The Ability of Exercise-Associated Oxidative Stress to Trigger Redox-Sensitive Signalling Responses

Abstract
:1. Introduction
2. Redox-Sensitive Signalling Effects
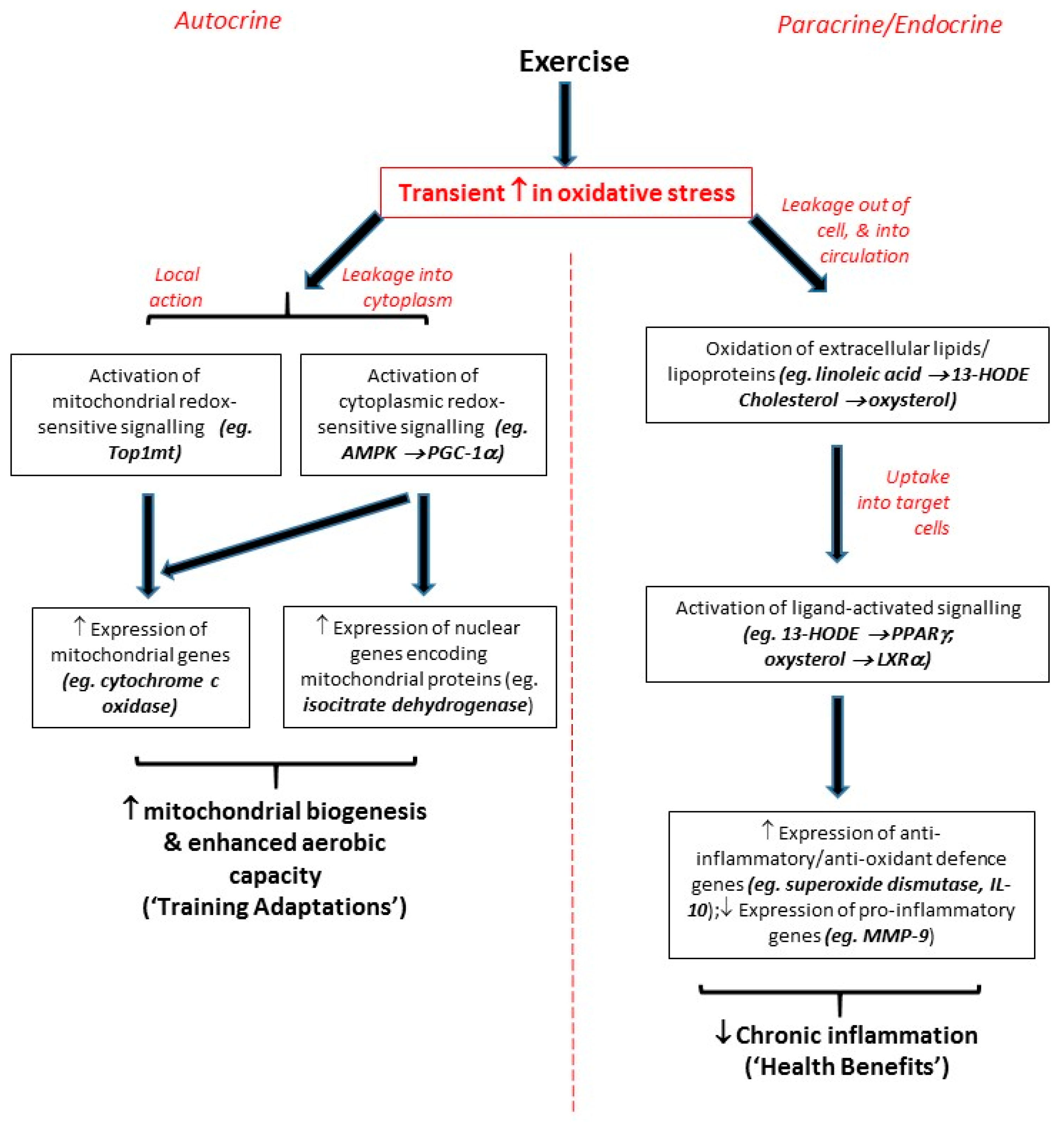
2.1. Endogenous ROS as Direct Triggers of Local Autocrine Signalling Effects
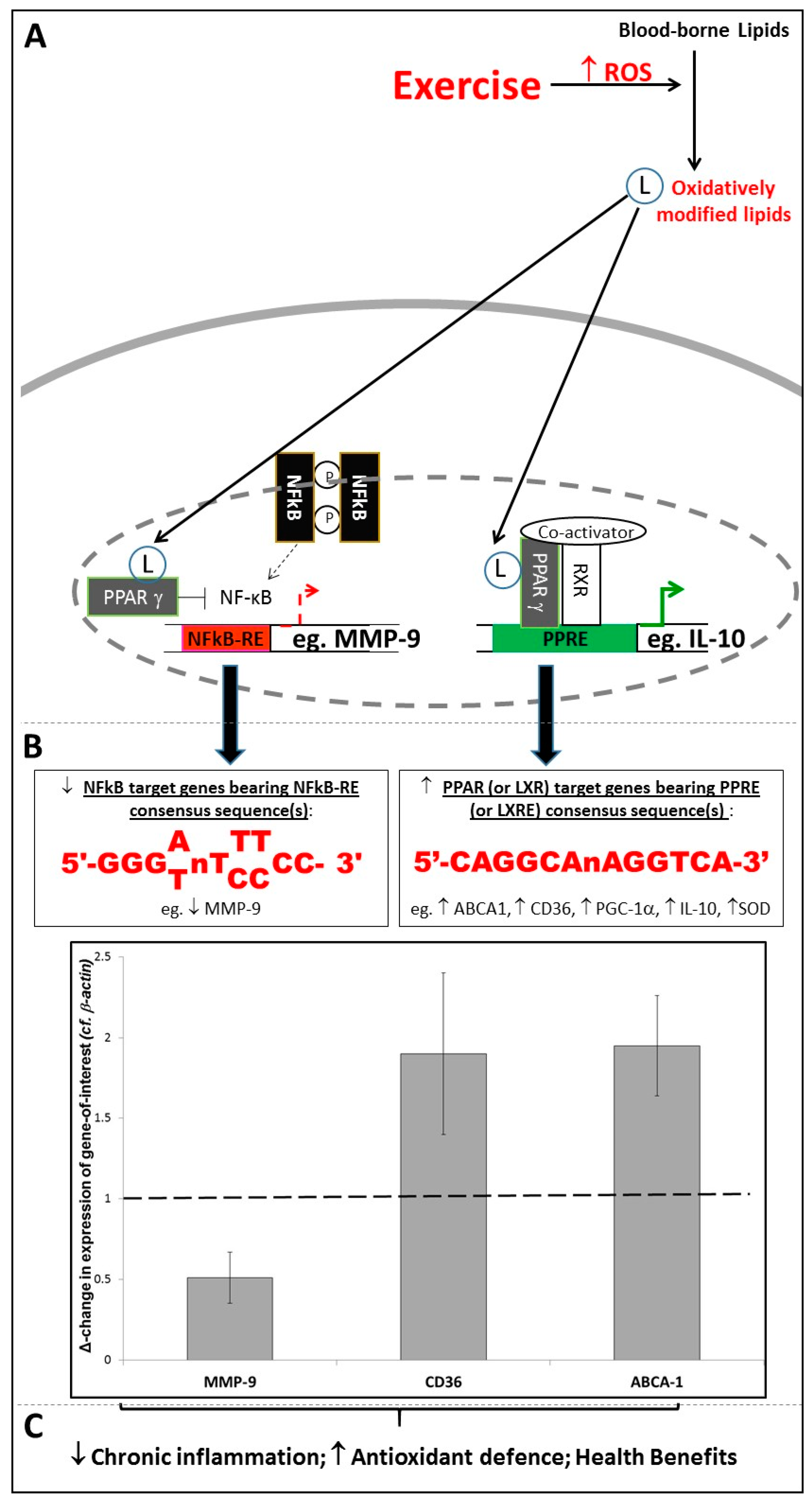
2.2. Extracellular ROS as Paracrine/Endocrine Signalling Molecules Exerting Distant Effects
3. Exercise as an Initiator of Redox-Sensitive Signaling Responses
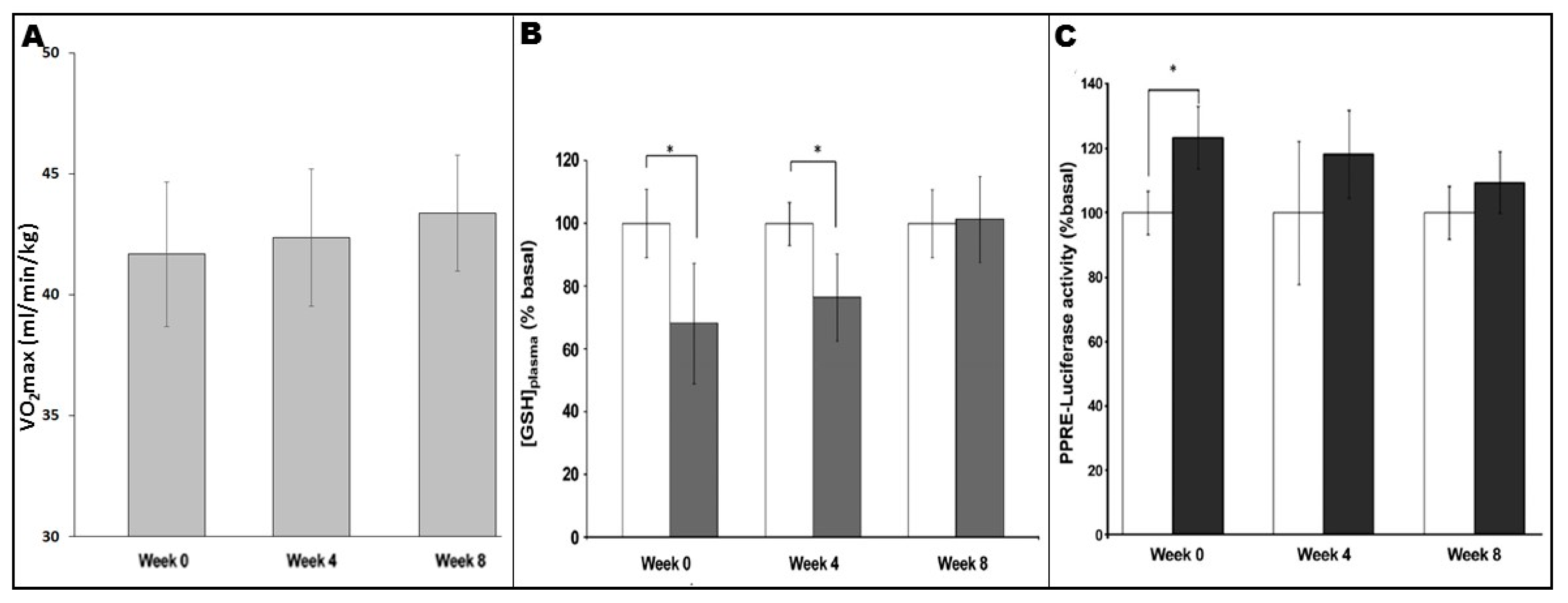
4. Exercise-Associated Redox-Sensitive Signalling Responses and Chronic Inflammatory Disease
5. Exercise-Associated Redox-Sensitive Signaling Responses and Design of Exercise Programmes
6. Conclusions
Author Contributions
Conflicts of Interest
References
- Evans, L.W.; Omaye, S.T. Use of Saliva Biomarkers to Monitor Efficacy of Vitamin C in Exercise-Induced Oxidative Stress. Antioxidants 2017, 6, 5. [Google Scholar] [CrossRef] [PubMed]
- McLeay, Y.; Stannard, S.; Houltham, S.; Starck, C. Dietary thiols in exercise: Oxidative stress defence, exercise performance, and adaptation. J. Int. Soc. Sports Nutr. 2017, 14, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Steinbacher, P. Impact of oxidative stress on exercising skeletal muscle. Biomolecules 2015, 5, 356–377. [Google Scholar] [CrossRef] [PubMed]
- Bild, W.; Ciobica, A.; Padurariu, M.; Bild, V. The interdependence of the reactive species of oxygen, nitrogen, and carbon. J. Physiol. Biochem. 2013, 69, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Harman, D. Aging: A theory based on free radical and radiation chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef] [PubMed]
- Desler, C.; Marcker, M.L.; Singh, K.K.; Rasmussen, L.J. The importance of mitochondrial DNA in aging and cancer. J. Aging Res. 2011, 407536, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Lagouge, M.; Larsson, N.G. The role of mitochondrial DNA mutations and free radicals in disease and ageing. J. Intern. Med. 2013, 273, 529–543. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.J. Control of reactive oxygen species production in contracting skeletal muscle. Antioxid. Redox Signal. 2011, 15, 2477–2486. [Google Scholar] [CrossRef] [PubMed]
- Powers, S.K.; Jackson, M.J. Exercise-induced oxidative stress: Cellular mechanisms and impact on muscle force production. Physiol. Rev. 2008, 88, 1243–1276. [Google Scholar] [CrossRef] [PubMed]
- Vasilaki, A.; Mansouri, A.; van Remmen, H.; van der Meulen, J.H.; Larkin, L.; Richardson, A.G.; McArdle, A.; Faulkner, J.A.; Jackson, M.J. Free radical generation by skeletal muscle of adult and old mice: Effect of contractile activity. Aging Cell 2006, 5, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Palomero, J.; Pye, D.; Kabayo, T.; Spiller, D.G.; Jackson, M.J. In situ detection and measurement of intracellular reactive oxygen species in single isolated mature skeletal muscle fibers by real time fluorescence microscopy. Antioxid. Redox Signal. 2008, 10, 1463–1474. [Google Scholar] [CrossRef] [PubMed]
- Teixeira de Lemos, E.; Pinto, R.; Oliveira, J.; Garrido, P.; Sereno, J.; Mascarenhas-Melo, F.; Páscoa-Pinheiro, J.; Teixeira, F.; Reis, F. Differential effects of acute (extenuating) and chronic (training) exercise on inflammation and oxidative stress status in an animal model of type 2 diabetes mellitus. Mediat. Inflamm. 2011, 2011, 253061. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Chiang, H.H.; Louw, M.; Susanto, A.; Chen, D. Nutrient Sensing and the Oxidative Stress Response. Trends Endocrinol. Metab. 2017, 28, 449–460. [Google Scholar] [CrossRef] [PubMed]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, oxidants, and aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Lane, N. The Vital Question: Why Is Life the Way It Is? 1st ed.; Profile Books Ltd.: London, UK, 2015; pp. 237–279. ISBN 9781781250365. [Google Scholar]
- Douarre, C.; Sourbier, C.; Dalla Rosa, I.; Brata Das, B.; Redon, C.E.; Zhang, H.; Neckers, L.; Pommier, Y. Mitochondrial topoisomerase I is critical for mitochondrial integrity and cellular energy metabolism. PLoS ONE 2012, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Khiati, S.; Baechler, S.A.; Factor, V.M.; Zhang, H.; Huang, S.Y.; Dalla Rosa, I.; Sourbier, C.; Neckers, L.; Thorgeirsson, S.S.; Pommier, Y. Lack of mitochondrial topoisomerase I (TOP1mt) impairs liver regeneration. Proc. Natl. Acad. Sci. USA 2015, 112, 11282–11287. [Google Scholar] [CrossRef] [PubMed]
