
Biological Chemistry of Hydrogen Selenide

Abstract
:1. Overview of Chalcogens in Biology
2. Chemistry of Sulfur vs. Selenium
2.1. Selenium Compounds Are More Reactive

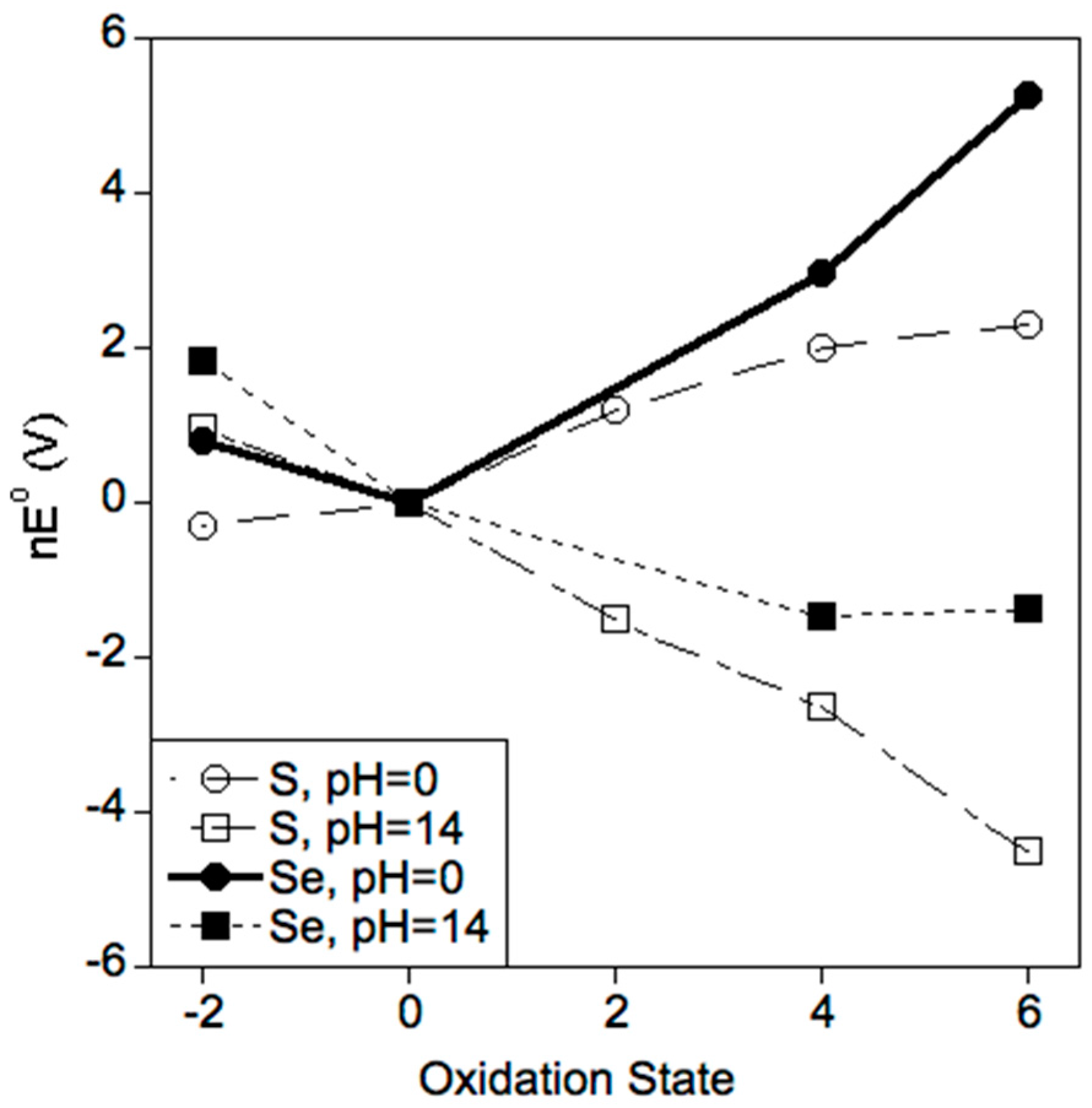
2.2. Selenium Favors Higher Oxidation States
3. Selenium in Biology
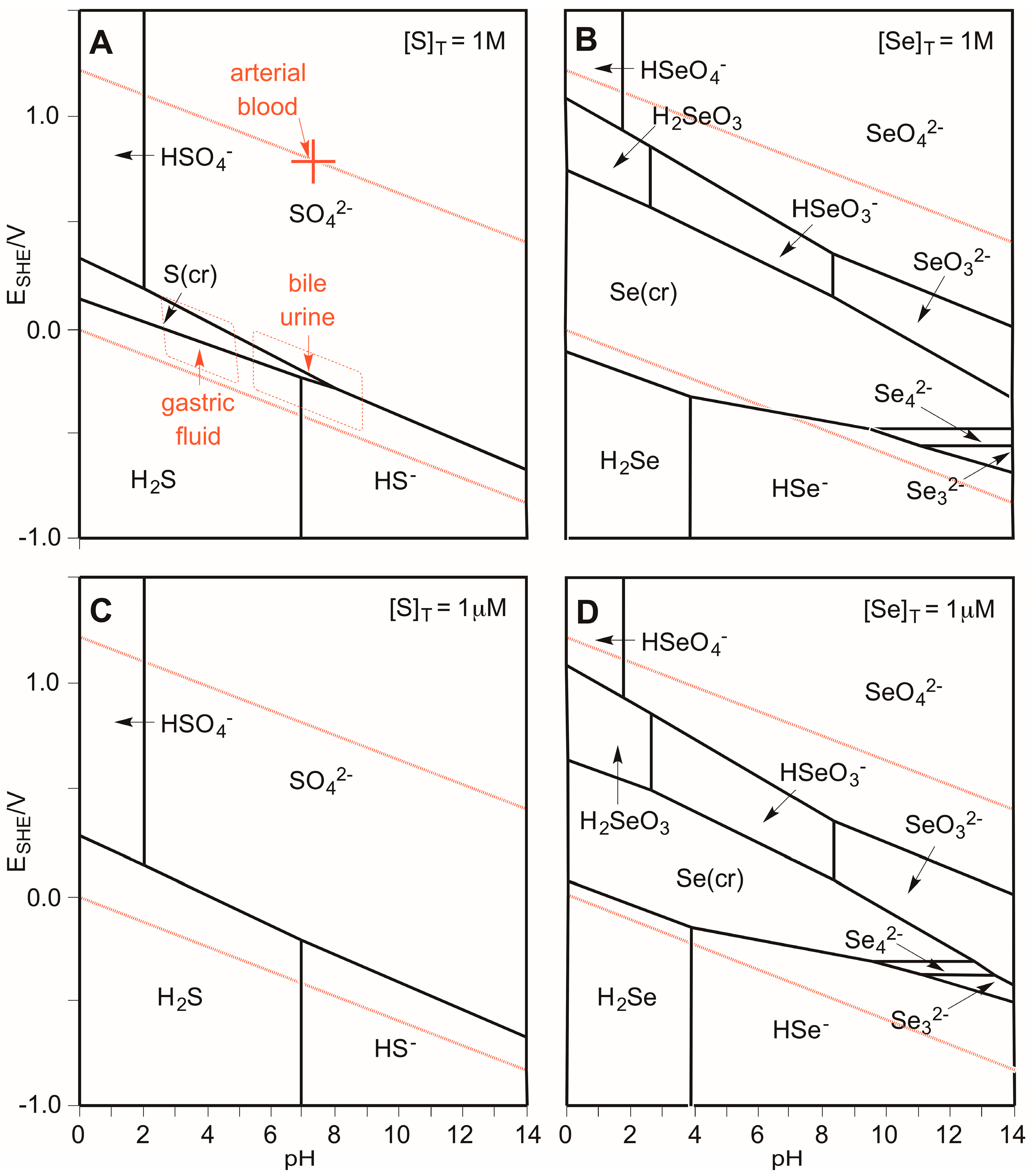
3.1. Which Oxidation States of Sulfur and Selenium Are (Most) Stable in Biology?
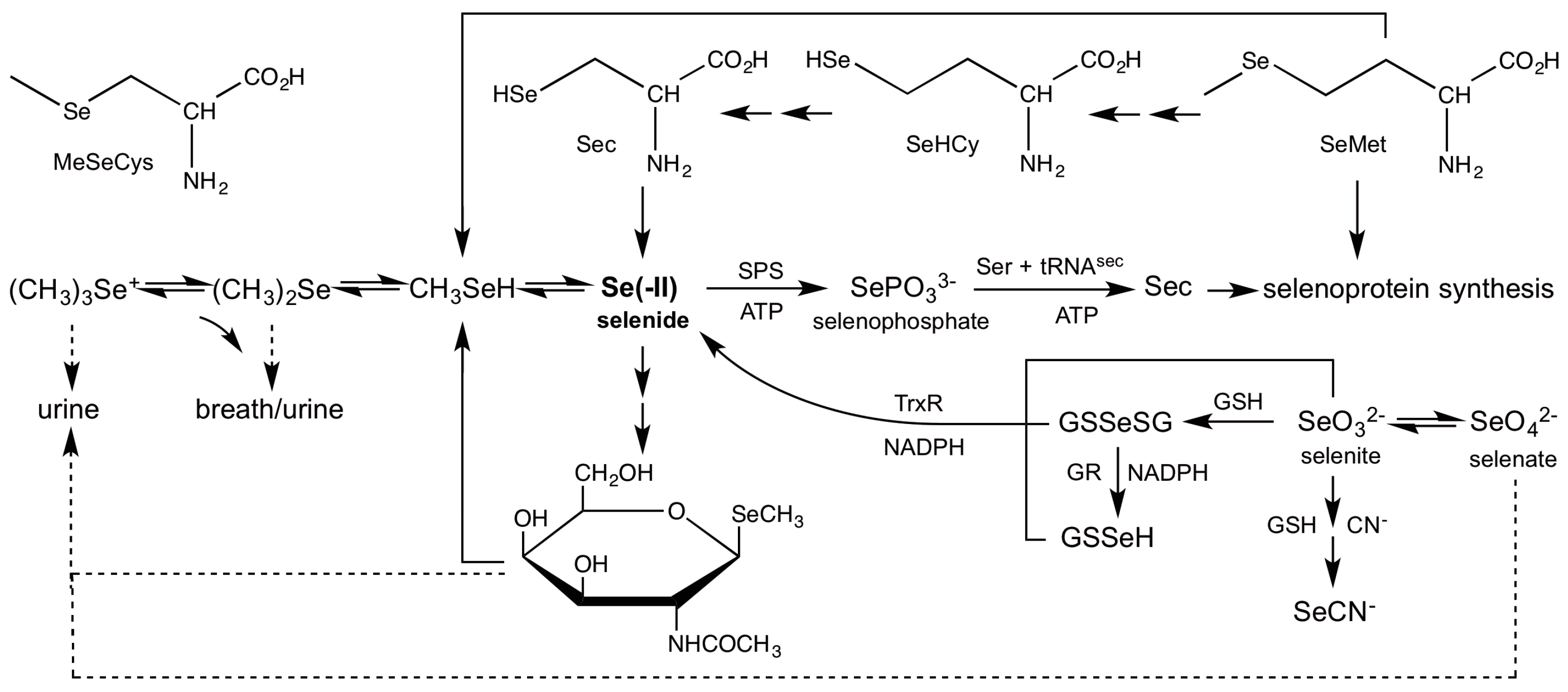
3.2. Role of Selenide In Vivo
3.3. Mechanism of Selenophosphate Synthetase (SPS)
3.4. Reduction of Inorganic Selenium
3.5. Mechanism of Selenocysteine β-Lyase
3.6. Demethylation of Methylselenol
3.7. Incorporation of Selenide into Selenosugars
4. Unanswered Questions
4.1. Is Se an Anti-oxidant or a Pro-Oxidant?
4.2. Why Is Inorganic Se More Toxic Than Organic Se?
4.3. Newly Discovered and Undiscovered Biological Selenium Compounds
5. Conclusions
Conflicts of Interest
References
- Schroeder, H.A.; Frost, D.V.; Balassa, J.J. Essential trace metals in man: Selenium. J. Chronic Dis. 1970, 23, 227–243. [Google Scholar] [CrossRef]
- Apel, K.; Hirt, H. Reactive oxygen species: Metabolism, oxidative stress, and signal transduction. Ann. Rev. Plant Biol. 2004, 55, 373–399. [Google Scholar] [CrossRef] [PubMed]
- Simon, H.U.; Haj-Yehia, A.; Levi-Schaffer, F. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis 2000, 5, 415–418. [Google Scholar] [CrossRef] [PubMed]
- Finkel, T.; Holbrook, N.J. Oxidants, oxidative stress and the biology of ageing. Nature 2000, 408, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Ashby, M.T. Hypothiocyanite. Adv. Inorg. Chem. 2012, 64, 263–303. [Google Scholar]
- Ashby, M.T. Inorganic chemistry of defensive peroxidases in the human oral cavity. J. Dent. Res. 2008, 87, 900–914. [Google Scholar] [CrossRef] [PubMed]
- Mishanina, T.V.; Libiad, M.; Banerjee, R. Biogenesis of reactive sulfur species for signaling by hydrogen sulfide oxidation pathways. Nat. Chem. Biol. 2015, 11, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Lin, V.S.; Chen, W.; Xian, M.; Chang, C.J. Chemical probes for molecular imaging and detection of hydrogen sulfide and reactive sulfur species in biological systems. Chem. Soc. Rev. 2015, 44, 4596–4618. [Google Scholar] [CrossRef] [PubMed]
- Gruhlke, M.C.H.; Slusarenko, A.J. The biology of reactive sulfur species (RSS). Plant Physiol. Biochem. 2012, 59, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Jacob, C.; Anwar, A. The chemistry behind redox regulation with a focus on sulphur redox systems. Physiol. Plant. 2008, 133, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Jacob, C.; Lancaster, J.R.; Giles, G.I. Reactive sulphur species in oxidative signal transduction. Biochem. Soc. Trans. 2004, 32, 1015–1017. [Google Scholar] [CrossRef] [PubMed]
- Giles Gregory, I.; Jacob, C. Reactive sulfur species: An emerging concept in oxidative stress. Biol. Chem. 2002, 383, 375–388. [Google Scholar]
