

Reactive Oxygen Species Signaling and Oxidative Stress: Transcriptional Regulation and Evolution


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
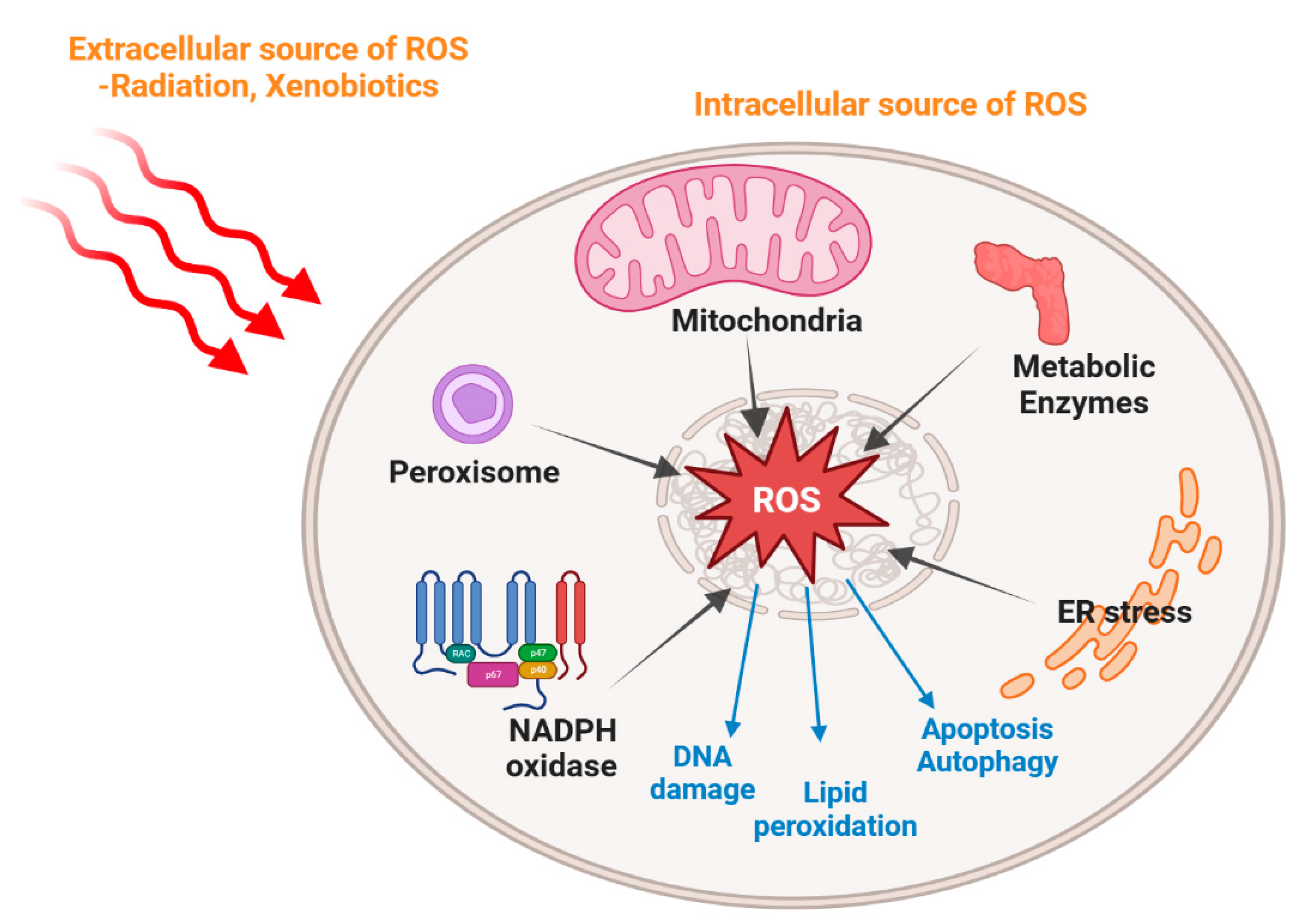
2. The Origins of ROS and Oxidative Stress
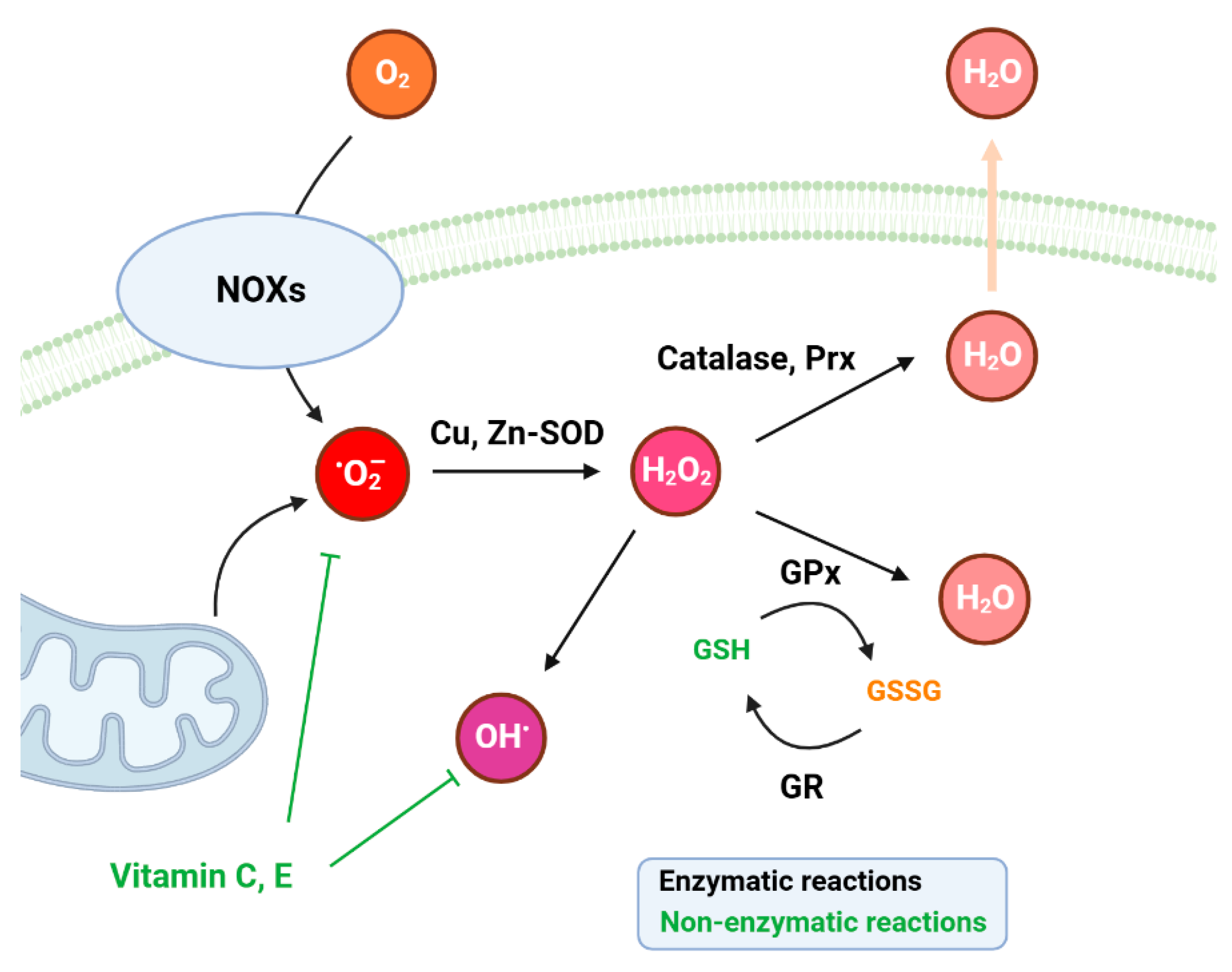
3. Intracellular ROS Balance and Antioxidant Systems
3.1. Cellular Antioxidative Systems
3.2. Reactive Sulfur Species as Antioxidants
3.3. Role of Nitric Oxide in Oxidative Stress
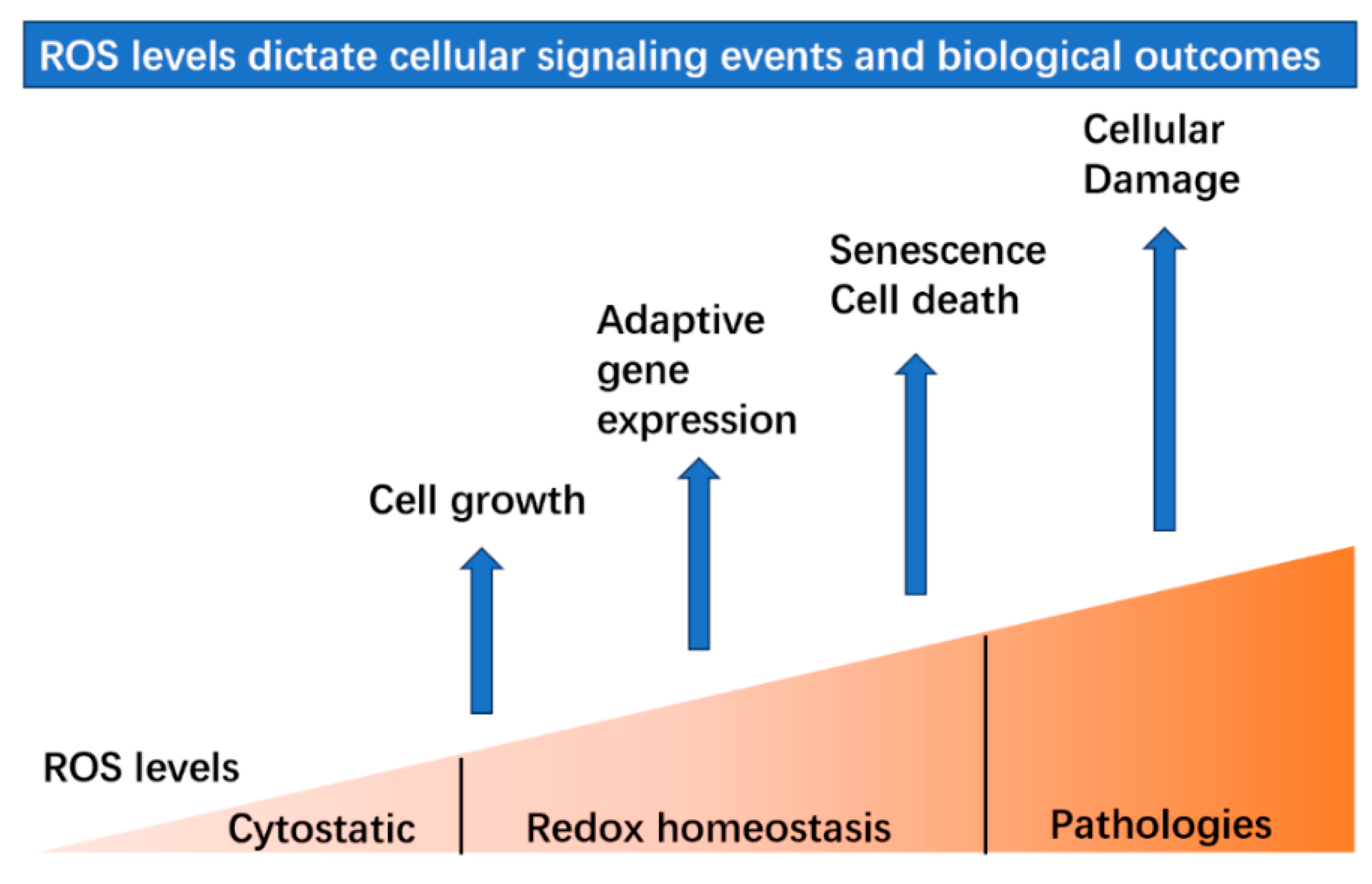
4. ROS Serve as Signaling Molecules
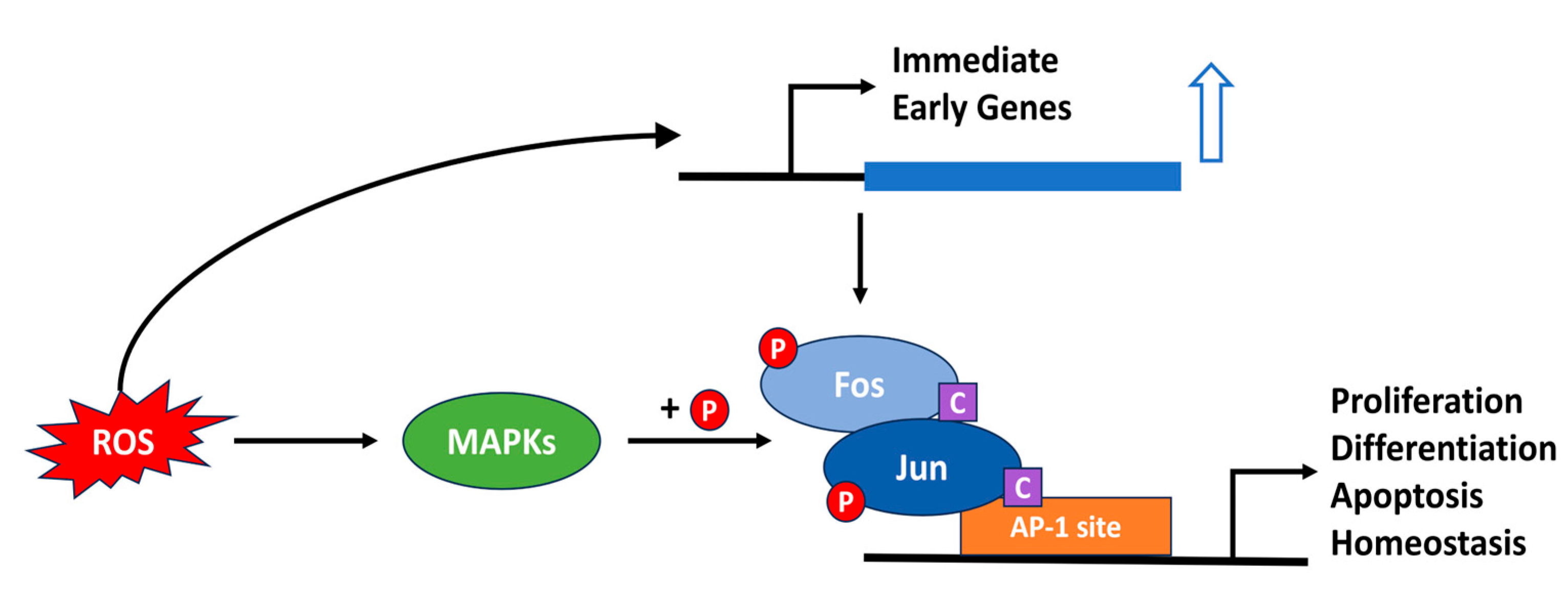
4.1. AP-1
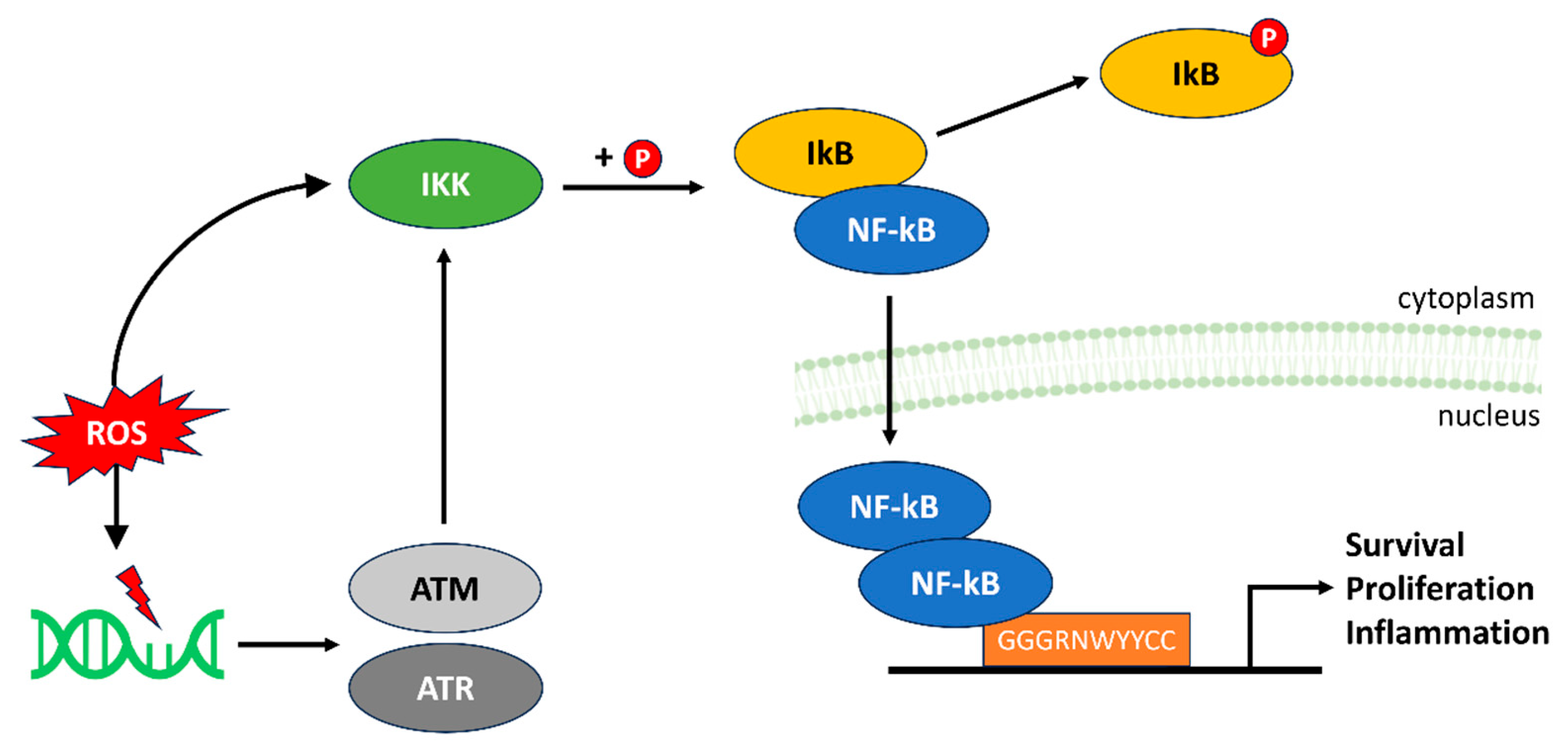
4.2. NF-kB
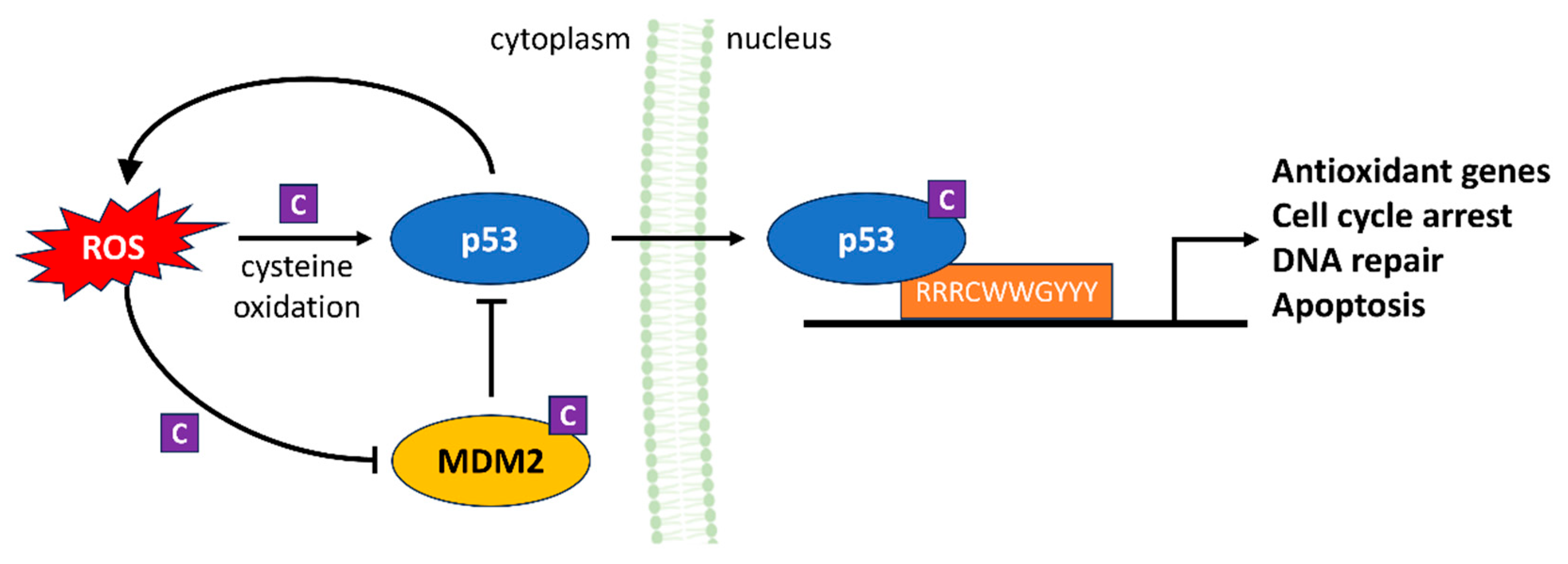
4.3. p53
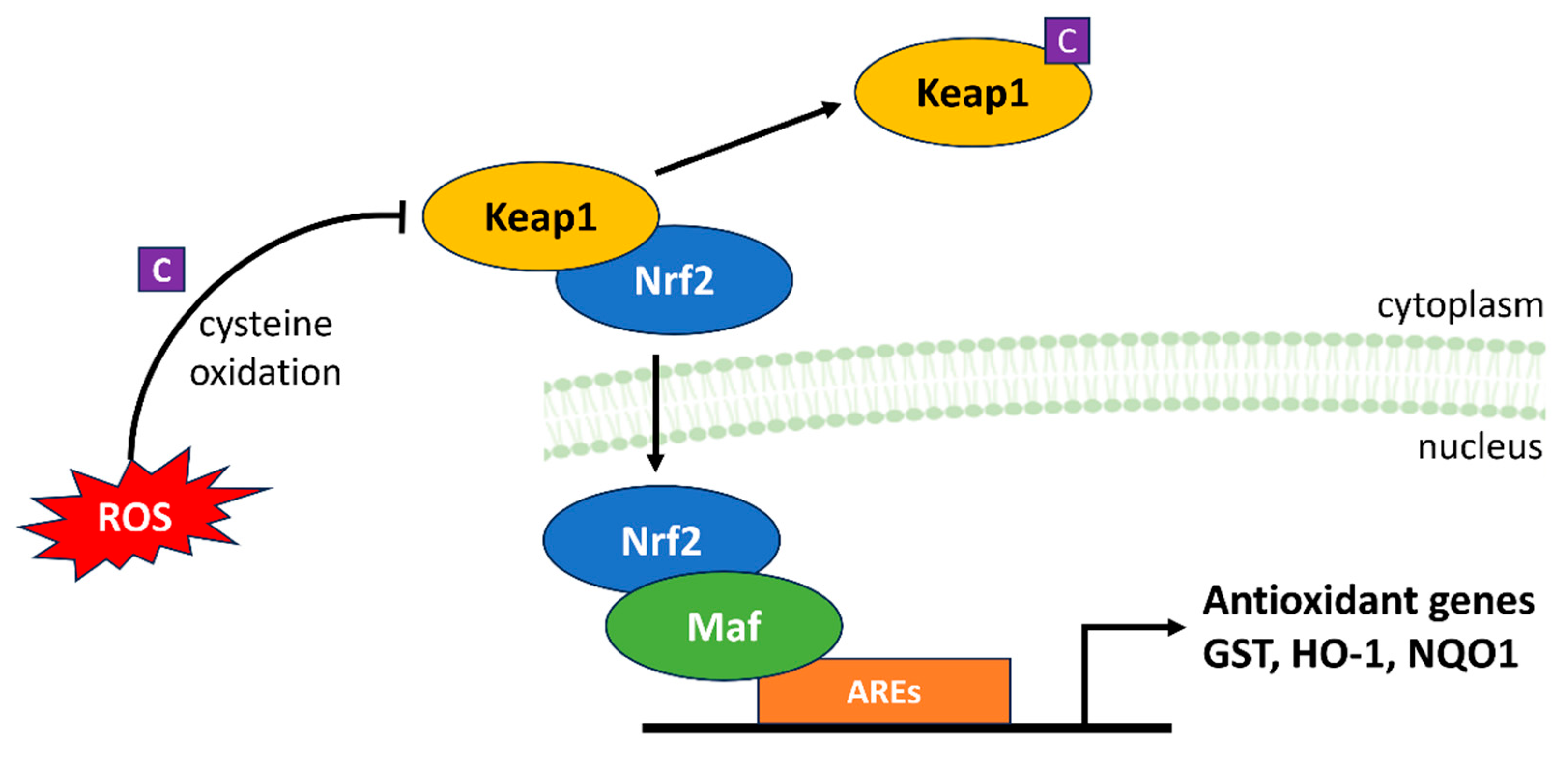
4.4. Keap1-Nrf2-ARE
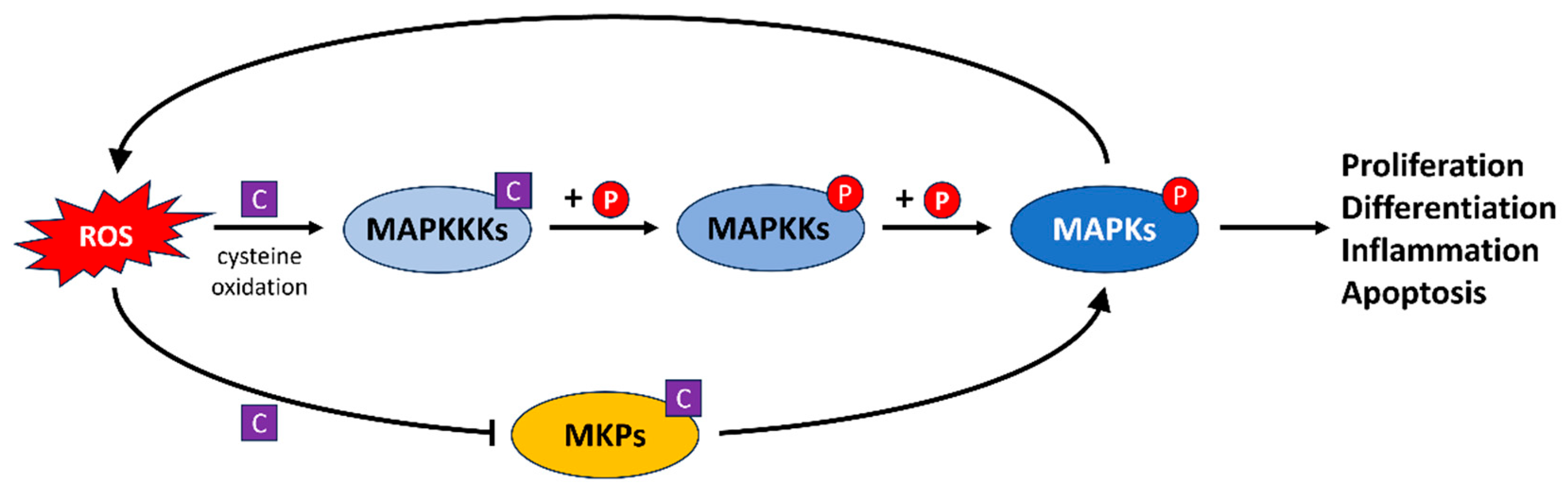
4.5. The MAPK Signaling Pathway
5. Role of ROS in DNA Damage Responses
6. Fish as Models to Study How ROS Contribute to Physiological Systems
6.1. Links between ROS, DNA Repair and the Circadian Clock
6.2. Role of the bZIP PAR/E4BP4 Factors in ROS- and Light-Regulated Transcription
6.3. Evolutionary Perspectives on ROS-Mediated Transcriptional Regulation in Fish
7. Conservation and Evolution of ROS Signaling
8. ROS in Human Disease
9. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bedard, K.; Krause, K.-H. The NOX Family of ROS-Generating NADPH Oxidases: Physiology and Pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- D’Autréaux, B.; Toledano, M.B. ROS as signalling molecules: Mechanisms that generate specificity in ROS homeostasis. Nat. Rev. Mol. Cell Biol. 2007, 8, 813–824. [Google Scholar] [CrossRef]
- Ray, P.D.; Huang, B.-W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-Mediated Cellular Signaling. Oxidative Med. Cell. Longev. 2016, 2016, 4350965. [Google Scholar] [CrossRef] [PubMed]
- Boveris, A.; Chance, B. The mitochondrial generation of hydrogen peroxide. General properties and effect of hyperbaric oxygen. Biochem. J. 1973, 134, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Kowaltowski, A.J.; de Souza-Pinto, N.C.; Castilho, R.F.; Vercesi, A.E. Mitochondria and reactive oxygen species. Free Radic. Biol. Med. 2009, 47, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q. Transcriptional responses to oxidative stress: Pathological and toxicological implications. Pharmacol. Ther. 2010, 125, 376–393. [Google Scholar] [CrossRef] [PubMed]
- Alfadda, A.A.; Sallam, R.M. Reactive Oxygen Species in Health and Disease. J. Biomed. Biotechnol. 2012, 2012, 936486. [Google Scholar] [CrossRef]
- Jones, D.P. Redefining Oxidative Stress. Antioxid. Redox Signal. 2006, 8, 1865–1879. [Google Scholar] [CrossRef]
- del Río, L.A.; López-Huertas, E. ROS Generation in Peroxisomes and its Role in Cell Signaling. Plant Cell Physiol. 2016, 57, 1364–1376. [Google Scholar] [CrossRef]
- Bhandary, B.; Marahatta, A.; Kim, H.-R.; Chae, H.-J. An Involvement of Oxidative Stress in Endoplasmic Reticulum Stress and Its Associated Diseases. Int. J. Mol. Sci. 2013, 14, 434–456. [Google Scholar] [CrossRef] [PubMed]
- Magnani, F.; Mattevi, A. Structure and mechanisms of ROS generation by NADPH oxidases. Curr. Opin. Struct. Biol. 2019, 59, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Klotz, L.-O.; Steinbrenner, H. Cellular adaptation to xenobiotics: Interplay between xenosensors, reactive oxygen species and FOXO transcription factors. Redox Biol. 2017, 13, 646–654. [Google Scholar] [CrossRef] [PubMed]
- Schieber, M.; Chandel, N.S. ROS Function in Redox Signaling and Oxidative Stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef]
- Reuter, S.; Gupta, S.C.; Chaturvedi, M.M.; Aggarwal, B.B. Oxidative stress, inflammation, and cancer: How are they linked? Free Radic. Biol. Med. 2010, 49, 1603–1616. [Google Scholar] [CrossRef] [PubMed]
- Ezraty, B.; Aussel, L.; Barras, F. Methionine sulfoxide reductases in prokaryotes. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2005, 1703, 221–229. [Google Scholar] [CrossRef]
- Valko, M.; Rhodes, C.J.; Moncol, J.; Izakovic, M.; Mazur, M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem.-Biol. Interact. 2006, 160, 1–40. [Google Scholar] [CrossRef]
- Okado-Matsumoto, A.; Fridovich, I. Subcellular Distribution of Superoxide Dismutases (SOD) in Rat Liver: Cu,Zn-SOD IN MITOCHONDRIA *. J. Biol. Chem. 2001, 276, 38388–38393. [Google Scholar] [CrossRef]
