
Simvastatin-Mediated Nrf2 Activation Induces Fetal Hemoglobin and Antioxidant Enzyme Expression to Ameliorate the Phenotype of Sickle Cell Disease

 ,
,
Abstract
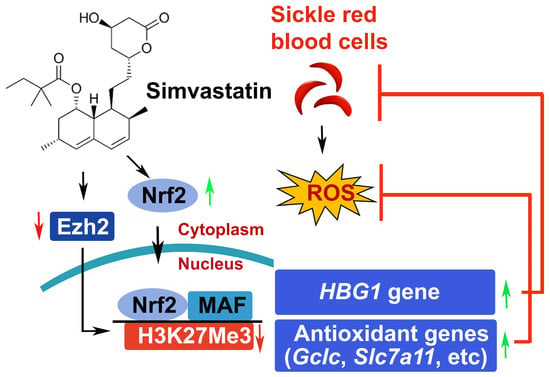
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Culture of Human SCD Patient Erythroblasts and H2O2 Exposure
2.3. Animal Treatment
2.4. Complete Blood Count and Differentials
2.5. Isolation of Mouse Spleen CD71+ Erythroblasts and Reticulocytes
2.6. Cell Proliferation and Viability
2.7. Flow Cytometry Analysis
2.8. RBC Sickling Analysis
2.9. Quantitative RT-PCR
2.10. Western Blot Analysis
2.11. GSH and GSSG Assay
2.12. NQO1 Activity Assay
2.13. Measurement of Intracellular Cysteine Levels
2.14. Cystine Uptake Assay
2.15. EZH2 Gene Knockdown Analysis
2.16. Chromatin Immunoprecipitation (ChIP)
2.17. NADP+/NADPH Levels
2.18. Histology, Immunohistochemistry, and Image Analysis
2.19. Iron Content
2.20. Statistical Analysis
3. Results
3.1. Simvastatin Activates γ-Globin Gene Expression and Reverses Sickling of SCD Erythroblasts
3.2. Simvastatin Increases the Expression of NRF2 and Antioxidant Proteins
3.3. Simvastatin Enhances SCD Erythroblasts’ Antioxidative Capacity
3.4. Simvastatin Attenuates EZH2 Expression and Histone H3K27Me3 to Modify Chromatin Structure in Gene Regulation
3.5. EZH2 Regulates NRF2 Expression to Modify ARE Motif Chromatin Structure on Target Genes
3.6. In Vivo Treatment with Simvastatin Suppresses H3K27Me3 Modification in SCD Mice
3.7. Simvastatin Protects against Organ Damage from SCD
3.8. Simvastatin Reduces ROS Levels and Inflammatory Stresses in Preclinical SCD Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rodgers, G.P.; Walker, E.C.; Podgor, M.J. Mortality in sickle cell disease. N. Engl. J. Med. 1994, 331, 1022–1023. [Google Scholar] [PubMed]
- Rees, D.C.; Williams, T.N.; Gladwin, M.T. Sickle-cell disease. Lancet 2010, 376, 2018–2031. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.J.; Krishnamurti, L.; Kutok, J.L.; Biernacki, M.; Rogers, S.; Zhang, W.; Antin, J.H.; Ritz, J. Evidence for ineffective erythropoiesis in severe sickle cell disease. Blood 2005, 106, 3639–3645. [Google Scholar] [CrossRef]
- Chirico, E.N.; Pialoux, V. Role of oxidative stress in the pathogenesis of sickle cell disease. IUBMB Life 2012, 64, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Silva, D.G.H.; Belini Junior, E.; de Almeida, E.A.; Bonini-Domingos, C.R. Oxidative stress in sickle cell disease: An overview of erythrocyte redox metabolism and current antioxidant therapeutic strategies. Free Radic. Biol. Med. 2013, 65, 1101–1109. [Google Scholar] [CrossRef]
- Nur, E.; Biemond, B.J.; Otten, H.M.; Brandjes, D.P.; Schnog, J.J.; CURAMA Study Group. Oxidative stress in sickle cell disease; pathophysiology and potential implications for disease management. Am. J. Hematol. 2011, 86, 484–489. [Google Scholar] [CrossRef]
- Zhao, B.; Mei, Y.; Yang, J.; Ji, P. Erythropoietin-regulated oxidative stress negatively affects enucleation during terminal erythropoiesis. Exp. Hematol. 2016, 44, 975–981. [Google Scholar] [CrossRef]