- Giles, G.I.; Tasker, K.M.; Jacob, C. Hypothesis: The role of reactive sulfur species in oxidative stress. Free Radic. Biol. Med. 2001, 31, 1279–1283. [Google Scholar] [CrossRef]
- Duntas, L.H.; Benvenga, S. Selenium: An element for life. Endocrine 2015, 48, 756–775. [Google Scholar] [CrossRef] [PubMed]
- Brigelius-Flohe, R. The evolving versatility of selenium in biology. Antioxid. Redox Signal. 2015, 23, 757–760. [Google Scholar] [CrossRef] [PubMed]
- Roman, M.; Jitaru, P.; Barbante, C. Selenium biochemistry and its role for human health. Metallomics Integr. Biomet. Sci. 2014, 6, 25–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rayman, M.P. Selenium and human health. Lancet 2012, 379, 1256–1268. [Google Scholar] [CrossRef]
- Terry, N.; Zayed, A.M.; De Souza, M.P.; Tarun, A.S. Selenium in higher plants. Ann. Rev. Plant Physiol. Plant Mol. Biol. 2000, 51, 401–432. [Google Scholar] [CrossRef] [PubMed]
- Boeck, A.; Thanbichler, M. Selenocysteine. EcoSal Plus 2014, 1–12. [Google Scholar]
- Block, E.; Birringer, M.; Jiang, W.; Nakahodo, T.; Thompson, H.J.; Toscano, P.J.; Uzar, H.; Zhang, X.; Zhu, Z. Allium chemistry: Synthesis, natural occurrence, biological activity, and chemistry of Se-alk(en)ylselenocysteines and their γ-glutamyl derivatives and oxidation products. J. Agric. Food Chem. 2001, 49, 458–470. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Holmgren, A. Selenoproteins. J. Biol. Chem. 2009, 284, 723–727. [Google Scholar] [CrossRef] [PubMed]
- Papp, L.V.; Lu, J.; Holmgren, A.; Khanna, K.K. From selenium to selenoproteins: Synthesis, identity, and their role in human health. Antioxid. Redox Signal. 2007, 9, 775–806. [Google Scholar] [CrossRef] [PubMed]
- Gromer, S.; Eubel, J.K.; Lee, B.L.; Jacob, J. Human selenoproteins at a glance. Cell. Mol. Life Sci. 2005, 62, 2414–2437. [Google Scholar] [CrossRef] [PubMed]
- Steinbrenner, H.; Speckmann, B.; Klotz, L.-O. Selenoproteins: Antioxidant selenoenzymes and beyond. Arch. Biochem. Biophys. 2016, 595, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.M.; Arthur, J.R. Selenium, selenoproteins and human health: A review. Public Health Nutr. 2001, 4, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Holben, D.H.; Smith, A.M. The diverse role of selenium within selenoproteins: A review. J. Am. Diet. Assoc. 1999, 99, 836–843. [Google Scholar] [CrossRef]
- Johansson, A.-L.; Collins, R.; Arner Elias, S.J.; Brzezinski, P.; Hogbom, M. Biochemical discrimination between selenium and sulfur 2: Mechanistic investigation of the selenium specificity of human selenocysteine lyase. PLoS ONE 2012, 7, e30528. [Google Scholar] [CrossRef] [PubMed]
- Haynes, W.M.; Lide, D.R.; Bruno, T.J. Crc Handbook of Chemistry and Physics, 94th ed.; CRC Press: Boca Raton, FL, USA, 2013; p. 2668. [Google Scholar]
- Greenwood, N.N.; Earnshaw, A. Chemistry of the Elements, 2nd ed.; Elsevier: New York, NY, USA, 1997. [Google Scholar]
- Slater, J.C. Atomic radii in crystals. J. Chem. Phys. 1964, 41, 3199–3204. [Google Scholar] [CrossRef]
