Kirkman, H.N.; Rolfo, M.; Ferraris, A.M.; Gaetani, G.F. Mechanisms of Protection of Catalase by NADPH: Kinetics and Stoichiometry. J. Biol. Chem. 1999, 274, 13908–13914. [Google Scholar] [CrossRef]
- Pigeolet, E.; Corbisier, P.; Houbion, A.; Lambert, D.; Michiels, C.; Raes, M.; Zachary, M.D.; Remacle, J. Glutathione peroxidase, superoxide dismutase, and catalase inactivation by peroxides and oxygen derived free radicals. Mech. Ageing Dev. 1990, 51, 283–297. [Google Scholar] [CrossRef]
- Escobar, J.A.; Rubio, M.A.; Lissi, E.A. Sod and catalase inactivation by singlet oxygen and peroxyl radicals. Free Radic. Biol. Med. 1996, 20, 285–290. [Google Scholar] [CrossRef]
- Kim, S.Y.; Kwon, O.J.; Park, J.W. Inactivation of catalase and superoxide dismutase by singlet oxygen derived from photoactivated dye. Biochimie 2001, 83, 437–444. [Google Scholar] [CrossRef]
- McCormick, R.; Pearson, T.; Vasilaki, A. Manipulation of environmental oxygen modifies reactive oxygen and nitrogen species generation during myogenesis. Redox Biol. 2016, 8, 243–251. [Google Scholar] [CrossRef]
- Ross, C.; Alston, M.; Bickenbach, J.R.; Aykin-Burns, N. Oxygen tension changes the rate of migration of human skin keratinocytes in an age-related manner. Exp. Dermatol. 2011, 20, 58. [Google Scholar] [CrossRef]
- Lages, Y.M.; Nascimento, J.M.; Lemos, G.A.; Galina, A.; Castilho, L.R.; Rehen, S.K. Low oxygen alters mitochondrial function and response to oxidative stress in human neural progenitor cells. PeerJ 2015, 3, 26713239. [Google Scholar] [CrossRef] [PubMed]
- Rafikov, R.; Kumar, S.; Aggarwal, S.; Hou, Y.; Kangath, A.; Pardo, D.; Fineman, J.R.; Black, S.M. Endothelin-1 stimulates catalase activity through the PKCδ mediated phosphorylation of Serine 167. Free Radic. Biol. Med. 2014, 67, 255. [Google Scholar] [CrossRef] [PubMed]
- Cao, C.; Leng, Y.; Kufe, D. Catalase activity is regulated by c-Abl and Arg in the oxidative stress response. J. Biol. Chem. 2003, 278, 29667–29675. [Google Scholar] [CrossRef] [PubMed]
- Higashimura, Y.; Nakajima, Y.; Yamaji, R.; Harada, N.; Shibasaki, F.; Nakano, Y.; Inui, H. Up-regulation of glyceraldehyde-3-phosphate dehydrogenase gene expression by HIF-1 activity depending on Sp1 in hypoxic breast cancer cells. Arch. Biochem. Biophys. 2011, 509, 1–8. [Google Scholar] [CrossRef]
- Anedda, A.; López-Bernardo, E.; Acosta-Iborra, B.; Suleiman, M.S.; Landázuri, M.O.; Cadenas; S. The transcription factor Nrf2 promotes survival by enhancing the expression of uncoupling protein 3 under conditions of oxidative stress. Free Radic. Biol. Med. 2013, 61, 395–407. [Google Scholar] [CrossRef]
- Suzuki, T.; Muramatsu, A.; Saito, R.; Uchida, K.; Baird, L.; Yamamoto, M. Molecular Mechanism of Cellular Oxidative Stress Sensing by Keap1. Cell Rep. 2019, 28, 746–758. [Google Scholar] [CrossRef]
- Harvey, C.J.; Thimmulappa, R.K.; Singh, A.; Blake, D.J.; Ling, G.; Wakabayashi, N.; Fujii, J.; Myers, A.; Biswal, S. Nrf2-regulated glutathione recycling independent of biosynthesis is critical for cell survival during oxidative stress. Free Radic. Biol. Med. 2009, 46, 443. [Google Scholar] [CrossRef]