- Iampietro, M.; Giovannetti, T.; Tarazi, R. Hypoxia and inflammation in children with sickle cell disease: Implications for hippocampal functioning and episodic memory. Neuropsychol. Rev. 2014, 24, 252–265. [Google Scholar] [CrossRef]
- Kaul, D.K.; Hebbel, R.P. Hypoxia/reoxygenation causes inflammatory response in transgenic sickle mice but not in normal mice. J. Clin. Investig. 2000, 106, 411–420. [Google Scholar] [CrossRef]
- Wu, H.; Bogdanov, M.; Zhang, Y.; Sun, K.; Zhao, S.; Song, A.; Luo, R.; Parchim, N.F.; Liu, H.; Huang, A.; et al. Hypoxia-mediated impaired erythrocyte Lands’ Cycle is pathogenic for sickle cell disease. Sci. Rep. 2016, 6, 29637. [Google Scholar] [CrossRef]
- Sun, K.; Xia, Y. New insights into sickle cell disease: A disease of hypoxia. Curr. Opin. Hematol. 2013, 20, 215–221. [Google Scholar] [CrossRef]
- Telen, M.J.; Malik, P.; Vercellotti, G.M. Therapeutic strategies for sickle cell disease: Towards a multi-agent approach. Nat. Rev. Drug Discov. 2019, 18, 139–158. [Google Scholar] [CrossRef]
- Charache, S.; Terrin, M.L.; Moore, R.D.; Dover, G.J.; Barton, F.B.; Eckert, S.V.; McMahon, R.P.; Bonds, D.R. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia. N. Engl. J. Med. 1995, 332, 1317–1322. [Google Scholar] [CrossRef]
- Niihara, Y.; Miller, S.T.; Kanter, J.; Lanzkron, S.; Smith, W.R.; Hsu, L.L.; Gordeuk, V.R.; Viswanathan, K.; Sarnaik, S.; Osunkwo, I.; et al. A Phase 3 Trial of l-Glutamine in Sickle Cell Disease. N. Engl. J. Med. 2018, 379, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Ataga, K.I.; Kutlar, A.; Kanter, J.; Liles, D.; Cancado, R.; Friedrisch, J.; Guthrie, T.H.; Knight-Madden, J.; Alvarez, O.A.; Gordeuk, V.R.; et al. Crizanlizumab for the Prevention of Pain Crises in Sickle Cell Disease. N. Engl. J. Med. 2017, 376, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Vichinsky, E.; Hoppe, C.C.; Ataga, K.I.; Ware, R.E.; Nduba, V.; El-Beshlawy, A.; Hassab, H.; Achebe, M.M.; Alkindi, S.; Brown, R.C.; et al. A Phase 3 Randomized Trial of Voxelotor in Sickle Cell Disease. N. Engl. J. Med. 2019, 381, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Niihara, Y.; Zerez, C.R.; Akiyama, D.S.; Tanaka, K.R. Oral L-glutamine therapy for sickle cell anemia: I. Subjective clinical improvement and favorable change in red cell NAD redox potential. Am. J. Hematol. 1998, 58, 117–121. [Google Scholar] [CrossRef]
- Klings, E.S.; Farber, H.W. Role of free radicals in the pathogenesis of acute chest syndrome in sickle cell disease. Respir. Res. 2001, 2, 280–285. [Google Scholar] [CrossRef]
- Queiroz, R.F.; Lima, E.S. Oxidative stress in sickle cell disease. Rev. Bras. Hematol. Hemoter. 2013, 35, 16–17. [Google Scholar] [CrossRef]
- Esperti, S.; Nader, E.; Stier, A.; Boisson, C.; Carin, R.; Marano, M.; Robert, M.; Martin, M.; Horand, F.; Cibiel, A.; et al. Increased retention of functional mitochondria in mature sickle red blood cells is associated with increased sickling tendency, hemolysis and oxidative stress. Haematologica 2023, 108, 3086–3094. [Google Scholar] [CrossRef]