- Bondi, A. Van der waals volumes and radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Allen, F.H. The cambridge structural database: A quarter of a million crystal structures and rising. Acta Crystallogr. B Struct. Sci. 2002, B58, 380–388. [Google Scholar] [CrossRef]
- Nicovich, J.M.; Kreutter, K.D.; van Dijk, C.A.; Wine, P.H. Temperature-dependent kinetics studies of the reactions bromine atom(2p3/2) + hydrogen sulfide ↔ mercapto + hydrogen bromide and bromine atom(2p3/2) + methanethiol ↔ methylthiol + hydrogen bromide. Heats of formation of mercapto and methylthio radicals. J. Phys. Chem. 1992, 96, 2518–2528. [Google Scholar] [CrossRef]
- Berkowitz, J.; Ellison, G.B.; Gutman, D. Three methods to measure RH bond energies. J. Phys. Chem. 1994, 98, 2744–2765. [Google Scholar] [CrossRef]
- Guziec, F.S., Jr. Seleno- and telluro-carbonyl compounds. Chem. Org. Selenium Tellurium Compd. 1987, 2, 215–273. [Google Scholar]
- Gonzales, J.M.; Musaev, D.G.; Morokuma, K. Theoretical studies of oxidative addition of E–E bonds (E = S, Se, Te) to palladium(0) and platinum(0) complexes. Organometallics 2005, 24, 4908–4914. [Google Scholar] [CrossRef]
- Moore, C.E. Ionization Potentials and Ionization Limits Derived from the Analyses of Optical Spectra; National Bureau of Standards: Washington, DC, USA, 1970; p. 22. [Google Scholar]
- Hotop, H.; Lineberger, W.C. Binding energies in atomic negative ions. J. Phys. Chem. Ref. Data 1975, 4, 539–576. [Google Scholar] [CrossRef]
- Allred, A.L. Electronegativity values from thermochemical data. J. Inorg. Nucl. Chem. 1961, 17, 215–221. [Google Scholar] [CrossRef]
- Andersson, K.; Sadlej, A.J. Electric dipole polarizabilities of atomic valence states. Phys. Rev. A Atomic Mol. Opt. Phys. 1992, 46, 2356–2362. [Google Scholar] [CrossRef]
- Alpher, R.A.; White, D.R. Optical refractivity of high-temperature gases. I. Effects resulting from dissociation of diatomic gases. Phys. Fluids 1959, 2, 153–161. [Google Scholar] [CrossRef]
- Widmer, M.; Schwarzenbach, G. Acidity of the hydrosulfide ion HS. Helv. Chim. Acta 1964, 47, 266–271. [Google Scholar] [CrossRef]
- Hagisawa, H. The glass electrode and its applications. XII. Dissociation constants of hydrogen selenide. Rikagaku Kenkyusho Iho 1941, 20, 384–389. [Google Scholar]
- Levy, D.E.; Myers, R.J. Spectroscopic determination of the second dissociation constant of hydrogen selenide and the activity coefficients and spectral shifts of its ions. J. Phys. Chem. 1990, 94, 7842–7847. [Google Scholar] [CrossRef]
- Byun, B.J.; Kang, Y.K. Conformational preferences and PKA value of selenocysteine residue. Biopolymers 2011, 95, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Nagy, P.; Ashby, M.T. Reactive sulfur species: Kinetics and mechanisms of the oxidation of cysteine by hypohalous acid to give cysteine sulfenic acid. J. Am. Chem. Soc. 2007, 129, 14082–14091. [Google Scholar] [CrossRef] [PubMed]
- Tajc, S.G.; Tolbert, B.S.; Basavappa, R.; Miller, B.L. Direct determination of thiol PKA by isothermal titration microcalorimetry. J. Am. Chem. Soc. 2004, 126, 10508–10509. [Google Scholar] [CrossRef] [PubMed]