- Vives-Bauza, C.; Starkov, A.; Garcia-Arumi, E. Measurements of the Antioxidant Enzyme Activities of Superoxide Dismutase, Catalase, and Glutathione Peroxidase. Methods Cell Biol. 2007, 80, 379–393. [Google Scholar]
- Cnubben, N.H.P.; Rietjens, I.M.C.M.; Wortelboer, H.; van Zanden, J.; van Bladeren, P.J. The interplay of glutathione-related processes in antioxidant defense. Environ. Toxicol. Pharmacol. 2001, 10, 141–152. [Google Scholar] [CrossRef]
- Davies, K.J.A. Intracellular proteolytic systems may function as secondary antioxidant defenses: An hypothesis. J. Free Radic. Biol. Med. 1986, 2, 155–173. [Google Scholar] [CrossRef] [PubMed]
- Loft, S.; Poulsen, H.E. Antioxidant intervention studies related to DNA damage, DNA repair and gene expression. Free Radic. Res. 2000, 33, S67–S83. [Google Scholar] [PubMed]
- Benzie, I.F.F.; Devaki, M. The ferric reducing/antioxidant power (FRAP) assay for non-enzymatic antioxidant capacity: Concepts, procedures, limitations and applications. In Measurement of Antioxidant Activity & Capacity; John Wiley & Sons Ltd.: Hoboken, NJ, USA, 2018; pp. 77–106. [Google Scholar] [CrossRef]
- Padayatty, S.J.; Katz, A.; Wang, Y.; Eck, P.; Kwon, O.; Lee, J.-H.; Chen, S.; Corpe, C.; Dutta, A.; Dutta, S.K.; et al. Vitamin C as an Antioxidant: Evaluation of Its Role in Disease Prevention. J. Am. Coll. Nutr. 2003, 22, 18–35. [Google Scholar] [CrossRef] [PubMed]
- Shakeri, M.; Oskoueian, E.; Le, H.H.; Shakeri, M. Strategies to Combat Heat Stress in Broiler Chickens: Unveiling the Roles of Selenium, Vitamin E and Vitamin C. Vet. Sci. 2020, 7, 71. [Google Scholar] [CrossRef] [PubMed]
- Pastori, G.M.; Kiddle, G.; Antoniw, J.; Bernard, S.; Veljovic-Jovanovic, S.; Verrier, P.J.; Noctor, G.; Foyer, C.H. Leaf Vitamin C Contents Modulate Plant Defense Transcripts and Regulate Genes That Control Development through Hormone Signaling. Plant Cell 2003, 15, 939–951. [Google Scholar] [CrossRef] [PubMed]
- Kaźmierczak-Barańska, J.; Boguszewska, K.; Adamus-Grabicka, A.; Karwowski, B.T. Two Faces of Vitamin C—Antioxidative and Pro-Oxidative Agent. Nutrients 2020, 12, 1501. [Google Scholar] [CrossRef] [PubMed]
- Augustyniak, A.; Bartosz, G.; Čipak, A.; Duburs, G.; Horáková, L.U.; Łuczaj, W.; Majekova, M.; Odysseos, A.D.; Rackova, L.; Skrzydlewska, E.; et al. Natural and synthetic antioxidants: An updated overview. Free Radic. Res. 2010, 44, 1216–1262. [Google Scholar] [CrossRef]
- Zamora, R.; Vodovotz, Y.; Billiar, T.R. Inducible Nitric Oxide Synthase and Inflammatory Diseases. Mol. Med. 2000, 6, 347–373. [Google Scholar] [CrossRef]
- Nowak, D. Vitamin C in Human Health and Disease. Nutrients 2021, 13, 1595. [Google Scholar] [CrossRef]
- Putchala, M.C.; Ramani, P.; Sherlin, H.J.; Premkumar, P.; Natesan, A. Ascorbic acid and its pro-oxidant activity as a therapy for tumours of oral cavity—A systematic review. Arch. Oral Biol. 2013, 58, 563–574. [Google Scholar] [CrossRef]
- Giles, G.I.; Nasim, M.J.; Ali, W.; Jacob, C. The Reactive Sulfur Species Concept: 15 Years On. Antioxidants 2017, 6, 38. [Google Scholar] [CrossRef] [PubMed]
- Olson, K.R. Are Reactive Sulfur Species the New Reactive Oxygen Species? Antioxid. Redox Signal. 2020, 33, 1125–1142. [Google Scholar] [CrossRef] [PubMed]
- Switzer, C.H.; Fukuto, J.M. The antioxidant and oxidant properties of hydropersulfides (RSSH) and polysulfide species. Redox Biol. 2022, 57, 102486. [Google Scholar] [CrossRef] [PubMed]
- Kolluru, G.K.; Shen, X.; Kevil, C.G. Reactive Sulfur Species. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 874–884. [Google Scholar] [CrossRef] [PubMed]
- Iciek, M.; Bilska-Wilkosz, A.; Kozdrowicki, M.; Górny, M. Reactive Sulfur Species in Human Diseases. Antioxid. Redox Signal. 2023, 39, 1000–1023. [Google Scholar] [CrossRef] [PubMed]
- Wink, D.A.; Miranda, K.M.; Espey, M.G.; Pluta, R.M.; Hewett, S.J.; Colton, C.; Vitek, M.; Feelisch, M.; Grisham, M.B. Mechanisms of the Antioxidant Effects of Nitric Oxide. Antioxid. Redox Signal. 2001, 3, 203–213. [Google Scholar] [CrossRef]
- Aktan, F. iNOS-mediated nitric oxide production and its regulation. Life Sci. 2004, 75, 639–653. [Google Scholar] [CrossRef] [PubMed]
- Grisham, M.B.; Jourd’Heuil, D.; Wink, D.A.I. Physiological chemistry of nitric oxide and its metabolites: Implications in inflammation. Am. J. Physiol.-Gastrointest. Liver Physiol. 1999, 276, G315–G321. [Google Scholar] [CrossRef]