- Irrcher, I.; Ljubicic, V.; Hood, D.A. Interactions between ROS and AMP kinase activity in the regulation of PGC-1alpha transcription in skeletal muscle cells. Am. J. Physiol. Cell Physiol. 2009, 296, C116–C123. [Google Scholar] [CrossRef] [PubMed]
- Safdar, A.; Little, J.P.; Stokl, A.J.; Hettinga, B.P.; Akhtar, M.; Tarnopolsky, M.A. Exercise increases mitochondrial PGC-1alpha content and promotes nuclear-mitochondrial cross-talk to coordinate mitochondrial biogenesis. J. Biol. Chem. 2011, 286, 10605–10617. [Google Scholar] [CrossRef] [PubMed]
- Russell, A.P.; Wada, S.; Vergani, L.; Hock, M.B.; Lamon, S.; Léger, B.; Ushida, T.; Cartoni, R.; Wadley, G.D.; Hespel, P.; et al. Disruption of skeletal muscle mitochondrial network genes and miRNAs in amyotrophic lateral sclerosis. Neurobiol. Dis. 2013, 49, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Wu, H.; Tarr, P.T.; Zhang, C.Y.; Wu, Z.; Boss, O.; Michael, L.F.; Puigserver, P.; Isotani, E.; Olson, E.N.; et al. Transcriptional co-activator PGC-1 alpha drives the formation of slow-twitch muscle fibres. Nature 2002, 418, 797–801. [Google Scholar] [CrossRef] [PubMed]
- Calvo, J.A.; Daniels, T.G.; Wang, X.; Paul, A.; Lin, J.; Spiegelman, B.M.; Stevenson, S.C.; Rangwala, S.M. Muscle-specific expression of PPARgamma coactivator-1alpha improves exercise performance and increases peak oxygen uptake. J. Appl. Physiol. 2008, 104, 1304–1312. [Google Scholar] [CrossRef] [PubMed]
- Russell, A.P.; Foletta, V.C.; Snow, R.J.; Wadley, G.D. Skeletal muscle mitochondria: A major player in exercise, health and disease. Biochim. Biophys. Acta 2014, 1840, 1276–1284. [Google Scholar] [CrossRef] [PubMed]
- Keller, P.; Vollaard, N.B.; Gustafsson, T.; Gallagher, I.J.; Sundberg, C.J.; Rankinen, T.; Britton, S.L.; Bouchard, C.; Koch, L.G.; Timmons, J.A. A transcriptional map of the impact of endurance exercise training on skeletal muscle phenotype. J. Appl. Physiol. 2011, 110, 46–59. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Cabrera, M.C.; Domenech, E.; Romagnoli, M.; Arduini, A.; Borras, C.; Pallardo, F.V.; Sastre, J.; Viña, J. Oral administration of Vitamin C decreases muscle mitochondrial biogenesis and hampers training-induced adaptations in endurance performance. Am. J. Clin. Nutr. 2008, 87, 142–149. [Google Scholar] [PubMed]
- Li, Y.; Zhu, H.; Kuppusamy, P.; Zweier, J.L.; Trush, M.A. Mitochondrial Electron Transport Chain-Derived Superoxide Exits Macrophages: Implications for Mononuclear Cell-Mediated Pathophysiological Processes. React. Oxyg. Species (Apex) 2016, 1, 81–98. [Google Scholar] [CrossRef] [PubMed]
- Nagy, L.; Tontonoz, P.; Alvarez, J.G.; Chen, H.; Evans, R.M. Oxidized LDL regulates macrophage gene expression through ligand activation of PPARgamma. Cell 1998, 93, 229–240. [Google Scholar] [CrossRef]
- Amanso, A.; Lyle, A.N.; Griendling, K.K. NADPH Oxidases and Measurement of Reactive Oxygen Species. Methods Mol. Biol. 2017, 1527, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Poli, G.; Biasi, F.; Leonarduzzi, G. Oxysterols in the pathogenesis of major chronic diseases. Redox Biol. 2013, 1, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Khassaf, M.; McArdle, A.; Esanu, C.; Vasilaki, A.; McArdle, F.; Griffiths, R.D.; Brodie, D.A.; Jackson, M.J. Effect of Vitamin C supplements on antioxidant defence and stress proteins in human lymphocytes and skeletal muscle. J. Physiol. 2003, 549 Pt 2, 645–652. [Google Scholar] [CrossRef] [PubMed]
- Davies, N.A.; Watkeys, L.; Butcher, L.; Potter, S.; Hughes, M.G.; Moir, H.; Morris, K.; Thomas, A.W.; Webb, R. The contributions of oxidative stress, oxidised lipoproteins and AMPK towards exercise-associated PPARγ signalling within human monocytic cells. Free Radic. Res. 2015, 49, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Butcher, L.; Backx, K.; Roberts, A.; Thomas, A.; Webb, R.; Morris, K. Low-intensity exercise exerts beneficial effects on plasma lipids via PPARγ. Med. Sci. Sports Exerc. 2008, 40, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Thompson, P.W.; Bayliffe, A.I.; Warren, A.P.; Lamb, J.R. Interleukin-10 is upregulated by nanomolar rosiglitazone treatment of mature dendritic cells and human CD4+ T cells. Cytokine 2007, 39, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.Y.; Chang, M.S.; Rho, H.M. Induction of the rat Cu/Zn superoxide dismutase gene through the peroxisome proliferator-responsive element by arachidonic acid. Gene 1999, 234, 87–91. [Google Scholar] [CrossRef]