- Jagadeeswaran, R.; Vazquez, B.A.; Thiruppathi, M.; Ganesh, B.B.; Ibanez, V.; Cui, S.; Engel, J.D.; Diamond, A.M.; Molokie, R.E.; DeSimone, J.; et al. Pharmacological inhibition of LSD1 and mTOR reduces mitochondrial retention and associated ROS levels in the red blood cells of sickle cell disease. Exp. Hematol. 2017, 50, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Antwi-Boasiako, C.; Dankwah, G.B.; Aryee, R.; Hayfron-Benjamin, C.; Donkor, E.S.; Campbell, A.D. Oxidative Profile of Patients with Sickle Cell Disease. Med. Sci. 2019, 7, 17. [Google Scholar] [CrossRef] [PubMed]
- Telen, M.J. Beyond hydroxyurea: New and old drugs in the pipeline for sickle cell disease. Blood 2016, 127, 810–819. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, S.; Little, J.A.; Pecker, L.H. Advances in the Treatment of Sickle Cell Disease. Mayo Clin. Proc. 2018, 93, 1810–1824. [Google Scholar] [CrossRef]
- Little, J.A.; McGowan, V.R.; Kato, G.J.; Partovi, K.S.; Feld, J.J.; Maric, I.; Martyr, S.; Taylor, J.G.; Machado, R.F.; Heller, T.; et al. Combination erythropoietin-hydroxyurea therapy in sickle cell disease: Experience from the National Institutes of Health and a literature review. Haematologica 2006, 91, 1076–1083. [Google Scholar]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef]
- Takaya, K.; Suzuki, T.; Motohashi, H.; Onodera, K.; Satomi, S.; Kensler, T.W.; Yamamoto, M. Validation of the multiple sensor mechanism of the Keap1-Nrf2 system. Free Radic. Biol. Med. 2012, 53, 817–827. [Google Scholar] [CrossRef]
- Cheng, X.; Qian, W.; Chen, F.; Jin, Y.; Wang, F.; Lu, X.; Lee, S.R.; Su, D.; Chen, B. ATRA protects skin fibroblasts against UV-induced oxidative damage through inhibition of E3 ligase Hrd1. Mol. Med. Rep. 2019, 20, 2294–2302. [Google Scholar] [CrossRef]
- Wu, T.; Zhao, F.; Gao, B.; Tan, C.; Yagishita, N.; Nakajima, T.; Wong, P.K.; Chapman, E.; Fang, D.; Zhang, D.D. Hrd1 suppresses Nrf2-mediated cellular protection during liver cirrhosis. Genes. Dev. 2014, 28, 708–722. [Google Scholar] [CrossRef] [PubMed]
- Rada, P.; Rojo, A.I.; Chowdhry, S.; McMahon, M.; Hayes, J.D.; Cuadrado, A. SCF/beta-TrCP promotes glycogen synthase kinase 3-dependent degradation of the Nrf2 transcription factor in a Keap1-independent manner. Mol. Cell Biol. 2011, 31, 1121–1133. [Google Scholar] [CrossRef] [PubMed]
- Gameiro, I.; Michalska, P.; Tenti, G.; Cores, A.; Buendia, I.; Rojo, A.I.; Georgakopoulos, N.D.; Hernandez-Guijo, J.M.; Teresa Ramos, M.; Wells, G.; et al. Discovery of the first dual GSK3beta inhibitor/Nrf2 inducer. A new multitarget therapeutic strategy for Alzheimer’s disease. Sci. Rep. 2017, 7, 45701. [Google Scholar] [CrossRef]
- Chorley, B.N.; Campbell, M.R.; Wang, X.; Karaca, M.; Sambandan, D.; Bangura, F.; Xue, P.; Pi, J.; Kleeberger, S.R.; Bell, D.A. Identification of novel NRF2-regulated genes by ChIP-Seq: Influence on retinoid X receptor alpha. Nucleic Acids Res. 2012, 40, 7416–7429. [Google Scholar] [CrossRef]