- Arnold, A.P.; Tan, K.S.; Rabenstein, D.L. Nuclear magnetic resonance studies of the solution chemistry of metal complexes. 23. Complexation of methylmercury by selenohydryl-containing amino acids and related molecules. Inorg. Chem. 1986, 25, 2433–2437. [Google Scholar] [CrossRef]
- Greig, I.R. The analysis of enzymic free energy relationships using kinetic and computational models. Chem. Soc. Rev. 2010, 39, 2272–2301. [Google Scholar] [CrossRef] [PubMed]
- Schmid, R.; Sapunov, V.N. Monographs in Modern Chemistry, Volume 14: Non-Formal Kinetics in Search for Chemical Reaction Pathways; Verlag Chemie: Weinheim, Germany, 1982; p. 199. [Google Scholar]
- Huber, R.; Criddle, R.S. Comparison of the chemical properties of selenocysteine and selenocystine with their sulfur analogs. Arch. Biochem. Biophys. 1967, 122, 164–173. [Google Scholar] [CrossRef]
- Steinmann, D.; Nauser, T.; Koppenol, W.H. Selenium and sulfur in exchange reactions: A comparative study. J. Org. Chem. 2010, 75, 6696–6699. [Google Scholar] [CrossRef] [PubMed]
- Seby, F.; Potin-Gautier, M.; Giffaut, E.; Borge, G.; Donard, O.F.X. A critical review of thermodynamic data for selenium species at 25 °C. Chem. Geol. 2001, 171, 173–194. [Google Scholar] [CrossRef]
- Frost, A.A. Oxidation potential-free energy diagrams. J. Am. Chem. Soc. 1951, 73, 2680–2682. [Google Scholar] [CrossRef]
- Black, J. Biological Performance of Materials: Fundamentals of Biocompatibility, 3rd ed.; Marcel Dekker: New York, NY, USA, 1999; p. 463. [Google Scholar]
- Yoshizawa, S.; Boeck, A. The many levels of control on bacterial selenoprotein synthesis. Biochim. Biophys. Acta Gen. Subj. 2009, 1790, 1404–1414. [Google Scholar] [CrossRef] [PubMed]
- Allmang, C.; Wurth, L.; Krol, A. The selenium to selenoprotein pathway in eukaryotes: More molecular partners than anticipated. Biochim. Biophys. Acta Gen. Subj. 2009, 1790, 1415–1423. [Google Scholar] [CrossRef] [PubMed]
- Kryukov, G.V.; Castellano, S.; Novoselov, S.V.; Lobanov, A.V.; Zehtab, O.; Guigo, R.; Gladyshev, V.N. Characterization of mammalian selenoproteomes. Science 2003, 300, 1439–1443. [Google Scholar] [CrossRef] [PubMed]
- Raman, A.V.; Berry, M.J. Selenoproteins in cellular redox regulation and signaling. Oxid. Stress Vertebr. Invertebr. 2012, 195–208. [Google Scholar]
- Steinbrenner, H.; Sies, H. Protection against reactive oxygen species by selenoproteins. Biochim. Biophys. Acta Gen. Subj. 2009, 1790, 1478–1485. [Google Scholar] [CrossRef] [PubMed]
- MacFarquhar Jennifer, K.; Broussard Danielle, L.; Melstrom, P.; Hutchinson, R.; Wolkin, A.; Martin, C.; Burk Raymond, F.; Dunn John, R.; Green Alice, L.; Hammond, R.; et al. Acute selenium toxicity associated with a dietary supplement. Arch. Intern. Med. 2010, 170, 256–261. [Google Scholar] [CrossRef] [PubMed]
- Puigdomenech, I. Hydra/medusa Chemical Equilibrium Database and Plotting Software. Available online: http://www.kth.se/che/medusa/ (accessed on 14 November 2016).