- Zhou, Y.; Jia, S.; Wang, C.; Chen, Z.; Chi, Y.; Li, J.; Xu, G.; Guan, Y.; Yang, J. FAM3A is a target gene of peroxisome proliferator-activated receptor gamma. Biochim. Biophys. Acta 2013, 1830, 4160–4170. [Google Scholar] [CrossRef] [PubMed]
- Nunn, A.V.; Bell, J.; Barter, P. The integration of lipid-sensing and anti-inflammatory effects: How the PPARs play a role in metabolic balance. Nucl. Recept. 2007, 5, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Janowski, B.A.; Willy, P.J.; Devi, T.R.; Falck, J.R.; Mangelsdorf, D.J. An oxysterol signalling pathway mediated by the nuclear receptor LXR alpha. Nature 1996, 383, 728–731. [Google Scholar] [CrossRef] [PubMed]
- Mitro, N.; Mak, P.A.; Vargas, L.; Godio, C.; Hampton, E.; Molteni, V.; Kreusch, A.; Saez, E. The nuclear receptor LXR is a glucose sensor. Nature 2007, 445, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Bolten, C.; Bhat, B.G.; Woodring-Dietz, J.; Li, S.; Prayaga, S.K.; Xia, C.; Lala, D.S. Induction of human liver X receptor alpha gene expression via an autoregulatory loop mechanism. Mol. Endocrinol. 2002, 16, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Pawar, A.; Botolin, D.; Mangelsdorf, D.J.; Jump, D.B. The role of liver X receptor-alpha in the fatty acid regulation of hepatic gene expression. J. Biol. Chem. 2003, 278, 40736–40743. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, M.A.; Venkateswaran, A.; Tarr, P.T.; Xenarios, I.; Kudoh, J.; Shimizu, N.; Edwards, P.A. Characterization of the human ABCG1 gene: Liver X receptor activates an internal promoter that produces a novel transcript encoding an alternative form of the protein. J. Biol. Chem. 2001, 276, 39438–39447, Erratum in: J. Biol. Chem. 2002, 277, 17375. [Google Scholar] [CrossRef] [PubMed]
- Li, A.C.; Glass, C.K. PPAR- and LXR-dependent pathways controlling lipid metabolism and the development of atherosclerosis. J. Lipid Res. 2004, 45, 2161–2173. [Google Scholar] [CrossRef] [PubMed]
- Hetzel, M.; Walcher, D.; Grüb, M.; Bach, H.; Hombach, V.; Marx, N. Inhibition of MMP-9 expression by PPARγ activators in human bronchial epithelial cells. Thorax 2003, 58, 778–783. [Google Scholar] [CrossRef] [PubMed]
- Schroen, D.J.; Brinckerhoff, C.E. Nuclear hormone receptors inhibit matrix metalloproteinase (MMP) gene expression through diverse mechanisms. Gene Expr. 1996, 6, 197–207. [Google Scholar] [PubMed]
- Novak, U.; Cocks, B.G.; Hamilton, J.A. A labile repressor acts through the NF-κB-like binding sites of the human urokinase gene. Nucleic Acids Res. 1991, 19, 3389–3393. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Mitchell, P.; Tjian, R. Purified transcription factor AP-1 interacts with TPA-inducible enhancer elements. Cell 1987, 49, 741–752. [Google Scholar] [CrossRef]
- Thompson, J.E.S.; Webb, R.; Hewlett, P.; Llewellyn, D.; McDonnell, B.J. Matrix metalloproteinase-9 and augmentation index are reduced with an 8-week green-exercise walking programme. J. Hypertens. 2013, 2, 127–133. [Google Scholar] [CrossRef]
- Calabrese, E.J.; Baldwin, L.A. Defining hormesis. Hum. Exp. Toxicol. 2002, 21, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Radak, Z.; Chung, H.Y.; Koltai, E.; Taylor, A.W.; Goto, S. Exercise, oxidative stress and hormesis. Ageing Res. Rev. 2007, 7, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Ristow, M.; Zarse, K.; Oberbach, A.; Klöting, N.; Birringer, M.; Kiehntopf, M.; Stumvoll, M.; Kahn, C.R.; Blüher, M. Antioxidants prevent health-promoting effects of physical exercise in humans. Proc. Natl. Acad. Sci. USA 2009, 106, 8665–8670. [Google Scholar] [CrossRef] [PubMed]
- Gems, D.; Partridge, L. Stress-Response Hormesis and Aging: That Which Dones Not Kill Us Makes Us Stronger. Cell Metabolism 2008, 7, 200–203. [Google Scholar] [CrossRef] [PubMed]