- Hamanaka, R.B.; Chandel, N.S. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem. Sci. 2010, 35, 505–513. [Google Scholar] [CrossRef]
- Giorgio, M.; Trinei, M.; Migliaccio, E.; Pelicci, P.G. Hydrogen peroxide: A metabolic by-product or a common mediator of ageing signals? Nat. Rev. Mol. Cell Biol. 2007, 8, 722–728. [Google Scholar] [CrossRef]
- Siauciunaite, R.; Foulkes, N.S.; Calabrò, V.; Vallone, D. Evolution Shapes the Gene Expression Response to Oxidative Stress. Int. J. Mol. Sci. 2019, 20, 3040. [Google Scholar] [CrossRef] [PubMed]
- Amoutzias, G.D.; Veron, A.S.; Weiner, J., III; Robinson-Rechavi, M.; Bornberg-Bauer, E.; Oliver, S.G.; Robertson, D.L. One Billion Years of bZIP Transcription Factor Evolution: Conservation and Change in Dimerization and DNA-Binding Site Specificity. Mol. Biol. Evol. 2007, 24, 827–835. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Liu, Z.-G.; Zandi, E. AP-1 function and regulation. Curr. Opin. Cell Biol. 1997, 9, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Shaulian, E.; Karin, M. AP-1 as a regulator of cell life and death. Nat. Cell Biol. 2002, 4, E131–E136. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.J.; Forman, H.J.; Sevanian, A. Oxidants as Stimulators of Signal Transduction. Free Radic. Biol. Med. 1997, 22, 269–285. [Google Scholar] [CrossRef] [PubMed]
- Toone, W.M.; Morgan, B.A.; Jones, N. Redox control of AP-1-like factors in yeast and beyond. Oncogene 2001, 20, 2336–2346. [Google Scholar] [CrossRef] [PubMed]
- Janssen, Y.M.W.; Matalon, S.; Mossman, B.T. Differential induction of c-fos, c-jun, and apoptosis in lung epithelial cells exposed to ROS or RNS. Am. J. Physiol.-Lung Cell. Mol. Physiol. 1997, 273, L789–L796. [Google Scholar] [CrossRef]
- Kramer-Stickland, K.; Edmonds, A.; Bair, W.B., III; Bowden, G.T. Inhibitory effects of deferoxamine on UVB-induced AP-1 transactivation. Carcinogenesis 1999, 20, 2137–2142. [Google Scholar] [CrossRef]
- Lu, J.; Guo, J.-H.; Tu, X.-L.; Zhang, C.; Zhao, M.; Zhang, Q.-W.; Gao, F.-H. Tiron Inhibits UVB-Induced AP-1 Binding Sites Transcriptional Activation on MMP-1 and MMP-3 Promoters by MAPK Signaling Pathway in Human Dermal Fibroblasts. PLoS ONE 2016, 11, e0159998. [Google Scholar] [CrossRef]
- Ding, M.; Li, J.-J.; Leonard, S.S.; Ye, J.-P.; Shi, X.; Colburn, N.H.; Castranova, V.; Vallyathan, V. Vanadate-induced activation of activator protein-1: Role of reactive oxygen species. Carcinogenesis 1999, 20, 663–668. [Google Scholar] [CrossRef]
- Lian, S.; Li, S.; Zhu, J.; Xia, Y.; Do Jung, Y. Nicotine stimulates IL-8 expression via ROS/NF-κB and ROS/MAPK/AP-1 axis in human gastric cancer cells. Toxicology 2022, 466, 153062. [Google Scholar] [CrossRef] [PubMed]
- Gius, D.; Botero, A.; Shah, S.; Curry, H.A. Intracellular oxidation/reduction status in the regulation of transcription factors NF-κB and AP-1. Toxicol. Lett. 1999, 106, 93–106. [Google Scholar] [CrossRef]
- Tanos, T.; Marinissen, M.J.; Leskow, F.C.; Hochbaum, D.; Martinetto, H.; Gutkind, J.S.; Coso, O.A. Phosphorylation of c-Fos by Members of the p38 MAPK Family: ROLE IN THE AP-1 RESPONSE TO UV LIGHT *. J. Biol. Chem. 2005, 280, 18842–18852. [Google Scholar] [CrossRef]
- Morgan, M.J.; Liu, Z.-G. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Perkins, N.D. Integrating cell-signalling pathways with NF-κB and IKK function. Nat. Rev. Mol. Cell Biol. 2007, 8, 49–62. [Google Scholar] [CrossRef]
- Raza, H.; John, A.; Brown, E.M.; Benedict, S.; Kambal, A. Alterations in mitochondrial respiratory functions, redox metabolism and apoptosis by oxidant 4-hydroxynonenal and antioxidants curcumin and melatonin in PC12 cells. Toxicol. Appl. Pharmacol. 2008, 226, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Karin, M. Is NF-κB the sensor of oxidative stress? FASEB J. 1999, 13, 1137–1143. [Google Scholar] [CrossRef]
- Gloire, G.; Legrand-Poels, S.; Piette, J. NF-κB activation by reactive oxygen species: Fifteen years later. Biochem. Pharmacol. 2006, 72, 1493–1505. [Google Scholar] [CrossRef]
- Lingappan, K. NF-κB in oxidative stress. Curr. Opin. Toxicol. 2018, 7, 81–86. [Google Scholar] [CrossRef]
- Turillazzi, E.; Neri, M.; Cerretani, D.; Cantatore, S.; Frati, P.; Moltoni, L.; Busardò, F.P.; Pomara, C.; Riezzo, I.; Fineschi, V. Lipid peroxidation and apoptotic response in rat brain areas induced by long-term administration of nandrolone: The mutual crosstalk between ROS and NF-kB. J. Cell. Mol. Med. 2016, 20, 601–612. [Google Scholar] [CrossRef]