- Brigelius-Flohé, R.; Flohé, L. Basic principles and emerging concepts in the redox control of transcription factors. Antioxid. Redox Signal 2011, 15, 2335–2381. [Google Scholar] [CrossRef]
- Kubo, E.; Chhunchha, B.; Singh, P.; Sasaki, H.; Singh, D.P. Sulforaphane reactivates cellular antioxidant defense by inducing Nrf2/ARE/Prdx6 activity during aging and oxidative stress. Sci. Rep. 2017, 7, 14130. [Google Scholar] [CrossRef]
- Warnatz, H.J.; Schmidt, D.; Manke, T.; Piccini, I.; Sultan, M.; Borodina, T.; Balzereit, D.; Wruck, W.; Soldatov, A.; Vingron, M.; et al. The BTB and CNC Homology 1 (BACH1) Target Genes Are Involved in the Oxidative Stress Response and in Control of the Cell Cycle. J. Biol. Chem. 2011, 286, 23521–23532. [Google Scholar] [CrossRef] [PubMed]
- Wiel, C.; Le Gal, K.; Ibrahim, M.X.; Jahangir, C.A.; Kashif, M.; Yao, H.; Ziegler, D.V.; Xu, X.; Ghosh, T.; Mondal, T.; et al. BACH1 Stabilization by Antioxidants Stimulates Lung Cancer Metastasis. Cell 2019, 178, 330–345.e22. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. On the history of oxidative stress: Concept and some aspects of current development. Curr. Opin. Toxicol. 2018, 7, 122–126. [Google Scholar] [CrossRef]
- Clément, M.V.; Ponton, A.; Pervaiz, S. Apoptosis induced by hydrogen peroxide is mediated by decreased superoxide anion concentration and reduction of intracellular milieu. FEBS Lett. 1998, 440, 13–18. [Google Scholar] [CrossRef]
- Troyano, A.; Sancho, P.; Fernández, C.; de Blas, E.; Bernardi, P.; Aller, P. The selection between apoptosis and necrosis is differentially regulated in hyrdrogen peroxide-treated and glutathione-depleted human promonocytic cells. Cell Death Differ. 2003, 10, 889–898. [Google Scholar] [CrossRef]
- Xiang, J.; Wan, C.; Guo, R.; Guo, D. Is Hydrogen Peroxide a Suitable Apoptosis Inducer for All Cell Types? Biomed. Res. Int. 2016, 2016, 7343965. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Nishio, K.; Ogawa, Y.; Kimata, J.; Kinumi, T.; Yoshida, Y.; Noguchi, N.; Niki, E. Turning point in apoptosis/necrosis induced by hydrogen peroxide. Free Radic. Res. 2006, 40, 619–630. [Google Scholar] [CrossRef] [PubMed]
- Whittemore, E.R.; Loo, D.T.; Cotman, C.W. Exposure to hydrogen peroxide induces cell death via apoptosis in cultured rat cortical neurons. Neuroreport 1994, 5, 1485–1488. [Google Scholar] [CrossRef] [PubMed]
- Ransy, C.; Vaz, C.; Lombès, A.; Bouillaud, F. Use of H2O2 to Cause Oxidative Stress, the Catalase Issue. Int. J. Mol. Sci. 2020, 21, 9149. [Google Scholar] [CrossRef] [PubMed]
- Gaikwad, R.; Thangaraj, P.R.; Sen, A.K. Direct and rapid measurement of hydrogen peroxide in human blood using a microfluidic device. Sci. Rep. 2021, 11, 2960. [Google Scholar] [CrossRef] [PubMed]