- McArdle, F.; Spiers, S.; Aldemir, H.; Vasilaki, A.; Beaver, A.; Iwanejko, L.; McArdle, A.; Jackson, M.J. Preconditioning of skeletal muscle against contraction-induced damage: The role of adaptations to oxidants in mice. J. Physiol. 2004, 561 Pt 1, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Mahoney, D.J.; Parise, G.; Melov, S.; Safdar, A.; Tarnopolsky, M.A. Analysis of global mRNA expression in human skeletal muscle during recovery from endurance exercise. FASEB J. 2005, 19, 1498–1500. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.W.; Davies, N.A.; Moir, H.; Watkeys, L.; Ruffino, J.-S.; Isa, S.A.; Butcher, L.R.; Hughes, M.G.; Morris, K.; Webb, R. Exercise-associated generation of PPARγ ligands activates PPARγ signalling events and upregulates genes related to lipid metabolism. J. Appl. Physiol. 2012, 112, 806–815. [Google Scholar] [CrossRef] [PubMed]
- Ruffino, J.-S.; Davies, N.A.; Morris, K.; Ludgate, M.; Zhang, L.; Webb, R.; Thomas, A.W. Moderate-intensity exercise alters monocyte M1 and M2 marker gene expression in sedentary females: Putative roles for PPARγ and IL-6. Eur. J. Appl. Physiol. 2016, 116, 1671–1682. [Google Scholar] [CrossRef] [PubMed]
- Webb, R.; Thompson, J.E.S.; Ruffino, J.-S.; Davies, N.A.; Watkeys, L.; Hooper, S.; Jones, P.M.; Walters, G.; Clayton, D.; Thomas, A.W.; et al. An Evaluation of Cardiovascular Risk-Lowering Health Benefits Accruing from Laboratory-based, Community-based and Exercise-Referral Exercise Programmes. BMJ Open Sport Exerc. Med. 2016, 2, e000089. [Google Scholar] [CrossRef] [PubMed]
- Yakeu, G.; Butcher, L.; Isa, S.; Webb, R.; Roberts, A.; Thomas, A.W.; Backx, K.; James, P.E.; Morris, K. Low-intensity exercise triggers monocyte polarisation into the M2 anti-inflammatory phenotype. Atherosclerosis 2010, 212, 668–673. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.A. Central role of the adipocyte in the insulin-sensitizing and cardiovascular risk modifying actions of the thiazolidinediones. Biochimie 2003, 85, 1219–1230. [Google Scholar] [CrossRef] [PubMed]
- Brewer, H.B.; Remaley, A.T.; Neufeld, E.B.; Basso, F.; Joyce, C. Regulation of plasma high-density lipoprotein levels by the ABCA1 transporter and the emerging role of high-density lipoprotein in the treatment of cardiovascular disease. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1755–1760. [Google Scholar] [CrossRef] [PubMed]
- Tsekouras, Y.E.; Yanni, A.E.; Bougatsas, D.; Kavouras, S.A.; Sidossis, L.S. A single bout of brisk walking increases basal very low-density lipoprotein triacylglycerol clearance in young men. Metabolism 2007, 56, 1037–1043. [Google Scholar] [CrossRef] [PubMed]
- Szanto, A.; Balint, B.L.; Nagy, Z.S.; Barta, E.; Dezso, B.; Pap, A.; Szeles, L.; Poliska, S.; Oros, M.; Evans, R.M.; et al. STAT6 transcription factor is a facilitator of the nuclear receptor PPARγ-regulated gene expression in macrophages and dendritic cells. Immunity 2010, 33, 699–712. [Google Scholar] [CrossRef] [PubMed]
- Katzmarzyk, P.T.; Church, T.S.; Blair, S.N. Cardiorespiratory fitness attenuates the effects of the metabolic syndrome on all-cause and cardiovascular disease mortality in men. Arch. Intern. Med. 2004, 164, 1092–1097. [Google Scholar] [CrossRef] [PubMed]
- Schuch, F.B.; Vancampfort, D.; Richards, J.; Rosenbaum, S.; Ward, P.B.; Stubbs, B. Exercise as a treatment for depression: A meta-analysis adjusting for publication bias. Psychiatr. Res. 2016, 77, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Monteiro-Junior, R.S.; Cevada, T.; Oliveira, B.R.; Lattari, E.; Portugal, E.M.; Carvalho, A.; Deslandes, A.C. We need to move more: Neurobiological hypotheses of physical exercise as a treatment for Parkinson’s disease. Med. Hypotheses 2015, 85, 537–541. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, S.; Vasileva, L. Current and emerging strategies in osteoporosis management. Curr. Pharm. Des. 2017. [Google Scholar] [CrossRef]
- Ruiz-Casado, A.; Martín-Ruiz, A.; Pérez, L.M.; Provencio, M.; Fiuza-Luces, C.; Lucia, A. Exercise and the Hallmarks of Cancer. Trends Cancer 2017, 3, 423–441. [Google Scholar] [CrossRef] [PubMed]