- Barnum, D.W. Potential-pH diagrams. J. Chem. Educ. 1982, 59, 809–812. [Google Scholar] [CrossRef]
- Campbell, J.A.; Whiteker, R.A. Periodic table based on potential-pH diagrams. J. Chem. Educ. 1969, 46, 90–92. [Google Scholar] [CrossRef]
- Delahay, P.; Pourbaix, M.; Van Rysselberghe, P. Potential-pH diagrams. J. Chem. Educ. 1950, 27, 683–688. [Google Scholar] [CrossRef]
- Pesterfield, L.L.; Maddox, J.B.; Crocker, M.S.; Schweitzer, G.K. Pourbaix (E-pH-M) diagrams in three dimensions. J. Chem. Educ. 2012, 89, 891–899. [Google Scholar] [CrossRef]
- Nuttall, K.L.; Allen, F.S. Kinetics of the reaction between hydrogen selenide ion and oxygen. Inorg. Chim. Acta 1984, 91, 243–246. [Google Scholar] [CrossRef]
- Kemp, M.; Go, Y.-M.; Jones, D.P. Nonequilibrium thermodynamics of thiol/disulfide redox systems: A perspective on redox systems biology. Free Radic. Biol. Med. 2008, 44, 921–937. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.P.; Carlson, J.L.; Mody, V.C.; Cai, J.; Lynn, M.J.; Sternberg, P. Redox state of glutathione in human plasma. Free Radic. Biol. Med. 2000, 28, 625–635. [Google Scholar] [CrossRef]
- Painter, E.P. The chemistry and toxicity of selenium compounds, with special reference to the selenium problem. Chem. Rev. 1941, 28, 179–213. [Google Scholar] [CrossRef]
- Ganther, H.E. Selenotrisulfides. Formation by the reaction of thiols with selenious acid. Biochemistry 1968, 7, 2898–2905. [Google Scholar] [CrossRef] [PubMed]
- Ganther, H.E. Reduction of the selenotrisulfide derivative of glutathione to a persulfide analog by glutathione reductase. Biochemistry 1971, 10, 4089–4098. [Google Scholar] [CrossRef] [PubMed]
- Kice, J.L.; Lee, T.W.S.; Pan, S.-T. Mechanism of the reaction of thiols with selenite. J. Am. Chem. Soc. 1980, 102, 4448–4455. [Google Scholar] [CrossRef]
- Tarze, A.; Dauplais, M.; Grigoras, I.; Lazard, M.; Ha-Duong, N.-T.; Barbier, F.; Blanquet, S.; Plateau, P. Extracellular production of hydrogen selenide accounts for thiol-assisted toxicity of selenite against saccharomyces cerevisiae. J. Biol. Chem. 2007, 282, 8759–8767. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Jhee, K.-H.; Kruger, W.D. Production of the neuromodulator H2S by cystathionine β-synthase via the condensation of cysteine and homocysteine. J. Biol. Chem. 2004, 279, 52082–52086. [Google Scholar] [CrossRef] [PubMed]
- Chiku, T.; Padovani, D.; Zhu, W.; Singh, S.; Vitvitsky, V.; Banerjee, R. H2S biogenesis by human cystathionine γ-lyase leads to the novel sulfur metabolites lanthionine and homolanthionine and is responsive to the grade of hyperhomocysteinemia. J. Biol. Chem. 2009, 284, 11601–11612. [Google Scholar] [CrossRef] [PubMed]
- Kimura, Y.; Toyofuku, Y.; Koike, S.; Shibuya, N.; Nagahara, N.; Lefer, D.; Ogasawara, Y.; Kimura, H. Identification of H2S3 and H2S produced by 3-mercaptopyruvate sulfurtransferase in the brain. Sci. Rep. 2015, 5, 14774. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, R.; Otsuguro, K.-I.; Yamaguchi, S.; Ito, S. Contribution of cysteine aminotransferase and mercaptopyruvate sulfurtransferase to hydrogen sulfide production in peripheral neurons. J. Neurochem. 2014, 130, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H. Hydrogen sulfide: Its production, release and functions. Amino Acids 2011, 41, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Ogasawara, Y.; Ishii, K.; Togawa, T.; Tanabe, S. Determination of bound sulfur in serum by gas dialysis/high-performance liquid chromatography. Anal. Biochem. 1993, 215, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Toohey, J.I. Sulphane sulphur in biological systems: A possible regulatory role. Biochem. J. 1989, 264, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Ishigami, M.; Hiraki, K.; Umemura, K.; Ogasawara, Y.; Ishii, K.; Kimura, H. A source of hydrogen sulfide and a mechanism of its release in the brain. Antioxid. Redox Signal. 2009, 11, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Ogasawara, Y.; Isoda, S.; Tanabe, S. Tissue and subcellular distribution of bound and acid-labile sulfur, and the enzymic capacity for sulfide production in the rat. Biol. Pharm. Bull. 1994, 17, 1535–1542. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Moore, P.K. Putative biological roles of hydrogen sulfide in health and disease: A breath of not so fresh air? Trends Pharmacol. Sci. 2008, 29, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Itoh, M.; Suzuki, K.T. Effects of dose on the methylation of selenium to monomethylselenol and trimethylselenonium ion in rats. Arch. Toxicol. 1997, 71, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Okuno, T.; Hasegawa, T.; Ueno, H.; Nakamuro, K. Study on detoxification and utilization of selenomethionine: A role of cystathionine γ-lyase in selenomethionine metabolism. Biomed. Res. Trace Elem. 2007, 18, 221–230. [Google Scholar]
- Ohta, Y.; Suzuki, K.T. Methylation and demethylation of intermediates selenide and methylselenol in the metabolism of selenium. Toxicol. Appl. Pharmacol. 2008, 226, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Lisk, D.; Block, E.; Ip, C. Characterization of the biological activity of γ-glutamyl-se-methylselenocysteine: A novel, naturally occurring anticancer agent from garlic. Cancer Res. 2001, 61, 2923–2928. [Google Scholar] [PubMed]
- Andreadou, I.; Menge, W.M.P.B.; Commandeur, J.N.M.; Worthington, E.A.; Vermeulen, N.P.E. Synthesis of novel Se-substituted selenocysteine derivatives as potential kidney selective prodrugs of biologically active selenol compounds: Evaluation of kinetics of β-elimination reactions in rat renal cytosol. J. Med. Chem. 1996, 39, 2040–2046. [Google Scholar] [CrossRef] [PubMed]
- Esaki, N.; Nakamura, T.; Tanaka, H.; Soda, K. Selenocysteine lyase, a novel enzyme that specifically acts on selenocysteine. Mammalian distribution and purification and properties of pig liver enzyme. J. Biol. Chem. 1982, 257, 4386–4391. [Google Scholar] [PubMed]