- Bensimon, A.; Aebersold, R.; Shiloh, Y. Beyond ATM: The protein kinase landscape of the DNA damage response. FEBS Lett. 2011, 585, 1625–1639. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Lu, X. Live or let die: The cell’s response to p53. Nat. Rev. Cancer 2002, 2, 594–604. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Chen, Y.; St. Clair, D.K. ROS and p53: A versatile partnership. Free Radic. Biol. Med. 2008, 44, 1529–1535. [Google Scholar] [CrossRef] [PubMed]
- Maillet, A.; Pervaiz, S. Redox Regulation of p53, Redox Effectors Regulated by p53: A Subtle Balance. Antioxid. Redox Signal. 2011, 16, 1285–1294. [Google Scholar] [CrossRef] [PubMed]
- Abbas, H.A.; Maccio, D.R.; Coskun, S.; Jackson, J.G.; Hazen, A.L.; Sills, T.M.; You, M.J.; Hirschi, K.K.; Lozano, G. Mdm2 Is Required for Survival of Hematopoietic Stem Cells/Progenitors via Dampening of ROS-Induced p53 Activity. Cell Stem Cell 2010, 7, 606–617. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, K.; Shi, Y.; Shao, C. The tango of ROS and p53 in tissue stem cells. Cell Death Differ. 2018, 25, 639–641. [Google Scholar] [CrossRef]
- Desaint, S.; Luriau, S.; Aude, J.-C.; Rousselet, G.; Toledano, M.B. Mammalian Antioxidant Defenses Are Not Inducible by H2O2*. J. Biol. Chem. 2004, 279, 31157–31163. [Google Scholar] [CrossRef]
- Hainaut, P.; Mann, K. Zinc Binding and Redox Control of p53 Structure and Function. Antioxid. Redox Signal. 2001, 3, 611–623. [Google Scholar] [CrossRef]
- Kensler, T.W.; Wakabayashi, N.; Biswal, S. Cell Survival Responses to Environmental Stresses Via the Keap1-Nrf2-ARE Pathway. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 89–116. [Google Scholar] [CrossRef]
- Aaen, S.M.; Horsberg, T.E. A screening of multiple classes of pharmaceutical compounds for effect on preadult salmon lice Lepeophtheirus salmonis. J. Fish Dis. 2016, 39, 1213–1223. [Google Scholar] [CrossRef]
- Rajendran, P.; Nandakumar, N.; Rengarajan, T.; Palaniswami, R.; Gnanadhas, E.N.; Lakshminarasaiah, U.; Gopas, J.; Nishigaki, I. Antioxidants and human diseases. Clin. Chim. Acta 2014, 436, 332–347. [Google Scholar] [CrossRef] [PubMed]
- Plotnikov, A.; Zehorai, E.; Procaccia, S.; Seger, R. The MAPK cascades: Signaling components, nuclear roles and mechanisms of nuclear translocation. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2011, 1813, 1619–1633. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.; Seger, R. The extracellular signal-regulated kinase: Multiple substrates regulate diverse cellular functions. Growth Factors 2006, 24, 21–44. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.J. MAPKs: New JNK expands the group. Trends Biochem. Sci. 1994, 19, 470–473. [Google Scholar] [CrossRef] [PubMed]
- Kyriakis, J.M.; Avruch, J. Mammalian Mitogen-Activated Protein Kinase Signal Transduction Pathways Activated by Stress and Inflammation. Physiol. Rev. 2001, 81, 807–869. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Karin, M. Mammalian MAP kinase signalling cascades. Nature 2001, 410, 37–40. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; He, C. A review of redox signaling and the control of MAP kinase pathway in plants. Redox Biol. 2017, 11, 192–204. [Google Scholar] [CrossRef]
- Matsuzawa, A.; Ichijo, H. Redox control of cell fate by MAP kinase: Physiological roles of ASK1-MAP kinase pathway in stress signaling. Biochim. Biophys. Acta (BBA) Gen. Subj. 2008, 1780, 1325–1336. [Google Scholar] [CrossRef]
- Chen, Y.-R.; Shrivastava, A.; Tan, T.-H. Down-regulation of the c-Jun N-terminal kinase (JNK) phosphatase M3/6 and activation of JNK by hydrogen peroxide and pyrrolidine dithiocarbamate. Oncogene 2001, 20, 367–374. [Google Scholar] [CrossRef]
- Kamata, H.; Honda, S.-i.; Maeda, S.; Chang, L.; Hirata, H.; Karin, M. Reactive Oxygen Species Promote TNFα-Induced Death and Sustained JNK Activation by Inhibiting MAP Kinase Phosphatases. Cell 2005, 120, 649–661. [Google Scholar] [CrossRef]
- Park, S.; Ahn, J.-Y.; Lim, M.-J.; Kim, M.-H.; Yun, Y.-S.; Jeong, G.; Song, J.-Y. Sustained expression of NADPH oxidase 4 by p38 MAPK-Akt signaling potentiates radiation-induced differentiation of lung fibroblasts. J. Mol. Med. 2010, 88, 807–816. [Google Scholar] [CrossRef] [PubMed]
- Filina, Y.; Gabdoulkhakova, A.; Rizvanov, A.; Safronova, V. MAP kinases in regulation of NOX activity stimulated through two types of formyl peptide receptors in murine bone marrow granulocytes. Cell. Signal. 2022, 90, 110205. [Google Scholar] [CrossRef] [PubMed]