- Olefsky, J.M.; Glass, C.K. Macrophages, inflammation, and insulin resistance. Annu. Rev. Physiol. 2010, 72, 219–246. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Chawla, A.; Pollard, J.W. Macrophage biology in development, homeostasis and disease. Nature 2013, 496, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Hevener, A.L.; Olefsky, J.M.; Reichart, D.; Nguyen, M.T.; Bandyopadyhay, G.; Leung, H.Y.; Watt, M.J.; Benner, C.; Febbraio, M.A.; Nguyen, A.K.; et al. Macrophage PPARγ is required for normal skeletal muscle and hepatic insulin sensitivity and full antidiabetic effects of thiazolidinediones. J. Clin. Investig. 2007, 117, 1658–1669. [Google Scholar] [CrossRef] [PubMed]
- Odegaard, J.I.; Ricardo-Gonzalez, R.R.; Goforth, M.H.; Morel, C.R.; Subramanian, V.; Mukundan, L.; Red Eagle, A.; Vats, D.; Brombacher, F.; Ferrante, A.W.; et al. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature 2007, 447, 1116–1120. [Google Scholar] [CrossRef] [PubMed]
- Meeusen, R. Exercise, nutrition and the brain. Sports Med. 2014, 1, S47–S56. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Zhang, X.; Huang, W.-J. Inflammation in neurodegenerative diseases. Mol. Med. Rep. 2016, 13, 3391–3396. [Google Scholar] [CrossRef] [PubMed]
- Noh, H.; Jeon, J.; Seo, H. Systemic injection of LPS induces region-specific neuroinflammation and mitochondrial dysfunction in normal mouse brain. Neurochem. Int. 2014, 69, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Waisman, A.; Ginhoux, F.; Greter, M. Homeostasis of Microglia in the Adult Brain: Review of Novel Microglia Depletion Systems. Trends Immunol. 2015, 36, 625–636. [Google Scholar] [CrossRef] [PubMed]
- Koizumi, S.; Shigemoto-Mogami, Y.; Nasu-Tada, K.; Shinozaki, Y.; Ohsawa, K.; Tsuda, M.; Joshi, B.V.; Jacobson, K.A.; Kohsaka, S.; Inoue, K. UDP acting at P2Y6 receptors is a mediator of microglial phagocytosis. Nature 2007, 446, 1091–1109. [Google Scholar] [CrossRef] [PubMed]
- Davalos, D.; Grutzendler, J.; Yang, G.; Kim, J.V.; Zuo, Y.; Jung, S.; Littman, D.R.; Dustin, M.L.; Gan, W.B. ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 2005, 8, 752–758. [Google Scholar] [CrossRef] [PubMed]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef] [PubMed]
- Orihuela, R.; McPherson, C.M.; Harry, G.J. Microglial M1/M2 polarization and metabolic states. Br. J. Pharmacol. 2016, 173, 649–665. [Google Scholar] [CrossRef] [PubMed]
- Faustino, J.V.; Wang, X.; Johnson, C.E.; Klibanov, A.; Derugin, N.; Wendland, M.F.; Vexler, Z.S. Microglial cells contribute to endogenous brain defenses after acute neonatal focal stroke. J. Neurosci. 2011, 31, 12992–13001. [Google Scholar] [CrossRef] [PubMed]
- Miró-Mur, F.; Pérez-de-Puig, I.; Ferrer-Ferrer, M.; Urra, X.; Justicia, C.; Chamorro, A.; Planas, A.M. Immature monocytes recruited to the ischemic mouse brain differentiate into macrophages with features of alternative activation. Brain Behav. Immun. 2016, 53, 18–33. [Google Scholar] [CrossRef] [PubMed]
- Wattananit, S.; Tornero, D.; Graubardt, N.; Memanishvili, T.; Monni, E.; Tatarishvili, J.; Miskinyte, G.; Ge, R.; Ahlenius, H.; Lindvall, O.; et al. Monocyte-Derived Macrophages Contribute to Spontaneous Long-Term Functional Recovery after Stroke in Mice. J. Neurosci. 2016, 36, 4182–4195. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Dong, Y.; Tucker, D.; Wang, R.; Ahmed, M.E.; Brann, D.; Zhang, Q. Treadmill Exercise Exerts Neuroprotection and Regulates Microglial Polarization and Oxidative Stress in a Streptozotocin-Induced Rat Model of Sporadic Alzheimer’s Disease. J. Alzheimers Dis. 2017, 56, 1469–1484. [Google Scholar] [CrossRef] [PubMed]
- Gleeson, M.; Bishop, N.C.; Stensel, D.J.; Lindley, M.R.; Mastana, S.S.; Nimmo, M.A. The anti-inflammatory effects of exercise: Mechanisms and implications for the prevention and treatment of disease. Nat. Rev. Immunol. 2011, 11, 607–615. [Google Scholar] [CrossRef] [PubMed]
- DREAM (Diabetes REduction Assessment with ramipril and rosiglitazone Medication) Trial Investigators. Effect of rosiglitazone on the frequency of diabetes in patients with impaired glucose tolerance or impaired fasting glucose: A randomised controlled trial. Lancet 2006, 368, 1096–1105. [Google Scholar] [CrossRef]
- Morton, J.P.; Kayani, A.C.; McArdle, A.; Drust, B. The exercise-induced stress response of skeletal muscle, with specific emphasis on humans. Sports Med. 2009, 39, 643–662. [Google Scholar] [CrossRef] [PubMed]