- Macari, E.R.; Schaeffer, E.K.; West, R.J.; Lowrey, C.H. Simvastatin and t-butylhydroquinone suppress KLF1 and BCL11A gene expression and additively increase fetal hemoglobin in primary human erythroid cells. Blood 2013, 121, 830–839. [Google Scholar] [CrossRef]
- Keleku-Lukwete, N.; Suzuki, M.; Otsuki, A.; Tsuchida, K.; Katayama, S.; Hayashi, M.; Naganuma, E.; Moriguchi, T.; Tanabe, O.; Engel, J.D.; et al. Amelioration of inflammation and tissue damage in sickle cell model mice by Nrf2 activation. Proc. Natl. Acad. Sci. USA 2015, 112, 12169–12174. [Google Scholar] [CrossRef] [PubMed]
- Keleku-Lukwete, N.; Suzuki, M.; Panda, H.; Otsuki, A.; Katsuoka, F.; Saito, R.; Saigusa, D.; Uruno, A.; Yamamoto, M. Nrf2 activation in myeloid cells and endothelial cells differentially mitigates sickle cell disease pathology in mice. Blood Adv. 2019, 3, 1285–1297. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Xi, C.; Thomas, B.; Pace, B.S. Loss of NRF2 function exacerbates the pathophysiology of sickle cell disease in a transgenic mouse model. Blood 2018, 131, 558–562. [Google Scholar] [CrossRef]
- Xi, C.; Pang, J.; Zhi, W.; Chang, C.S.; Siddaramappa, U.; Shi, H.; Horuzsko, A.; Pace, B.S.; Zhu, X. Nrf2 sensitizes ferroptosis through l-2-hydroxyglutarate-mediated chromatin modifications in sickle cell disease. Blood 2023, 142, 382–396. [Google Scholar] [CrossRef] [PubMed]
- Krishnamoorthy, S.; Pace, B.; Gupta, D.; Sturtevant, S.; Li, B.; Makala, L.; Brittain, J.; Moore, N.; Vieira, B.F.; Thullen, T.; et al. Dimethyl fumarate increases fetal hemoglobin, provides heme detoxification, and corrects anemia in sickle cell disease. JCI Insight 2017, 2, e96409. [Google Scholar] [CrossRef]
- Zhu, X.; Li, B.; Pace, B.S. NRF2 mediates gamma-globin gene regulation and fetal hemoglobin induction in human erythroid progenitors. Haematologica 2017, 102, e285–e288. [Google Scholar] [CrossRef]
- Chin, M.P.; Wrolstad, D.; Bakris, G.L.; Chertow, G.M.; de Zeeuw, D.; Goldsberry, A.; Linde, P.G.; McCullough, P.A.; McMurray, J.J.; Wittes, J.; et al. Risk factors for heart failure in patients with type 2 diabetes mellitus and stage 4 chronic kidney disease treated with bardoxolone methyl. J. Card. Fail. 2014, 20, 953–958. [Google Scholar] [CrossRef]
- Chin, M.P.; Reisman, S.A.; Bakris, G.L.; O’Grady, M.; Linde, P.G.; McCullough, P.A.; Packham, D.; Vaziri, N.D.; Ward, K.W.; Warnock, D.G.; et al. Mechanisms contributing to adverse cardiovascular events in patients with type 2 diabetes mellitus and stage 4 chronic kidney disease treated with bardoxolone methyl. Am. J. Nephrol. 2014, 39, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Macari, E.R.; Lowrey, C.H. Induction of human fetal hemoglobin via the NRF2 antioxidant response signaling pathway. Blood 2011, 117, 5987–5997. [Google Scholar] [CrossRef] [PubMed]
- Salazar, M.; Rojo, A.I.; Velasco, D.; de Sagarra, R.M.; Cuadrado, A. Glycogen synthase kinase-3beta inhibits the xenobiotic and antioxidant cell response by direct phosphorylation and nuclear exclusion of the transcription factor Nrf2. J. Biol. Chem. 2006, 281, 14841–14851. [Google Scholar] [CrossRef]