- Yousefzadeh, M.; Henpita, C.; Vyas, R.; Soto-Palma, C.; Robbins, P.; Niedernhofer, L. DNA damage—How and why we age? eLife 2021, 10, e62852. [Google Scholar] [CrossRef] [PubMed]
- Eyfjord, J.E.; Bodvarsdottir, S.K. Genomic instability and cancer: Networks involved in response to DNA damage. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2005, 592, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Kryston, T.B.; Georgiev, A.B.; Pissis, P.; Georgakilas, A.G. Role of oxidative stress and DNA damage in human carcinogenesis. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2011, 711, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Sorrell, M.; Berman, Z. Functional interplay between ATM/ATR-mediated DNA damage response and DNA repair pathways in oxidative stress. Cell. Mol. Life Sci. 2014, 71, 3951–3967. [Google Scholar] [CrossRef] [PubMed]
- Poetsch, A.R. The genomics of oxidative DNA damage, repair, and resulting mutagenesis. Comput. Struct. Biotechnol. J. 2020, 18, 207–219. [Google Scholar] [CrossRef]
- Bauer, N.C.; Corbett, A.H.; Doetsch, P.W. The current state of eukaryotic DNA base damage and repair. Nucleic Acids Res. 2015, 43, 10083–10101. [Google Scholar] [CrossRef]
- Rosenquist, T.A.; Zaika, E.; Fernandes, A.S.; Zharkov, D.O.; Miller, H.; Grollman, A.P. The novel DNA glycosylase, NEIL1, protects mammalian cells from radiation-mediated cell death. DNA Repair 2003, 2, 581–591. [Google Scholar] [CrossRef]
- Cline, S.D.; Hanawalt, P.C. Who’s on first in the cellular response to DNA damage? Nat. Rev. Mol. Cell Biol. 2003, 4, 361–373. [Google Scholar] [CrossRef]
- Marteijn, J.A.; Lans, H.; Vermeulen, W.; Hoeijmakers, J.H.J. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat. Rev. Mol. Cell Biol. 2014, 15, 465–481. [Google Scholar] [CrossRef] [PubMed]
- Chavanne, F.; Broughton, B.C.; Pietra, D.; Nardo, T.; Browitt, A.; Lehmann, A.R.; Stefanini, M. Mutations in the XPC Gene in Families with Xeroderma Pigmentosum and Consequences at the Cell, Protein, and Transcript Levels1. Cancer Res. 2000, 60, 1974–1982. [Google Scholar]
- Liu, K.; Sun, Z.; Yang, C.; Lo, L.J.; Chen, J. Loss-of-Function of xpc Sensitizes Zebrafish to Ultraviolet Irradiation. Fishes 2023, 8, 191. [Google Scholar] [CrossRef]
- Zhao, H.; Di Mauro, G.; Lungu-Mitea, S.; Negrini, P.; Guarino, A.M.; Frigato, E.; Braunbeck, T.; Ma, H.; Lamparter, T.; Vallone, D.; et al. Modulation of DNA Repair Systems in Blind Cavefish during Evolution in Constant Darkness. Curr. Biol. 2018, 28, 3229–3243.e3224. [Google Scholar] [CrossRef] [PubMed]
- Christmann, M.; Kaina, B. Transcriptional regulation of human DNA repair genes following genotoxic stress: Trigger mechanisms, inducible responses and genotoxic adaptation. Nucleic Acids Res. 2013, 41, 8403–8420. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Hazra, T.K.; Boldogh, I.; Mitra, S.; Bhakat, K.K. Induction of the Human Oxidized Base-specific DNA Glycosylase NEIL1 by Reactive Oxygen Species *. J. Biol. Chem. 2005, 280, 35272–35280. [Google Scholar] [CrossRef] [PubMed]
- Lisse, T.S.; King, B.L.; Rieger, S. Comparative transcriptomic profiling of hydrogen peroxide signaling networks in zebrafish and human keratinocytes: Implications toward conservation, migration and wound healing. Sci. Rep. 2016, 6, 20328. [Google Scholar] [CrossRef]
- Saleem, S.; Kannan, R.R. Zebrafish: An emerging real-time model system to study Alzheimer’s disease and neurospecific drug discovery. Cell Death Discov. 2018, 4, 45. [Google Scholar] [CrossRef]
- Razaghi, B.; Steele, S.L.; Prykhozhij, S.V.; Stoyek, M.R.; Hill, J.A.; Cooper, M.D.; McDonald, L.; Lin, W.; Daugaard, M.; Crapoulet, N.; et al. hace1 Influences zebrafish cardiac development via ROS-dependent mechanisms. Dev. Dyn. 2018, 247, 289–303. [Google Scholar] [CrossRef]
- Rieger, S.; Sagasti, A. Hydrogen Peroxide Promotes Injury-Induced Peripheral Sensory Axon Regeneration in the Zebrafish Skin. PLoS Biol. 2011, 9, e1000621. [Google Scholar] [CrossRef]
- Meda, F.; Gauron, C.; Rampon, C.; Teillon, J.; Volovitch, M.; Vriz, S. Nerves Control Redox Levels in Mature Tissues Through Schwann Cells and Hedgehog Signaling. Antioxid. Redox Signal. 2015, 24, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Loudon, A.S.I. Circadian Biology: A 2.5 Billion Year Old Clock. Curr. Biol. 2012, 22, R570–R571. [Google Scholar] [CrossRef] [PubMed]