- Marinangeli, C.P.; Varady, K.A.; Jones, P.J. Plant sterols combined with exercise for the treatment of hypercholesterolemia: Overview of independent and synergistic mechanisms of action. J. Nutr. Biochem. 2006, 17, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.; O’Moore, K.M.; Dickman, J.R.; Ji, L.L. Exercise activation of muscle peroxisome proliferator-activated receptor-gamma coactivator-1alpha signaling is redox sensitive. Free Radic. Biol. Med. 2009, 47, 1394–1400. [Google Scholar] [CrossRef] [PubMed]
- Petersen, A.C.; McKenna, M.J.; Medved, I.; Murphy, K.T.; Brown, M.J.; Della Gatta, P.; Cameron-Smith, D. Infusion with the antioxidant N-acetylcysteine attenuates early adaptive responses to exercise in human skeletal muscle. Acta Physiol. (Oxf.) 2012, 204, 382–392. [Google Scholar] [CrossRef] [PubMed]
- Strobel, N.A.; Peake, J.M.; Matsumoto, A.; Marsh, S.A.; Coombes, J.S.; Wadley, G.D. Antioxidant supplementation reduces skeletal muscle mitochondrial biogenesis. Med. Sci. Sports Exerc. 2011, 43, 1017–1024. [Google Scholar] [CrossRef] [PubMed]
- Cardenia, V.; Rodriguez-Estrada, M.T.; Lorenzini, A.; Bandini, E.; Angeloni, C.; Hrelia, S.; Malaguti, M. Effect of broccoli extract enriched diet on liver cholesterol oxidation in rats subjected to exhaustive exercise. J. Steroid. Biochem. Mol. Biol. 2017, 169, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Ryan, M.J.; Dudash, H.J.; Docherty, M.; Geronilla, K.B.; Baker, B.A.; Haff, G.G.; Cutlip, R.G.; Alway, S.E. Vitamin E and C supplementation reduces oxidative stress, improves antioxidant enzymes and positive muscle work in chronically loaded muscles of aged rats. Exp. Gerontol. 2010, 45, 882–895. [Google Scholar] [CrossRef] [PubMed]
- Medved, I.; Brown, M.J.; Bjorksten, A.R.; Murphy, K.T.; Petersen, A.C.; Sostaric, S.; Gong, X.; McKenna, M.J. N-acetylcysteine enhances muscle cysteine and glutathione availability and attenuates fatigue during prolonged exercise in endurance-trained individuals. J. Appl. Physiol. 2004, 97, 1477–1485. [Google Scholar] [CrossRef] [PubMed]
- Reid, M.B.; Stokić, D.S.; Koch, S.M.; Khawli, F.A.; Leis, A.A. N-acetylcysteine inhibits muscle fatigue in humans. J. Clin. Investig. 1994, 94, 2468–2474. [Google Scholar] [CrossRef] [PubMed]
- Yfanti, C.; Akerström, T.; Nielsen, S.; Nielsen, A.R.; Mounier, R.; Mortensen, O.H.; Lykkesfeldt, J.; Rose, A.J.; Fischer, C.P.; Pedersen, B.K. Antioxidant supplementation does not alter endurance training adaptation. Med. Sci. Sports Exerc. 2010, 42, 1388–1395. [Google Scholar] [CrossRef] [PubMed]
- Petersen, E.W.; Ostrowski, K.; Ibfelt, T.; Richelle, M.; Offord, E.; Halkjaer-Kristensen, J.; Pedersen, B.K. Effect of vitamin supplementation on cytokine response and on muscle damage after strenuous exercise. Am. J. Physiol. Cell Physiol. 2001, 280, C1570–C1575. [Google Scholar] [PubMed]
- Higashida, K.; Kim, S.H.; Higuchi, M.; Holloszy, J.O.; Han, D.H. Normal adaptations to exercise despite protection against oxidative stress. Am. J. Physiol. Endocrinol. Metab. 2011, 301, E779–E784. [Google Scholar] [CrossRef] [PubMed]
- Roberts, L.A.; Beattie, K.; Close, G.L.; Morton, J.P. Vitamin C consumption does not impair training-induced improvements in exercise performance. Int. J. Sports Physiol. Perform. 2011, 6, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Theodorou, A.A.; Nikolaidis, M.G.; Paschalis, V.; Koutsias, S.; Panayiotou, G.; Fatouros, I.G.; Koutedakis, Y.; Jamurtas, A.Z. No effect of antioxidant supplementation on muscle performance and blood redox status adaptations to eccentric training. Am. J. Clin. Nutr. 2011, 93, 1373–1383. [Google Scholar] [CrossRef] [PubMed]
- Nikolaidis, M.G.; Kerksick, C.M.; Lamprecht, M.; McAnulty, S.R. Does Vitamin C and E supplementation impair the favorable adaptations of regular exercise? Oxid. Med. Cell. Longev. 2012, 707941, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Cabrera, M.C.; Ristow, M.; Vina, J. Antioxidant supplements in exercise: Worse than useless? Am. J. Physiol. Endocrinol. Metab. 2012, 302, E476–E477. [Google Scholar] [CrossRef] [PubMed]
- Sobal, J.; Marquart, L.F. Vitamin/mineral supplement use among athletes: A review of the literature. Int. J. Sport Nutr. 1994, 4, 320–334. [Google Scholar] [CrossRef] [PubMed]
- National Institutes of Health (NIH) Office of Dietary Supplements Fact Sheets. Available online: https://ods.od.nih.gov/factsheets/list-all/ (accessed on 30 June 2017).