- Jain, A.K.; Jaiswal, A.K. GSK-3beta acts upstream of Fyn kinase in regulation of nuclear export and degradation of NF-E2 related factor 2. J. Biol. Chem. 2007, 282, 16502–16510. [Google Scholar] [CrossRef] [PubMed]
- Mohammadzadeh, N.; Montecucco, F.; Carbone, F.; Xu, S.; Al-Rasadi, K.; Sahebkar, A. Statins: Epidrugs with effects on endothelial health? Eur. J. Clin. Investig. 2020, 50, e13388. [Google Scholar] [CrossRef]
- Liu, C.; Shen, M.; Tan, W.L.W.; Chen, I.Y.; Liu, Y.; Yu, X.; Yang, H.; Zhang, A.; Liu, Y.; Zhao, M.T.; et al. Statins improve endothelial function via suppression of epigenetic-driven EndMT. Nat. Cardiovasc. Res. 2023, 2, 467–485. [Google Scholar] [CrossRef]
- Lin, Y.C.; Lin, J.H.; Chou, C.W.; Chang, Y.F.; Yeh, S.H.; Chen, C.C. Statins increase p21 through inhibition of histone deacetylase activity and release of promoter-associated HDAC1/2. Cancer Res. 2008, 68, 2375–2383. [Google Scholar] [CrossRef]
- Tikoo, K.; Patel, G.; Kumar, S.; Karpe, P.A.; Sanghavi, M.; Malek, V.; Srinivasan, K. Tissue specific up regulation of ACE2 in rabbit model of atherosclerosis by atorvastatin: Role of epigenetic histone modifications. Biochem. Pharmacol. 2015, 93, 343–351. [Google Scholar] [CrossRef]
- Ochoa-Rosales, C.; Portilla-Fernandez, E.; Nano, J.; Wilson, R.; Lehne, B.; Mishra, P.P.; Gao, X.; Ghanbari, M.; Rueda-Ochoa, O.L.; Juvinao-Quintero, D.; et al. Epigenetic Link Between Statin Therapy and Type 2 Diabetes. Diabetes Care 2020, 43, 875–884. [Google Scholar] [CrossRef]
- Feig, J.E.; Shang, Y.; Rotllan, N.; Vengrenyuk, Y.; Wu, C.; Shamir, R.; Torra, I.P.; Fernandez-Hernando, C.; Fisher, E.A.; Garabedian, M.J. Statins promote the regression of atherosclerosis via activation of the CCR7-dependent emigration pathway in macrophages. PLoS ONE 2011, 6, e28534. [Google Scholar] [CrossRef]
- Kim, Y.C.; Kim, K.K.; Shevach, E.M. Simvastatin induces Foxp3+ T regulatory cells by modulation of transforming growth factor-beta signal transduction. Immunology 2010, 130, 484–493. [Google Scholar] [CrossRef]
- Makino, N.; Maeda, T.; Abe, N. Short telomere subtelomeric hypomethylation is associated with telomere attrition in elderly diabetic patients (1). Can. J. Physiol. Pharmacol. 2019, 97, 335–339. [Google Scholar] [CrossRef]
- Zhu, X.; Wang, Y.; Pi, W.; Liu, H.; Wickrema, A.; Tuan, D. NF-Y recruits both transcription activator and repressor to modulate tissue- and developmental stage-specific expression of human gamma-globin gene. PLoS ONE 2012, 7, e47175. [Google Scholar]
- Ryan, T.M.; Ciavatta, D.J.; Townes, T.M. Knockout-transgenic mouse model of sickle cell disease. Science 1997, 278, 873–876. [Google Scholar] [CrossRef]
- Zhang, Y.; Dai, Y.; Wen, J.; Zhang, W.; Grenz, A.; Sun, H.; Tao, L.; Lu, G.; Alexander, D.C.; Milburn, M.V.; et al. Detrimental effects of adenosine signaling in sickle cell disease. Nat. Med. 2011, 17, 79–86. [Google Scholar] [CrossRef]
- Zhu, X.; Ling, J.; Zhang, L.; Pi, W.; Wu, M.; Tuan, D. A facilitated tracking and transcription mechanism of long-range enhancer function. Nucleic Acids Res. 2007, 35, 5532–5544. [Google Scholar] [CrossRef]