- Rosbash, M. The Implications of Multiple Circadian Clock Origins. PLoS Biol. 2009, 7, e1000062. [Google Scholar] [CrossRef] [PubMed]
- Sancar, A.; Lindsey-Boltz, L.A.; Kang, T.-H.; Reardon, J.T.; Lee, J.H.; Ozturk, N. Circadian clock control of the cellular response to DNA damage. FEBS Lett. 2010, 584, 2618–2625. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, J.S. Transcriptional architecture of the mammalian circadian clock. Nat. Rev. Genet. 2017, 18, 164–179. [Google Scholar] [CrossRef] [PubMed]
- Lai, A.G.; Doherty, C.J.; Mueller-Roeber, B.; Kay, S.A.; Schippers, J.H.M.; Dijkwel, P.P. Circadian clock-associated 1 regulates ROS homeostasis and oxidative stress responses. Proc. Natl. Acad. Sci. USA 2012, 109, 17129–17134. [Google Scholar] [CrossRef] [PubMed]
- Pei, J.-F.; Li, X.-K.; Li, W.-Q.; Gao, Q.; Zhang, Y.; Wang, X.-M.; Fu, J.-Q.; Cui, S.-S.; Qu, J.-H.; Zhao, X.; et al. Diurnal oscillations of endogenous H2O2 sustained by p66Shc regulate circadian clocks. Nat. Cell Biol. 2019, 21, 1553–1564. [Google Scholar] [CrossRef]
- Paul, M.K.; Bisht, B.; Darmawan, D.O.; Chiou, R.; Ha, V.L.; Wallace, W.D.; Chon, A.T.; Hegab, A.E.; Grogan, T.; Elashoff, D.A.; et al. Dynamic Changes in Intracellular ROS Levels Regulate Airway Basal Stem Cell Homeostasis through Nrf2-Dependent Notch Signaling. Cell Stem Cell 2014, 15, 199–214. [Google Scholar] [CrossRef]
- Stangherlin, A.; Reddy, A.B. Regulation of Circadian Clocks by Redox Homeostasis *. J. Biol. Chem. 2013, 288, 26505–26511. [Google Scholar] [CrossRef]
- Patel, S.A.; Velingkaar, N.S.; Kondratov, R.V. Transcriptional Control of Antioxidant Defense by the Circadian Clock. Antioxid. Redox Signal. 2013, 20, 2997–3006. [Google Scholar] [CrossRef]
- Yamajuku, D.; Shibata, Y.; Kitazawa, M.; Katakura, T.; Urata, H.; Kojima, T.; Takayasu, S.; Nakata, O.; Hashimoto, S. Cellular DBP and E4BP4 proteins are critical for determining the period length of the circadian oscillator. FEBS Lett. 2011, 585, 2217–2222. [Google Scholar] [CrossRef] [PubMed]
- Ben-Moshe, Z.; Vatine, G.; Alon, S.; Tovin, A.; Mracek, P.; Foulkes, N.S.; Gothilf, Y. Multiple par and E4BP4 bZIP Transcription Factors in Zebrafish: Diverse Spatial and Temporal Expression Patterns. Chronobiol. Int. 2010, 27, 1509–1531. [Google Scholar] [CrossRef] [PubMed]
- Ripperger, J.A.; Shearman, L.P.; Reppert, S.M.; Schibler, U. CLOCK, an essential pacemaker component, controls expression of the circadian transcription factor DBP. Genes Dev. 2000, 14, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Vatine, G.; Vallone, D.; Appelbaum, L.; Mracek, P.; Ben-Moshe, Z.; Lahiri, K.; Gothilf, Y.; Foulkes, N.S. Light Directs Zebrafish period2 Expression via Conserved D and E Boxes. PLoS Biol. 2009, 7, e1000223. [Google Scholar] [CrossRef] [PubMed]
- Pagano, C.; Siauciunaite, R.; Idda, M.L.; Ruggiero, G.; Ceinos, R.M.; Pagano, M.; Frigato, E.; Bertolucci, C.; Foulkes, N.S.; Vallone, D. Evolution shapes the responsiveness of the D-box enhancer element to light and reactive oxygen species in vertebrates. Sci. Rep. 2018, 8, 13180. [Google Scholar] [CrossRef] [PubMed]
- Stemmer, M.; Schuhmacher, L.-N.; Foulkes, N.S.; Bertolucci, C.; Wittbrodt, J. Cavefish eye loss in response to an early block in retinal differentiation progression. Development 2015, 142, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Lagunas-Rangel, F.A.; Chávez-Valencia, V. Learning of nature: The curious case of the naked mole rat. Mech. Ageing Dev. 2017, 164, 76–81. [Google Scholar] [CrossRef]
- Yoshizawa, M.; Yamamoto, Y.; O’Quin, K.E.; Jeffery, W.R. Evolution of an adaptive behavior and its sensory receptors promotes eye regression in blind cavefish. BMC Biol. 2012, 10, 108. [Google Scholar] [CrossRef]
- Howarth, F.G.; Moldovan, O.T. The Ecological Classification of Cave Animals and Their Adaptations. In Cave Ecology; Moldovan, O.T., Kováč, Ľ., Halse, S., Eds.; Springer International Publishing: Cham, Switzerland, 2018; pp. 41–67. [Google Scholar] [CrossRef]
- Soares, D.; Niemiller, M.L. Sensory Adaptations of Fishes to Subterranean Environments. BioScience 2013, 63, 274–283. [Google Scholar] [CrossRef]
- Jeffery, W.R. Cavefish as a Model System in Evolutionary Developmental Biology. Dev. Biol. 2001, 231, 1–12. [Google Scholar] [CrossRef]