- UK National Health Service ”NHS Choices” Website. Available online: http://www.nhs.uk/Conditions/vitamins-minerals/Pages/Vitamin-E.aspx (accessed on 30 June 2017).
- Bendich, A. Food and Nutrition Board Panel on Antioxidants and Related Compounds, Institute of Medicine and National Academy of Sciences. In Dietary Reference Intakes for Vitamin C, Vitamin E, Selenium, and Carotenoids, 1st ed.; Glade, M.J., Ed.; National Academies Press: Washington, DC, USA, 2000; Volume 17, p. 364. ISBN 978-0-309-06935-9. [Google Scholar]
- Novotny, J.A.; Fadel, J.G.; Holstege, D.M.; Furr, H.C.; Clifford, A.J. This kinetic, bioavailability, and metabolism study of RRR-α-tocopherol in healthy adults suggests lower intake requirements than previous estimates. J. Nutr. 2012, 142, 2105–2111. [Google Scholar] [CrossRef] [PubMed]
- Jubri, Z.; Latif, A.A.; Top, A.G.; Ngah, W.Z. Perturbation of cellular immune functions in cigarette smokers and protection by palm oil Vitamin E supplementation. Nutr. J. 2013, 12, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Baker, H.; DeAngelis, B.; Baker, E.; Khalil, M.; Frank, O. Human plasma patterns during 14 days ingestion of Vitamin E, beta-carotene, ascorbic acid, and their various combinations. J. Am. Coll. Nutr. 1996, 15, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Levine, M.; Conry-Cantilena, C.; Wang, Y.; Welch, R.W.; Washko, P.W.; Dhariwal, K.R.; Park, J.B.; Lazarev, A.; Graumlich, J.F.; King, J.; et al. Vitamin C pharmacokinetics in healthy volunteers: Evidence for a recommended dietary allowance. Proc. Natl. Acad. Sci. USA 1996, 93, 3704–3709. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Moreno, C.; Cano, M.P.; de Ancos, B.; Plaza, L.; Olmedilla, B.; Granado, F.; Martín, A. Effect of orange juice intake on Vitamin C concentrations and biomarkers of antioxidant status in humans. Am. J. Clin. Nutr. 2003, 78, 454–460. [Google Scholar] [PubMed]
- Sánchez-Moreno, C.; Cano, M.P.; Ancos, B.; Plaza, L.; Olmedilla, B.; Granado, F.; Martín, A. High-pressurized orange juice consumption affects plasma Vitamin C, antioxidative status and inflammatory markers in healthy humans. J. Nutr. 2003, 133, 2204–2209. [Google Scholar] [PubMed]
- Njus, D.; Kelley, P.M. Vitamins C and E donate single hydrogen atoms in vivo. FEBS Lett. 1991, 284, 147–151. [Google Scholar] [CrossRef]
- Wolber, F.M.; McGrath, M.; Jackson, F.; Wylie, K.; Broomfield, A. Cysteic Acid in Dietary Keratin is Metabolized to Glutathione and Liver Taurine in a Rat Model of Human Digestion. Nutrients 2016, 8, 104. [Google Scholar] [CrossRef] [PubMed]
- American College of Sports Medicine and the American Diabetes Association. Exercise and Type 2 Diabetes. Joint Position Statement. Med. Sci. Sports Exerc. 2010, 42, 2282–2303. [Google Scholar] [CrossRef]
- Moore, G.F.; Raisanen, L.; Moore, L.; Ud Din, N.; Murphy, S. Mixed-method process evaluation of the Welsh National Exercise Referral Scheme. Health Educ. 2013, 113, 476–501. [Google Scholar] [CrossRef]
- Hagger, M.S.; Wood, C.W.; Stiff, C.; Chatzisarantis, N.L.D. Self-regulation and self-control in exercise: The strength-energy model. Int. Rev. Sport Exerc. Psychol. 2010, 3, 62–86. [Google Scholar] [CrossRef]
- Tierney, S.; Mamas, M.; Woods, S.; Rutter, M.K.; Gibson, M.; Neyses, L.; Deaton, C. What strategies are effective for exercise adherence in heart failure? A systematic review of controlled studies. Heart Fail Rev. 2012, 17, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Nissen, S.E.; Wolski, K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N. Engl. J. Med. 2007, 356, 2457–2471. [Google Scholar] [CrossRef] [PubMed]
- Leal, I.; Romio, S.A.; Schuemie, M.; Oteri, A.; Sturkenboom, M.; Trifirò, G. Prescribing pattern of glucose lowering drugs in the United Kingdom in the last decade: A focus on the effects of safety warnings about rosiglitazone. Br. J. Clin. Pharmacol. 2013, 75, 861–868. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Study [Reference] | Exercise Protocol | Cohort Characteristics (Species/Age/Gender/‘n’) | Type/Dose of Antioxidant Supplementatn | Oxidative Stress Analysis | Endpoints (Training or Biomarker-related) | Impact of Supplementatn? |
|---|---|---|---|---|---|---|
| Strobel, 2011 [89] | 14 weeks running (4 sessions, 90 min/week) | Untrained male rats (2.5 months; n = 12) | Vit E (1000 IU/kg diet) plus α-lipoic acid (1.6 g/kg diet) | SOD, GPx, Xanthine Oxidase, MDA (muscle) | Mitochondrial biogenesis PPARγ target gene expressn | Suppressn by antiox Suppressn by antiox |
| Higashida, 2011 [96] | 3 weeks swimming (6 sessions of 6 h/week) | Untrained male rats (3 months, n = 3–6) | Vit C (750 mg/kg bw/day) plus Vit E (150 mg/kg bw/day) | TBARS, MnSOD, CuZn SOD (muscle) | Mitochondrial proteins GLUT4 glucose transport | No Impact of antiox No Impact of antiox |