- Armstrong, J.S.; Whiteman, M.; Yang, H.; Jones, D.P.; Sternberg, P., Jr. Cysteine starvation activates the redox-dependent mitochondrial permeability transition in retinal pigment epithelial cells. Investig. Ophthalmol. Vis. Sci. 2004, 45, 4183–4189. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, S.; Hayashi, H.; Kinoshita, K.; Abe, M.; Kuroki, H.; Tokunaga, R.; Tomiyasu, S.; Tanaka, H.; Sugita, H.; Arita, T.; et al. Statins inhibit tumor progression via an enhancer of zeste homolog 2-mediated epigenetic alteration in colorectal cancer. Int. J. Cancer 2014, 135, 2528–2536. [Google Scholar] [CrossRef]
- Okubo, K.; Miyai, K.; Kato, K.; Asano, T.; Sato, A. Simvastatin-romidepsin combination kills bladder cancer cells synergistically. Transl. Oncol. 2021, 14, 101154. [Google Scholar] [CrossRef] [PubMed]
- Greer, E.L.; Shi, Y. Histone methylation: A dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 2012, 13, 343–357. [Google Scholar] [CrossRef] [PubMed]
- Belcher, J.D.; Chen, C.; Nguyen, J.; Zhang, P.; Abdulla, F.; Nguyen, P.; Killeen, T.; Xu, P.; O’Sullivan, G.; Nath, K.A.; et al. Control of Oxidative Stress and Inflammation in Sickle Cell Disease with the Nrf2 Activator Dimethyl Fumarate. Antioxid. Redox Signal. 2017, 26, 748–762. [Google Scholar] [CrossRef]
- Liang, H.; Feng, Y.; Cui, R.; Qiu, M.; Zhang, J.; Liu, C. Simvastatin protects against acetaminophen-induced liver injury in mice. Biomed. Pharmacother. 2018, 98, 916–924. [Google Scholar] [CrossRef]
- McGregor, G.H.; Campbell, A.D.; Fey, S.K.; Tumanov, S.; Sumpton, D.; Blanco, G.R.; Mackay, G.; Nixon, C.; Vazquez, A.; Sansom, O.J.; et al. Targeting the Metabolic Response to Statin-Mediated Oxidative Stress Produces a Synergistic Antitumor Response. Cancer Res. 2020, 80, 175–188. [Google Scholar] [CrossRef]
- Rasmussen, S.T.; Andersen, J.T.; Nielsen, T.K.; Cejvanovic, V.; Petersen, K.M.; Henriksen, T.; Weimann, A.; Lykkesfeldt, J.; Poulsen, H.E. Simvastatin and oxidative stress in humans: A randomized, double-blinded, placebo-controlled clinical trial. Redox Biol. 2016, 9, 32–38. [Google Scholar] [CrossRef]
- Kumar, V.; Joshi, T.; Vatsa, N.; Singh, B.K.; Jana, N.R. Simvastatin Restores HDAC1/2 Activity and Improves Behavioral Deficits in Angelman Syndrome Model Mouse. Front. Mol. Neurosci. 2019, 12, 289. [Google Scholar] [CrossRef] [PubMed]
- Mattioli, E.; Andrenacci, D.; Mai, A.; Valente, S.; Robijns, J.; De Vos, W.H.; Capanni, C.; Lattanzi, G. Statins and Histone Deacetylase Inhibitors Affect Lamin A/C—Histone Deacetylase 2 Interaction in Human Cells. Front. Cell Dev. Biol. 2019, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Schmeck, B.; Beermann, W.; N’Guessan, P.D.; Hocke, A.C.; Opitz, B.; Eitel, J.; Dinh, Q.T.; Witzenrath, M.; Krull, M.; Suttorp, N.; et al. Simvastatin reduces Chlamydophila pneumoniae-mediated histone modifications and gene expression in cultured human endothelial cells. Circ. Res. 2008, 102, 888–895. [Google Scholar] [CrossRef]