- Lu, J.; Berndt, C.; Holmgren, A. Metabolism of selenium compounds catalyzed by the mammalian selenoprotein thioredoxin reductase. Biochim. Biophys. Acta Gen. Subj. 2009, 1790, 1513–1519. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, H.S.; Ganther, H.E. Biosynthesis of dimethyl selenide from sodium selenite in rat liver and kidney cell-free systems. Biochim. Biophys. Acta Gen. Subj. 1977, 497, 205–217. [Google Scholar] [CrossRef]
- Wolfe, M.D. Selenophosphate synthetase. EcoSal Plus 2014, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Francesconi Kevin, A.; Pannier, F. Selenium metabolites in urine: A critical overview of past work and current status. Clin. Chem. 2004, 50, 2240–2253. [Google Scholar] [CrossRef] [PubMed]
- Kremer, D.; Ilgen, G.; Feldmann, J. GC-ICP-MS determination of dimethylselenide in human breath after ingestion of 77Se-enriched selenite: Monitoring of in vivo methylation of selenium. Anal. Bioanal. Chem. 2005, 383, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Lajin, B.; Kuehnelt, D.; Francesconi, K.A. Exploring the urinary selenometabolome following a multi-phase selenite administration regimen in humans. Metallomics Integr. Biomet. Sci. 2016, 8, 774–781. [Google Scholar] [CrossRef] [PubMed]
- Stadtman, T.C. Selenocysteine lyase. EcoSal Plus 2014, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Veres, Z.; Tsai, L.; Scholz, T.D.; Politino, M.; Balaban, R.S.; Stadtman, T.C. Synthesis of 5-methylaminomethyl-2-selenouridine in tRNAs: Phosphorus-31 NMR studies show the labile selenium donor synthesized by the seld gene product contains selenium bonded to phosphorus. Proc. Natl. Acad. Sci. USA 1992, 89, 2975–2979. [Google Scholar] [CrossRef] [PubMed]
- Glass, R.S.; Singh, W.P.; Jung, W.; Veres, Z.; Scholz, T.D.; Stadtman, T. Monoselenophosphate: Synthesis, characterization, and identity with the prokaryotic biological selenium donor, compound sepx. Biochemistry 1993, 32, 12555–12559. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, A.P.; Wallenberg, M.; Gandin, V.; Misra, S.; Tisato, F.; Marzano, C.; Rigobello, M.P.; Kumar, S.; Bjoernstedt, M. Methylselenol formed by spontaneous methylation of selenide is a superior selenium substrate to the thioredoxin and glutaredoxin systems. PLoS ONE 2012, 7, e50727. [Google Scholar] [CrossRef] [PubMed]
- Stadtman, T.C. Selenocysteine. Ann. Rev. Biochem. 1996, 65, 83–100. [Google Scholar] [CrossRef] [PubMed]
- Anan, Y.; Kimura, M.; Hayashi, M.; Koike, R.; Ogra, Y. Detoxification of selenite to form selenocyanate in mammalian cells. Chem. Res. Toxicol. 2015, 28, 1803–1814. [Google Scholar] [CrossRef] [PubMed]
- Lacourciere, G.M. Selenium is mobilized in vivo from free selenocysteine and is incorporated specifically into formate dehydrogenase H and tRNA nucleosides. J. Bacteriol. 2002, 184, 1940–1946. [Google Scholar] [CrossRef] [PubMed]
- Lacourciere, G.M.; Stadtman, T.C. Utilization of selenocysteine as a source of selenium for selenophosphate biosynthesis. BioFactors 2001, 14, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Chocat, P.; Esaki, N.; Tanizawa, K.; Nakamura, K.; Tanaka, H.; Soda, K. Purification and characterization of selenocysteine beta-lyase from citrobacter freundii. J. Bacteriol. 1985, 163, 669–676. [Google Scholar] [PubMed]
- Ganther, H.E. Enzymic synthesis of dimethyl selenide from sodium selenite in mouse liver extracts. Biochemistry 1966, 5, 1089–1098. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.T.; Kurasaki, K.; Suzuki, N. Selenocysteine β-lyase and methylselenol demethylase in the metabolism of Se-methylated selenocompounds into selenide. Biochim. Biophys. Acta Gen. Subj. 2007, 1770, 1053–1061. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.T.; Kurasaki, K.; Ogawa, S.; Suzuki, N. Metabolic transformation of methylseleninic acid through key selenium intermediate selenide. Toxicol. Appl. Pharmacol. 2006, 215, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.T.; Doi, C.; Suzuki, N. Metabolism of 76Se-methylselenocysteine compared with that of 77Se-selenomethionine and 82Se-selenite. Toxicol. Appl. Pharmacol. 2006, 217, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Ogra, Y.; Ishiwata, K.; Takayama, H.; Aimi, N.; Suzuki, K.T. Selenosugars are key and urinary metabolites for selenium excretion within the required to low-toxic range. Proc. Natl. Acad. Sci. USA 2002, 99, 15932–15936. [Google Scholar] [CrossRef] [PubMed]
- Ogra, Y. Selenium metabolism. In Diversity of Selenium Functions in Health and Disease; Regina, B.-F., Sies, H., Eds.; CRC Press: Boca Raton, FL, USA, 2016; pp. 19–30. [Google Scholar]
- Weekley, C.M.; Harris, H.H. Which form is that? The importance of selenium speciation and metabolism in the prevention and treatment of disease. Chem. Soc. Rev. 2013, 42, 8870–8894. [Google Scholar] [CrossRef] [PubMed]
- Rahmanto Aldwin, S.; Davies Michael, J. Selenium-containing amino acids as direct and indirect antioxidants. IUBMB Life 2012, 64, 863–871. [Google Scholar] [CrossRef] [PubMed]
- Rahmanto, A.S.; Davies, M.J. Catalytic activity of selenomethionine in removing amino acid, peptide, and protein hydroperoxides. Free Radic. Biol. Med. 2011, 51, 2288–2299. [Google Scholar] [CrossRef] [PubMed]
- Krause, R.J.; Elfarra, A.A. Reduction of l-methionine selenoxide to seleno-l-methionine by endogenous thiols, ascorbic acid, or methimazole. Biochem. Pharmacol. 2009, 77, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Nagy, P.; Becker, J.D.; Mallo, R.C.; Ashby, M.T. The jekyll and hyde roles of cysteine derivatives during oxidative stress. ACS Symp. Ser. 2007, 967, 193–212. [Google Scholar]
- Buettner, G.R. The pecking order of free radicals and antioxidants: Lipid peroxidation, alpha-tocopherol, and ascorbate. Arch. Biochem. Biophys. 1993, 300, 535–543. [Google Scholar] [CrossRef] [PubMed]
- Smirnova, G.V.; Muzyka, N.G.; Oktyabrsky, O.N. Effects of cystine and hydrogen peroxide on glutathione status and expression of antioxidant genes in escherichia coli. Biochemistry 2005, 70, 926–934. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Imlay, J.A. High levels of intracellular cysteine promote oxidative DNA damage by driving the fenton reaction. J. Bacteriol. 2003, 185, 1942–1950. [Google Scholar] [CrossRef] [PubMed]