| Ryan, 2010 [91] | 4.5 weeks maximal contractions (3 sessions of 80 contractions/week) | Untrained male rats (3 or 30 months, n = 7) | Vit C (20 g/kg bw/day) plus Vit E (30 g/kg bw/day) | H2O2, MDA, SOD, Glutathione, GPx, catalase (muscle) | Muscle function (positive work) | Improvement in aged rats (beyond ex alone) by antiox |
| Cardenia, 2017 [84] | 1 week running (1 session of 10 min/week → single exhaustive exercise bout) | Trained female rats (4 months; n = 32) | Broccoli extract (2.5 mg/g of diet) | Glutathione, GPx, catalase, oxysterols (liver) | Tissue damage | Prevention of ex-induced tissue damage by antiox |
| Kang, 2009 [81] | 3 weeks running (3 sessions of 15 min/week → single exhaustive exercise bout) | Trained female rats (4 months; n = 9) | Allopurinol (0.4 mmol/kg) | H2DCFDA, xanthine oxidase, glutathione (muscle) | PPARγ target gene expressn NFkB signaling activation Mitochondrial transcriptn | Suppressn by antiox Suppressn by antiox Suppressn by antiox |
| Gomez-Cabrera, 2008 [80] | 3–6 weeks running (5 sessions, 25–85 min/week) 8 weeks cycling (3 sessions of 40 min/week) | Untrained male rats (3 months, n = 6) Untrained male humans (29–31 years, n = 5–9) | Vit C (500 mg/kg bw/day) Vit C (1000 mg/day) | SOD, GPx (muscle) | VO2max Running time Cytochrome C expressn | No Impact of antiox Suppressn by antiox Suppressn by antiox |
| Ristow, 2009 [50] | 4 weeks circuit training (5 sessions, 65 min/week) | Untrained/moderately trained male humans (26 years; n = 10) | Vit C (1000 mg/day) plus Vit E (400 IU/day) | TBARS (plasma/muscle), SOD, GPx, catalase (muscle) | Insulin sensitivity PPARγ target gene expressn | Suppressn by antiox Suppressn by antiox |
| Davies, 2015 [6] | Cycling (1 bout of 45 min at 70% VO2max) | Moderately trained male humans (32 years; n = 5) | Vit C (1000 mg/day) plus Vit E (400 IU/day) | H2DCFDA (monocytes) | Monocyte [ROS]cyto PPARγ target gene expressn | No Impact of antiox Suppressn by antiox |
| Khassaf, 2003 [34] | Cycling (1 bout of 45 min at 70% VO2max) | Untrained male humans (28 years; n = 16) | Vit C (500 mg/day) | HSP (muscle); SOD, CAT, HSP (lymphocytes) | Heat-Shock Protein expressn | Suppressn by antiox |
| Petersen, 2001 [107] | Running (1 bout of 90 min at 75% VO2max) | Moderately trained male humans (26–28 years; n = 20) | Vit C (500 mg/day) plus Vit E (400 mg/day) | HPLC quantitation of Vit C and Vit E (plasma) | IL-6 and IL-1RA expressn Muscle damage Lymphocyte counts | No Impact of antiox No Impact of antiox No Impact of antiox |
| Medved, 2004 [87] | Cycling (1 session of 45 min → exhaustive exercise bout) | Untrained male humans (27 years; n = 8) | N-acetyl cysteine (infusion at 25–125 mg/kg/h) | N-acetyl cysteine, cystine, glutathione cysteine (muscle) | Time to fatigue | Extension by antiox |
| Reid, 1994 [88] | Electrical muscle stimulatn (1–120 Hz; 0.2 msec pulses) | Untrained male humans (32 years; n = 10) | N-acetyl cysteine (infusion at 150 mg/kg/h) | - | Time to fatigue; force generation when fatigued | Improvement by antiox |
| Yfanti, 2010 [90] | 12 weeks cycling (5 sessions of 40–60 min/week) | Moderately trained male humans (29–31 years; n = 10–11) | Vit C (500 mg/day) plus Vit E (400 IU/day) | MDA, carbonyls, SOD, GPx, catalase (muscle) | VO2max/Body compositn/Glycogen content/Mito-chondrial proteins/Insulin sensitivity/Plasma lipids | No Impact of antiox in all cases |
| Roberts, 2011 [97] | 4 weeks 50–90% VO2max interval running (4 sessions of 50 min/week) | Moderately trained male humans (21–23 years; n = 7–8) | Vit C (1000 mg/day) | - | Performance tests Substrate metabolism | No Impact of antiox No Impact of antiox |
| Theodorou, 2011 [98] | 16 weeks resistance training (2 sessions of 75 contractions/week) | Moderately trained male humans (26 years; n = 14) | Vit C (1000 mg/day) plus Vit E (400 IU/day) | TBARS, carbonyls, glutathione, uric acid, catalase, TAC (plasma) | Muscle function Muscle damage | No Impact of antiox No Impact of antiox |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Webb, R.; Hughes, M.G.; Thomas, A.W.; Morris, K. The Ability of Exercise-Associated Oxidative Stress to Trigger Redox-Sensitive Signalling Responses. Antioxidants 2017, 6, 63. https://doi.org/10.3390/antiox6030063
Webb R, Hughes MG, Thomas AW, Morris K. The Ability of Exercise-Associated Oxidative Stress to Trigger Redox-Sensitive Signalling Responses. Antioxidants. 2017; 6(3):63. https://doi.org/10.3390/antiox6030063
Chicago/Turabian StyleWebb, Richard, Michael G. Hughes, Andrew W. Thomas, and Keith Morris. 2017. "The Ability of Exercise-Associated Oxidative Stress to Trigger Redox-Sensitive Signalling Responses" Antioxidants 6, no. 3: 63. https://doi.org/10.3390/antiox6030063
APA StyleWebb, R., Hughes, M. G., Thomas, A. W., & Morris, K. (2017). The Ability of Exercise-Associated Oxidative Stress to Trigger Redox-Sensitive Signalling Responses. Antioxidants, 6(3), 63. https://doi.org/10.3390/antiox6030063