- Forman, H.J.; Bernardo, A.; Davies, K.J.A. What is the concentration of hydrogen peroxide in blood and plasma? Arch. Biochem. Biophys. 2016, 603, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Forman, H.J.; Skelton, D.C.; Loeb, G.A.; Dorio, R.J. Membrane Permeability and Oxidant Induced Injury; Springer: Boston, MA, USA, 1988; Volume 49. [Google Scholar]
- Hempel, S.L.; Buettner, G.R.; Wessels, D.A.; Galvan, G.M.; O’Malley, Y.Q. Extracellular iron(II) can protect cells from hydrogen peroxide. Arch. Biochem. Biophys. 1996, 330, 401–408. [Google Scholar] [CrossRef]
- Buettner, G.R. The Pecking Order of Free Radicals and Antioxidants: Lipid Peroxidation, α-Tocopherol, and Ascorbate. Arch. Biochem. Biophys. 1993, 300, 535–543. [Google Scholar] [CrossRef]
- Kakuta, K.; Orino, K.; Yamamoto, S.; Watanabe, K. High levels of ferritin and its iron in fetal bovine serum. Comp. Biochem. Physiol. A Physiol. 1997, 118, 165–169. [Google Scholar] [CrossRef]
- Pilgrim, C.R.; McCahill, K.A.; Rops, J.G.; Dufour, J.M.; Russell, K.A.; Koch, T.G. A Review of Fetal Bovine Serum in the Culture of Mesenchymal Stromal Cells and Potential Alternatives for Veterinary Medicine. Front. Vet. Sci. 2022, 9, 859025. [Google Scholar] [CrossRef]
- Halliwell, B. Cell culture, oxidative stress, and antioxidants: Avoiding pitfalls. Biomed. J. 2014, 37, 99–105. [Google Scholar] [CrossRef] [PubMed]
- di Simplicio, P.; Cheeseman, K.H.; Slater, T.F. The reactivity of the SH group of bovine serum albumin with free radicals. Free Radic. Res. Commun. 1991, 14, 253–262. [Google Scholar] [CrossRef]
- Kouoh, F.; Gressier, B.; Luyckx, M.; Brunet, C.; Dine, T.; Cazin, M.; Cazin, J.C. Antioxidant properties of albumin: Effect on oxidative metabolism of human neutrophil granulocytes. Farmaco 1999, 54, 695–699. [Google Scholar] [CrossRef]
- Wang, P.; Wu, J.; Gao, Z.; Li, H. Tyrosine residues of bovine serum albumin play an important role in protecting SH-SY5Y cells against heme/H2O2/NO2—induced damage. Mol. Cell Biochem. 2019, 454, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B.; Gutteridge, J.M.C. The antioxidants of human extracellular fluids. Arch. Biochem. Biophys. 1990, 280, 1–8. [Google Scholar] [CrossRef]
- Halliwell, B. Oxidative stress in cell culture: An under-appreciated problem? FEBS Lett. 2003, 540, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Koo, B.K.; Knoblich, J.A. Human organoids: Model systems for human biology and medicine. Nat. Rev. Mol. Cell Biol. 2020, 21, 571–584. [Google Scholar] [CrossRef] [PubMed]
- Kasurinen, S.; Happo, M.S.; Rönkkö, T.J.; Orasche, J.; Jokiniemi, J.; Kortelainen, M.; Tissari, J.; Zimmermann, R.; Hirvonen, M.R.; Jalava, P.I. Differences between co-cultures and monocultures in testing the toxicity of particulate matter derived from log wood and pellet combustion. PLoS ONE 2018, 13, e0192453. [Google Scholar] [CrossRef]
- Koobotse, M.O.; Schmidt, D.; Holly, J.M.P.; Perks, C.M. Glucose Concentration in Cell Culture Medium Influences the BRCA1-Mediated Regulation of the Lipogenic Action of IGF-I in Breast Cancer Cells. Int. J. Mol. Sci. 2020, 21, 8674. [Google Scholar] [CrossRef]