- Kodach, L.L.; Jacobs, R.J.; Voorneveld, P.W.; Wildenberg, M.E.; Verspaget, H.W.; van Wezel, T.; Morreau, H.; Hommes, D.W.; Peppelenbosch, M.P.; van den Brink, G.R.; et al. Statins augment the chemosensitivity of colorectal cancer cells inducing epigenetic reprogramming and reducing colorectal cancer cell ‘stemness’ via the bone morphogenetic protein pathway. Gut 2011, 60, 1544–1553. [Google Scholar] [CrossRef]
- Alves, N.; Neuparth, T.; Barros, S.; Santos, M.M. The anti-lipidemic drug simvastatin modifies epigenetic biomarkers in the amphipod Gammarus locusta. Ecotoxicol. Environ. Saf. 2021, 209, 111849. [Google Scholar] [CrossRef]
- Chen, W.W.; Qi, J.W.; Hang, Y.; Wu, J.X.; Zhou, X.X.; Chen, J.Z.; Wang, J.; Wang, H.H. Simvastatin is beneficial to lung cancer progression by inducing METTL3-induced m6A modification on EZH2 mRNA. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 4263–4270. [Google Scholar] [PubMed]
- Bridgeman, S.; Northrop, W.; Ellison, G.; Sabapathy, T.; Melton, P.E.; Newsholme, P.; Mamotte, C.D.S. Statins Do Not Directly Inhibit the Activity of Major Epigenetic Modifying Enzymes. Cancers 2019, 11, 516. [Google Scholar] [CrossRef]
- Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Hayashi, M.; Sekine, H.; Tanaka, N.; Moriguchi, T.; Motohashi, H.; Nakayama, K.; et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat. Commun. 2016, 7, 11624. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Bauer, D.E.; Kerenyi, M.A.; Vo, T.D.; Hou, S.; Hsu, Y.J.; Yao, H.; Trowbridge, J.J.; Mandel, G.; Orkin, S.H. Corepressor-dependent silencing of fetal hemoglobin expression by BCL11A. Proc. Natl. Acad. Sci. USA 2013, 110, 6518–6523. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.W.; Kim, A. Characterization of histone H3K27 modifications in the beta-globin locus. Biochem. Biophys. Res. Commun. 2011, 405, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Myers, G.; Schneider, E.; Wang, Y.; Mathews, R.; Lim, K.C.; Siemieniak, D.; Tang, V.; Ginsburg, D.; Balbin-Cuesta, G.; et al. Identification of novel gamma-globin inducers among all potential erythroid druggable targets. Blood Adv. 2022, 6, 3280–3285. [Google Scholar] [CrossRef] [PubMed]
- Hoppe, C.; Kuypers, F.; Larkin, S.; Hagar, W.; Vichinsky, E.; Styles, L. A pilot study of the short-term use of simvastatin in sickle cell disease: Effects on markers of vascular dysfunction. Br. J. Haematol. 2011, 153, 655–663. [Google Scholar] [CrossRef] [PubMed]
- Hoppe, C.; Jacob, E.; Styles, L.; Kuypers, F.; Larkin, S.; Vichinsky, E. Simvastatin reduces vaso-occlusive pain in sickle cell anaemia: A pilot efficacy trial. Br. J. Haematol. 2017, 177, 620–629. [Google Scholar] [CrossRef]
- Biswas, S.K. Does the Interdependence between Oxidative Stress and Inflammation Explain the Antioxidant Paradox? Oxidative Med. Cell. Longev. 2016, 2016, 5698931. [Google Scholar] [CrossRef]
- Pedersen, T.R.; Faergeman, O.; Kastelein, J.J.; Olsson, A.G.; Tikkanen, M.J.; Holme, I.; Larsen, M.L.; Bendiksen, F.S.; Lindahl, C.; Szarek, M.; et al. High-dose atorvastatin vs usual-dose simvastatin for secondary prevention after myocardial infarction: The IDEAL study: A randomized controlled trial. JAMA 2005, 294, 2437–2445. [Google Scholar] [CrossRef]