- Beale, A.; Guibal, C.; Tamai, T.K.; Klotz, L.; Cowen, S.; Peyric, E.; Reynoso, V.H.; Yamamoto, Y.; Whitmore, D. Circadian rhythms in Mexican blind cavefish Astyanax mexicanus in the lab and in the field. Nat. Commun. 2013, 4, 2769. [Google Scholar] [CrossRef] [PubMed]
- Ercolini, A.; Berti, R.; Chelazzi, L.; Messana, G. Researches on the Phreatobic Fishes of Somalia: Achievements and Prospects. Monit. Zool. Ital. Suppl. 1982, 17, 219–241. [Google Scholar] [CrossRef]
- Cavallari, N.; Frigato, E.; Vallone, D.; Fröhlich, N.; Lopez-Olmeda, J.F.; Foà, A.; Berti, R.; Sánchez-Vázquez, F.J.; Bertolucci, C.; Foulkes, N.S. A Blind Circadian Clock in Cavefish Reveals that Opsins Mediate Peripheral Clock Photoreception. PLoS Biol. 2011, 9, e1001142. [Google Scholar] [CrossRef]
- Bailey, D.M. Oxygen, evolution and redox signalling in the human brain; quantum in the quotidian. J. Physiol. 2019, 597, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.L.; Bada, J.L. Submarine hot springs and the origin of life. Nature 1988, 334, 609–611. [Google Scholar] [CrossRef] [PubMed]
- Lyons, T.W.; Reinhard, C.T.; Planavsky, N.J. The rise of oxygen in Earth’s early ocean and atmosphere. Nature 2014, 506, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Schippers, J.H.M.; Nguyen, H.M.; Lu, D.; Schmidt, R.; Mueller-Roeber, B. ROS homeostasis during development: An evolutionary conserved strategy. Cell. Mol. Life Sci. 2012, 69, 3245–3257. [Google Scholar] [CrossRef]
- Meyer, Y.; Belin, C.; Delorme-Hinoux, V.; Reichheld, J.-P.; Riondet, C. Thioredoxin and Glutaredoxin Systems in Plants: Molecular Mechanisms, Crosstalks, and Functional Significance. Antioxid. Redox Signal. 2012, 17, 1124–1160. [Google Scholar] [CrossRef]
- Matsui, M.; Oshima, M.; Oshima, H.; Takaku, K.; Maruyama, T.; Yodoi, J.; Taketo, M.M. Early Embryonic Lethality Caused by Targeted Disruption of the Mouse Thioredoxin Gene. Dev. Biol. 1996, 178, 179–185. [Google Scholar] [CrossRef]
- Riondet, C.; Desouris, J.P.; Montoya, J.G.; Chartier, Y.; Meyer, Y.; Reichheld, J.-P. A dicotyledon-specific glutaredoxin GRXC1 family with dimer-dependent redox regulation is functionally redundant with GRXC2. Plant Cell Environ. 2012, 35, 360–373. [Google Scholar] [CrossRef]
- Hewitt, O.H.; Degnan, S.M. Distribution and diversity of ROS-generating enzymes across the animal kingdom, with a focus on sponges (Porifera). BMC Biol. 2022, 20, 212. [Google Scholar] [CrossRef] [PubMed]
- Galina, A.F.; Ahmet, E.K.; Boris, E.A.; Tamara, A.S.; Abolghaseem, R.; Mojgan, R.T.; Hosseinali, R.; Siamak, B. Invasive ctenophore Mnemiopsis leidyi in the Caspian Sea: Feeding, respiration, reproduction and predatory impact on the zooplankton community. Mar. Ecol. Prog. Ser. 2006, 314, 171–185. [Google Scholar]
- Ryan, J.F.; Pang, K.; Schnitzler, C.E.; Nguyen, A.-D.; Moreland, R.T.; Simmons, D.K.; Koch, B.J.; Francis, W.R.; Havlak, P.; Program, N.C.S.; et al. The Genome of the Ctenophore Mnemiopsis leidyi and Its Implications for Cell Type Evolution. Science 2013, 342, 1242592. [Google Scholar] [CrossRef] [PubMed]
- Timme-Laragy, A.R.; Hahn, M.E.; Hansen, J.M.; Rastogi, A.; Roy, M.A. Redox stress and signaling during vertebrate embryonic development: Regulation and responses. Semin. Cell Dev. Biol. 2018, 80, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Yumimoto, K.; Sugiyama, S.; Motomura, S.; Takahashi, D.; Nakayama, K.I. Molecular evolution of Keap1 was essential for adaptation of vertebrates to terrestrial life. Sci. Adv. 2023, 9, eadg2379. [Google Scholar] [CrossRef] [PubMed]
- Pitoniak, A.; Bohmann, D. Mechanisms and functions of Nrf2 signaling in Drosophila. Free Radic. Biol. Med. 2015, 88, 302–313. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.; Lu, R.; Chang, J.C.; Kan, Y.W. NRF2, a member of the NFE2 family of transcription factors, is not essential for murine erythropoiesis, growth, and development. Proc. Natl. Acad. Sci. USA 1996, 93, 13943–13948. [Google Scholar] [CrossRef]
- Derjuga, A.; Gourley, T.S.; Holm, T.M.; Heng, H.H.Q.; Shivdasani, R.A.; Ahmed, R.; Andrews, N.C.; Blank, V. Complexity of CNC Transcription Factors As Revealed by Gene Targeting of the Nrf3 Locus. Mol. Cell. Biol. 2004, 24, 3286–3294. [Google Scholar] [CrossRef]
- Mohler, J.; Mahaffey, J.W.; Deutsch, E.; Vani, K. Control of Drosophila head segment identity by the bZIP homeotic gene cnc. Development 1995, 121, 237–247. [Google Scholar] [CrossRef]