- Berglin, E.H.; Edlund, M.B.K.; Nyberg, G.K.; Carlsson, J. Potentiation by l-cysteine of the bactericidal effect of hydrogen peroxide in escherichia coli. J. Bacteriol. 1982, 152, 81–88. [Google Scholar] [PubMed]
- Singh, R.; Whitesides, G.M. Selenols catalyze the interchange reactions of dithiols and disulfides in water. J. Org. Chem. 1991, 56, 6931–6933. [Google Scholar] [CrossRef]
- Hafeman, D.G.; Sunde, R.A.; Hoekstra, W.G. Effect of dietary selenium on erythrocyte and liver glutathione peroxidase in the rat. J. Nutr. 1974, 104, 580–587. [Google Scholar] [PubMed]
- Csallany, A.S.; Menken, B.Z. Effect of dietary selenite on hepatic organic solvent-soluble lipofuscin pigments. J. Am. Coll. Toxicol. 1986, 5, 79–85. [Google Scholar] [CrossRef]
- Spallholz, J.E. Free radical generation by selenium compounds and their prooxidant toxicity. Biomed. Environ. Sci. 1997, 10, 260–270. [Google Scholar] [PubMed]
- Terada, A.; Yoshida, M.; Seko, Y.; Kobayashi, T.; Yoshida, K.; Nakada, M.; Nakada, K.; Echizen, H.; Ogata, H.; Rikihisa, T. Active oxygen species generation and cellular damage by additives of parenteral preparations: Selenium and sulfhydryl compounds. Nutrition 1999, 15, 651–655. [Google Scholar] [CrossRef]
- Raisbeck, M.F. Selenosis. Vet. Clin. N. Am. Food Anim. Pract. 2000, 16, 465–480. [Google Scholar] [CrossRef]
- Spallholz, J.E. On the nature of selenium toxicity and carcinostatic activity. Free Radic Biol Med 1994, 17, 45–64. [Google Scholar] [CrossRef]
- Bierla, K.; Bianga, J.; Ouerdane, L.; Szpunar, J.; Yiannikouris, A.; Lobinski, R. A comparative study of the Se/S substitution in methionine and cysteine in Se-enriched yeast using an inductively coupled plasma mass spectrometry (ICP MS)-assisted proteomics approach. J. Proteom. 2013, 87, 26–39. [Google Scholar] [CrossRef] [PubMed]
- Ip, C. Lessons from basic research in selenium and cancer prevention. J. Nutr. 1998, 128, 1845–1854. [Google Scholar] [PubMed]
- Combs, G.F., Jr.; Gray, W.P. Chemopreventive agents: Selenium. Pharmacol. Ther. 1998, 79, 179–192. [Google Scholar] [CrossRef]
- Rayman, M.P. Selenium in cancer prevention: A review of the evidence and mechanism of action. Proc. Nutr. Soc. 2005, 64, 527–542. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Combs, G.F., Jr. Selenium as an anticancer nutrient: Roles in cell proliferation and tumor cell invasion. J. Nutr. Biochem. 2008, 19, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Schrauzer, G.N. Anticarcinogenic effects of selenium. Cell. Mol. Life Sci. 2000, 57, 1864–1873. [Google Scholar] [CrossRef] [PubMed]
- Weekley, C.M.; Jeong, G.; Tierney, M.E.; Hossain, F.; Maw, A.M.; Shanu, A.; Harris, H.H.; Witting, P.K. Selenite-mediated production of superoxide radical anions in A549 cancer cells is accompanied by a selective increase in SOD1 concentration, enhanced apoptosis and Se-Cu bonding. J. Biol. Inorg. Chem. 2014, 19, 813–828. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-J.; Boylan, L.M.; Wu, C.-K.; Spallholz, J.E. Oxidation of glutathione and superoxide generation by inorganic and organic selenium compounds. BioFactors 2007, 31, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.-M.; Yang, C.-F.; Ong, C.-N. Sodium selenite-induced oxidative stress and apoptosis in human hepatoma HepG2 cells. Int. J. Cancer 1999, 81, 820–828. [Google Scholar] [CrossRef]
- Stewart, M.S.; Spallholz, J.E.; Neldner, K.H.; Pence, B.C. Selenium compounds have disparate abilities to impose oxidative stress and induce apoptosis. Free Radic. Biol. Med. 1998, 26, 42–48. [Google Scholar] [CrossRef]
- Kim, E.H.; Sohn, S.; Kwon, H.J.; Kim, S.U.; Kim, M.-J.; Lee, S.-J.; Choi, K.S. Sodium selenite induces superoxide-mediated mitochondrial damage and subsequent autophagic cell death in malignant glioma cells. Cancer Res. 2007, 67, 6314–6324. [Google Scholar] [CrossRef] [PubMed]
- Zhou, N.; Xiao, H.; Li, T.-K.; Nur-E-Kamal, A.; Liu, L.F. DNA damage-mediated apoptosis induced by selenium compounds. J. Biol. Chem. 2003, 278, 29532–29537. [Google Scholar] [CrossRef] [PubMed]
- Thompson, H.J.; Wilson, A.; Lu, J.; Singh, M.; Jiang, C.; Upadhyaya, P.; El-Bayoumy, K.; Ip, C. Comparison of the effects of an organic and an inorganic form of selenium on a mammary carcinoma cell line. Carcinogenesis 1994, 15, 183–186. [Google Scholar] [CrossRef] [PubMed]
- Drake, E.N. Cancer chemoprevention: Selenium as a prooxidant, not an antioxidant. Med. Hypotheses 2006, 67, 318–322. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.M.; Yang, C.F.; Ding, W.X.; Liu, J.; Ong, C.N. Superoxide radical-initiated apoptotic signalling pathway in selenite-treated HepG2 cells: Mitochondria serve as the main target. Free Radic. Biol. Med. 2001, 30, 9–21. [Google Scholar] [CrossRef]
- Shen, H.M.; Yang, C.F.; Liu, J.; Ong, C.N. Dual role of glutathione in selenite-induced oxidative stress and apoptosis in human hepatoma cells. Free Radic. Biol. Med. 2000, 28, 1115–1124. [Google Scholar] [CrossRef]
- Fisher, B.; Yarmolinsky, D.; Abdel-Ghany, S.; Pilon, M.; Pilon-Smits, E.A.; Sagi, M.; Van Hoewyk, D. Superoxide generated from the glutathione-mediated reduction of selenite damages the iron-sulfur cluster of chloroplastic ferredoxin. Plant Physiol. Biochem. 2016, 106, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Seko, Y.; Saito, Y.; Kitahara, J.; Imura, N. Active oxygen generation by the reaction of selenite with reduced glutathione in vitro. In Selenium in Biology and Medicine; Springer: Berlin, Germany, 1989; pp. 70–73. [Google Scholar]
- Pilon, M.; Owen Jennifer, D.; Garifullina Gulnara, F.; Kurihara, T.; Mihara, H.; Esaki, N.; Pilon-Smits Elizabeth, A.H. Enhanced selenium tolerance and accumulation in transgenic arabidopsis expressing a mouse selenocysteine lyase. Plant Physiol. 2003, 131, 1250–1257. [Google Scholar] [CrossRef] [PubMed]
- Kessi, J.; Hanselmann, K.W. Similarities between the abiotic reduction of selenite with glutathione and the dissimilatory reaction mediated by rhodospirillum rubrum and escherichia coli. J. Biol. Chem. 2004, 279, 50662–50669. [Google Scholar] [CrossRef] [PubMed]