- Xu, L.; Dong, X.W.; Shen, L.L.; Li, F.F.; Jiang, J.X.; Cao, R.; Yao, H.Y.; Shen, H.J.; Sun, Y.; Xie, Q.M. Simvastatin delivery via inhalation attenuates airway inflammation in a murine model of asthma. Int. Immunopharmacol. 2012, 12, 556–564. [Google Scholar] [CrossRef]
- Yuan, W.; Hai, B.; Ren, X.; Zhu, J.; Zhang, C.; Guan, Z.; Jia, J.; Wang, H.; Cao, B.; Song, C. Single-dose local intraosseous injection of simvastatin suppresses breast cancer with tumor vascular normalization. Transl. Oncol. 2020, 13, 100867. [Google Scholar] [CrossRef] [PubMed]
- Manci, E.A.; Hillery, C.A.; Bodian, C.A.; Zhang, Z.G.; Lutty, G.A.; Coller, B.S. Pathology of Berkeley sickle cell mice: Similarities and differences with human sickle cell disease. Blood 2006, 107, 1651–1658. [Google Scholar] [CrossRef] [PubMed]
- Jamialahmadi, T.; Abbasifard, M.; Reiner, Z.; Rizzo, M.; Eid, A.H.; Sahebkar, A. The Effects of Statin Treatment on Serum Ferritin Levels: A Systematic Review and Meta-Analysis. J. Clin. Med. 2022, 11, 5251. [Google Scholar] [CrossRef] [PubMed]
- Coates, T.D.; Wood, J.C. How we manage iron overload in sickle cell patients. Br. J. Haematol. 2017, 177, 703–716. [Google Scholar] [CrossRef]
- Ahn, J.; Lee, S.; Won, S. Possible link between statin and iron deficiency anemia: A South Korean nationwide population-based cohort study. Sci. Adv. 2023, 9, eadg6194. [Google Scholar] [CrossRef]
- Jang, H.J.; Hong, E.M.; Kim, M.; Kim, J.H.; Jang, J.; Park, S.W.; Byun, H.W.; Koh, D.H.; Choi, M.H.; Kae, S.H.; et al. Simvastatin induces heme oxygenase-1 via NF-E2-related factor 2 (Nrf2) activation through ERK and PI3K/Akt pathway in colon cancer. Oncotarget 2016, 7, 46219–46229. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xi, C.; Palani, C.; Takezaki, M.; Shi, H.; Horuzsko, A.; Pace, B.S.; Zhu, X. Simvastatin-Mediated Nrf2 Activation Induces Fetal Hemoglobin and Antioxidant Enzyme Expression to Ameliorate the Phenotype of Sickle Cell Disease. Antioxidants 2024, 13, 337. https://doi.org/10.3390/antiox13030337
Xi C, Palani C, Takezaki M, Shi H, Horuzsko A, Pace BS, Zhu X. Simvastatin-Mediated Nrf2 Activation Induces Fetal Hemoglobin and Antioxidant Enzyme Expression to Ameliorate the Phenotype of Sickle Cell Disease. Antioxidants. 2024; 13(3):337. https://doi.org/10.3390/antiox13030337
Chicago/Turabian StyleXi, Caixia, Chithra Palani, Mayuko Takezaki, Huidong Shi, Anatolij Horuzsko, Betty S. Pace, and Xingguo Zhu. 2024. "Simvastatin-Mediated Nrf2 Activation Induces Fetal Hemoglobin and Antioxidant Enzyme Expression to Ameliorate the Phenotype of Sickle Cell Disease" Antioxidants 13, no. 3: 337. https://doi.org/10.3390/antiox13030337
APA StyleXi, C., Palani, C., Takezaki, M., Shi, H., Horuzsko, A., Pace, B. S., & Zhu, X. (2024). Simvastatin-Mediated Nrf2 Activation Induces Fetal Hemoglobin and Antioxidant Enzyme Expression to Ameliorate the Phenotype of Sickle Cell Disease. Antioxidants, 13(3), 337. https://doi.org/10.3390/antiox13030337