- Hughes, P.; Marshall, D.; Reid, Y.; Parkes, H.; Gelber, C. The costs of using unauthenticated, over-passaged cell lines: How much more data do we need? Biotechniques 2007, 43, 575–586. [Google Scholar] [CrossRef] [PubMed]
- Place, T.L.; Domann, F.E.; Case, A.J. Limitations of oxygen delivery to cells in culture: An underappreciated problem in basic and translational research. Free Radic. Biol. Med. 2017, 113, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Shah, Y.M.; Xie, L. Hypoxia-Inducible Factors Link Iron Homeostasis and Erythropoiesis. Gastroenterology 2014, 146, 630. [Google Scholar] [CrossRef] [PubMed]
- Gaschler, M.M.; Stockwell, B.R. Lipid peroxidation in cell death. Biochem. Biophys. Res. Commun. 2017, 482, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Peng, F.; Li, J.; Cui, X.; Qiao, X.; Zhu, W. Ferroptosis-Specific Inhibitor Ferrostatin-1 Relieves H2O2-Induced Redox Imbalance in Primary Cardiomyocytes through the Nrf2/ARE Pathway. Dis. Markers 2022, 2022, 4539932. [Google Scholar] [CrossRef]
- Yu, Y.; Yan, Y.; Niu, F.; Wang, Y.; Chen, X.; Su, G.; Liu, Y.; Zhao, X.; Qian, L.; Liu, P.; et al. Ferroptosis: A cell death connecting oxidative stress, inflammation and cardiovascular diseases. Cell Death Discov. 2021, 7, 193. [Google Scholar] [CrossRef]
- Choi, S.Y.; Kwon, H.Y.; Kwon, O.B.; Eum, W.S.; Kang, J.H. Fragmentation of human ceruloplasmin induced by hydrogen peroxide. Biochimie 2000, 82, 175–180. [Google Scholar] [CrossRef]
- Pham, A.N.; Xing, G.; Miller, C.J.; Waite, T.D. Fenton-like copper redox chemistry revisited: Hydrogen peroxide and superoxide mediation of copper-catalyzed oxidant production. J. Catal. 2013, 301, 54–64. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, L.; Zhou, F. Cuproptosis: A new form of programmed cell death. Cell. Mol. Immunol. 2022, 19, 867–868. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jordan, J.B.; Smallwood, M.J.; Smerdon, G.R.; Winyard, P.G. Cellular Pre-Adaptation to the High O2 Concentration Used in Standard Cell Culture Confers Resistance to Subsequent H2O2-Induced Cell Death. Antioxidants 2024, 13, 269. https://doi.org/10.3390/antiox13030269
Jordan JB, Smallwood MJ, Smerdon GR, Winyard PG. Cellular Pre-Adaptation to the High O2 Concentration Used in Standard Cell Culture Confers Resistance to Subsequent H2O2-Induced Cell Death. Antioxidants. 2024; 13(3):269. https://doi.org/10.3390/antiox13030269
Chicago/Turabian StyleJordan, Jack B., Miranda J. Smallwood, Gary R. Smerdon, and Paul G. Winyard. 2024. "Cellular Pre-Adaptation to the High O2 Concentration Used in Standard Cell Culture Confers Resistance to Subsequent H2O2-Induced Cell Death" Antioxidants 13, no. 3: 269. https://doi.org/10.3390/antiox13030269
APA StyleJordan, J. B., Smallwood, M. J., Smerdon, G. R., & Winyard, P. G. (2024). Cellular Pre-Adaptation to the High O2 Concentration Used in Standard Cell Culture Confers Resistance to Subsequent H2O2-Induced Cell Death. Antioxidants, 13(3), 269. https://doi.org/10.3390/antiox13030269