- Oka, K.; Yamakawa, M.; Kawamura, Y.; Kutsukake, N.; Miura, K. The Naked Mole-Rat as a Model for Healthy Aging. Annu. Rev. Anim. Biosci. 2023, 11, 207–226. [Google Scholar] [CrossRef]
- Salmon, A.B.; Akha, A.A.S.; Buffenstein, R.; Miller, R.A. Fibroblasts From Naked Mole-Rats Are Resistant to Multiple Forms of Cell Injury, But Sensitive to Peroxide, Ultraviolet Light, and Endoplasmic Reticulum Stress. J. Gerontol. Ser. A 2008, 63, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Takasugi, M.; Firsanov, D.; Tombline, G.; Ning, H.; Ablaeva, J.; Seluanov, A.; Gorbunova, V. Naked mole-rat very-high-molecular-mass hyaluronan exhibits superior cytoprotective properties. Nat. Commun. 2020, 11, 2376. [Google Scholar] [CrossRef] [PubMed]
- Munro, D.; Baldy, C.; Pamenter, M.E.; Treberg, J.R. The exceptional longevity of the naked mole-rat may be explained by mitochondrial antioxidant defenses. Aging Cell 2019, 18, e12916. [Google Scholar] [CrossRef] [PubMed]
- Lewis, K.N.; Wason, E.; Edrey, Y.H.; Kristan, D.M.; Nevo, E.; Buffenstein, R. Regulation of Nrf2 signaling and longevity in naturally long-lived rodents. Proc. Natl. Acad. Sci. USA 2015, 112, 3722–3727. [Google Scholar] [CrossRef] [PubMed]
- Hajam, Y.A.; Rani, R.; Ganie, S.Y.; Sheikh, T.A.; Javaid, D.; Qadri, S.S.; Pramodh, S.; Alsulimani, A.; Alkhanani, M.F.; Harakeh, S.; et al. Oxidative Stress in Human Pathology and Aging: Molecular Mechanisms and Perspectives. Cells 2022, 11, 552. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, S.; Abramov, A.Y. Mechanism of Oxidative Stress in Neurodegeneration. Oxidative Med. Cell. Longev. 2012, 2012, 428010. [Google Scholar] [CrossRef] [PubMed]
- Moloney, J.N.; Cotter, T.G. ROS signalling in the biology of cancer. Semin. Cell Dev. Biol. 2018, 80, 50–64. [Google Scholar] [CrossRef] [PubMed]
- Malla, R.; Surepalli, N.; Farran, B.; Malhotra, S.V.; Nagaraju, G.P. Reactive oxygen species (ROS): Critical roles in breast tumor microenvironment. Crit. Rev. Oncol. Hematol. 2021, 160, 103285. [Google Scholar] [CrossRef]
- Kim, S.J.; Kim, H.S.; Seo, Y.R. Understanding of ROS-Inducing Strategy in Anticancer Therapy. Oxidative Med. Cell. Longev. 2019, 2019, 5381692. [Google Scholar] [CrossRef]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef]
- Buytaert, E.; Dewaele, M.; Agostinis, P. Molecular effectors of multiple cell death pathways initiated by photodynamic therapy. Biochim. Biophys. Acta (BBA) Rev. Cancer 2007, 1776, 86–107. [Google Scholar] [CrossRef]
- Sznarkowska, A.; Kostecka, A.; Meller, K.; Bielawski, K.P. Inhibition of cancer antioxidant defense by natural compounds. Oncotarget 2016, 8, 15996. [Google Scholar] [CrossRef]
- Panieri, E.; Santoro, M.M. ROS homeostasis and metabolism: A dangerous liason in cancer cells. Cell Death Dis. 2016, 7, e2253. [Google Scholar] [CrossRef]
- Jiang, H.; Zuo, J.; Li, B.; Chen, R.; Luo, K.; Xiang, X.; Lu, S.; Huang, C.; Liu, L.; Tang, J.; et al. Drug-induced oxidative stress in cancer treatments: Angel or devil? Redox Biol. 2023, 63, 102754. [Google Scholar] [CrossRef]
- Gabizon, A.A.; Patil, Y.; La-Beck, N.M. New insights and evolving role of pegylated liposomal doxorubicin in cancer therapy. Drug Resist. Updates 2016, 29, 90–106. [Google Scholar] [CrossRef]
- Li, Z.; Zou, J.; Chen, X. In Response to Precision Medicine: Current Subcellular Targeting Strategies for Cancer Therapy. Adv. Mater. 2023, 35, 2209529. [Google Scholar] [CrossRef] [PubMed]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hong, Y.; Boiti, A.; Vallone, D.; Foulkes, N.S. Reactive Oxygen Species Signaling and Oxidative Stress: Transcriptional Regulation and Evolution. Antioxidants 2024, 13, 312. https://doi.org/10.3390/antiox13030312
Hong Y, Boiti A, Vallone D, Foulkes NS. Reactive Oxygen Species Signaling and Oxidative Stress: Transcriptional Regulation and Evolution. Antioxidants. 2024; 13(3):312. https://doi.org/10.3390/antiox13030312
Chicago/Turabian StyleHong, Yuhang, Alessandra Boiti, Daniela Vallone, and Nicholas S. Foulkes. 2024. "Reactive Oxygen Species Signaling and Oxidative Stress: Transcriptional Regulation and Evolution" Antioxidants 13, no. 3: 312. https://doi.org/10.3390/antiox13030312
APA StyleHong, Y., Boiti, A., Vallone, D., & Foulkes, N. S. (2024). Reactive Oxygen Species Signaling and Oxidative Stress: Transcriptional Regulation and Evolution. Antioxidants, 13(3), 312. https://doi.org/10.3390/antiox13030312