- Peyroche, G.; Saveanu, C.; Dauplais, M.; Lazard, M.; Beuneu, F.; Decourty, L.; Malabat, C.; Jacquier, A.; Blanquet, S.; Plateau, P. Sodium selenide toxicity is mediated by O2-dependent DNA breaks. PLoS ONE 2012, 7, e36343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrero, E.; Weilinger, R.E. Yeast as a model system to study metabolic impact of selenium compounds. Microb. Cell 2015, 2, 139–149. [Google Scholar] [CrossRef]
- Bockhorn, J.; Balar, B.; He, D.; Seitomer, E.; Copeland, P.R.; Kinzy, T.G. Genome-wide screen of saccharomyces cerevisiae null allele strains identifies genes involved in selenomethionine resistance. Proc. Natl. Acad. Sci. USA 2008, 105, 17682–17687. [Google Scholar] [CrossRef] [PubMed]
- Meyer, J.; Moulis, J.M.; Gaillard, J.; Lutz, M. Replacement of sulfur by selenium in iron-sulfur proteins. Adv. Inorg. Chem. 1992, 38, 73–115. [Google Scholar]
- Spatzal, T.; Rees Douglas, C.; Spatzal, T.; Perez Kathryn, A.; Howard James, B.; Rees Douglas, C.; Howard James, B. Catalysis-dependent selenium incorporation and migration in the nitrogenase active site iron-molybdenum cofactor. eLife 2015, 4, e11620. [Google Scholar] [CrossRef] [PubMed]
- Wrobel, M.; Jurkowska, H.; Sliwa, L.; Srebro, Z. Sulfurtransferases and cyanide detoxification in mouse liver, kidney, and brain. Toxicol. Mech. Methods 2004, 14, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Bordo, D.; Bork, P. The rhodanese/Cdc25 phosphatase superfamily. Sequence-structure-function relations. EMBO Rep. 2002, 3, 741–746. [Google Scholar] [CrossRef] [PubMed]
- Westley, J. Rhodanese. Adv. Enzymol. Relat. Areas Mol. Biol. 1973, 39, 327–368. [Google Scholar] [PubMed]
- Mueller, E.G. Trafficking in persulfides: Delivering sulfur in biosynthetic pathways. Nat. Chem. Biol. 2006, 2, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Weuffen, W.; Franzke, C.; Thuerkow, B. Alimentary ingestion, analysis and biological significance of thiocyanate. Nahrung 1984, 28, 341–355. [Google Scholar] [CrossRef] [PubMed]
- Moir, D.; Rickert, W.S.; Levasseur, G.; Larose, Y.; Maertens, R.; White, P.; Desjardins, S. A comparison of mainstream and sidestream marijuana and tobacco cigarette smoke produced under two machine smoking conditions. Chem. Res. Toxicol. 2008, 21, 494–502. [Google Scholar] [CrossRef] [PubMed]
- Zgliczynski, J.M.; Stelmaszynska, T. Hydrogen cyanide and cyanogen chloride formation by the myeloperoxidase hydrogen peroxide-chloride ion system. Biochim. Biophys. Acta Enzymol. 1979, 567, 309–314. [Google Scholar] [CrossRef]
- Pessi, G.; Haas, D. Transcriptional control of the hydrogen cyanide biosynthetic genes hcnABC by the anaerobic regulator ANR and the quorum-sensing regulators LasR and RhlR in pseudomonas aeruginosa. J. Bacteriol. 2000, 182, 6940–6949. [Google Scholar] [CrossRef] [PubMed]
- Ryall, B.; Davies, J.C.; Wilson, R.; Shoemark, A.; Williams, H.D. Pseudomonas aeruginosa, cyanide accumulation and lung function in CF and non-CF bronchiectasis patients. Eur. Respir. J. 2008, 32, 740–747. [Google Scholar] [CrossRef] [PubMed]
- Broderick, K.E.; Chan, A.; Balasubramanian, M.; Feala, J.; Reed, S.L.; Panda, M.; Sharma, V.S.; Pilz, R.B.; Bigby, T.D.; Boss, G.R. Cyanide produced by human isolates of pseudomonas aeruginosa contributes to lethality in drosophila melanogaster. J. Infect. Dis. 2007, 197, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Askeland, R.A.; Morrison, S.M. Cyanide production by pseudomonas fluorescens and pseudomonas aeruginosa. Appl. Environ. Microbiol. 1983, 45, 1802–1807. [Google Scholar] [PubMed]
- Conner, G.E.; Wijkstrom-Frei, C.; Randell, S.H.; Fernandez, V.E.; Salathe, M. The lactoperoxidase system links anion transport to host defense in cystic fibrosis. FEBS Lett. 2007, 581, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Fragoso, M.A.; Fernandez, V.; Forteza, R.; Randell, S.H.; Salathe, M.; Conner, G.E. Transcellular thiocyanate transport by human airway epithelia. J. Physiol. 2004, 561, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Eskandari, S.; Loo, D.D.F.; Dai, G.; Levy, O.; Wright, E.M.; Carrasco, N. Thyroid NA+/I− symporter. Mechanism, stoichiometry, and specificity. J. Biol. Chem. 1997, 272, 27230–27238. [Google Scholar] [CrossRef] [PubMed]
- Wolff, J.; Maurey, J.R. Thyroidal iodide transport. IX. The accumulation and metabolism of selenocyanate. Endocrinology 1966, 79, 795–800. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-L.; Zhao, Y.-B. CdS quantum dots as fluorescence probes for the sensitive and selective detection of highly reactive HSe− ions in aqueous solution. Anal. Bioanal. Chem. 2007, 388, 717–722. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Property | S | Se | |
|---|---|---|---|
| Electron configuration | [Ne] 3s23p4 | [Ar] 3d104s24p4 | |
| Average atomic mass (amu) | 32.06 | 78.96 | |
| Covalent radius (pm) [30] | 100 | 115 | |
| van der Waals radius (pm) [31] | 180 | 190 | |
| Bond length (pm) [32] | 134 (S–H) | 146 (Se–H) | |
| 180 (S–C) | 196 (Se–C) | ||
| 205 (S–S) | 232 (Se–Se) | ||
| Bond energy (kJ·mol−1) | HX–H | 381.6 (S–H) [33] | 334.9 (Se–H) [34] |
| CH3X–CH3 | 307.9 (S–C) [33] | 234 (Se–C) [35] | |
| CH3X–XCH3 | 273.6 (S–S) [33] | 197.6 (Se–Se) [36] | |
| Ionization Energies (kJ·mol−1) [37] | 1st | 999.6 | 940.9 |
| 2nd | 2251 | 2045 | |
| 3rd | 3361 | 2974 | |
| Electron affinity (kJ·mol−1) [38] | 200 | 195 | |
| Pauling electronegativity [39] | 2.58 | 2.55 | |
| Polarizability (in Å3) | 2.90 [40] | 3.89 [41] | |
| pKa1, (H2X) | 6.88 [42] | 3.89 [43] | |
| pKa2, (HX−) | 14.15 [42] | 15.1 [44] | |
| pKa2, (Cys/Sec) [45] | 8.22 [46,47] | 5.43 [48] | |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cupp-Sutton, K.A.; Ashby, M.T. Biological Chemistry of Hydrogen Selenide. Antioxidants 2016, 5, 42. https://doi.org/10.3390/antiox5040042
Cupp-Sutton KA, Ashby MT. Biological Chemistry of Hydrogen Selenide. Antioxidants. 2016; 5(4):42. https://doi.org/10.3390/antiox5040042
Chicago/Turabian StyleCupp-Sutton, Kellye A., and Michael T. Ashby. 2016. "Biological Chemistry of Hydrogen Selenide" Antioxidants 5, no. 4: 42. https://doi.org/10.3390/antiox5040042
APA StyleCupp-Sutton, K. A., & Ashby, M. T. (2016). Biological Chemistry of Hydrogen Selenide. Antioxidants, 5(4), 42. https://doi.org/10.3390/antiox5040042